Play all audios:
ABSTRACT Down syndrome (DS) is caused by the trisomy of chromosome 21. Among the many disabilities found in individuals with DS is an increased risk of early-onset Alzheimer's disease
(AD). Although higher oxidative stress and an upregulation of amyloid β (Aβ) peptides from an extra copy of the _APP_ gene are attributed to the AD susceptibility, the relationship between
the two factors is unclear. To address this issue, we established an in vitro cellular model using neurons differentiated from DS patient-derived induced pluripotent stem cells (iPSCs) and
isogenic euploid iPSCs. Neurons differentiated from DS patient-derived iPSCs secreted more Aβ compared to those differentiated from the euploid iPSCs. Treatment of the neurons with an
antioxidant, N-acetylcysteine, significantly suppressed the Aβ secretion. These findings suggest that oxidative stress has an important role in controlling the Aβ level in neurons
differentiated from DS patient-derived iPSCs and that N-acetylcysteine can be a potential therapeutic option to ameliorate the Aβ secretion. SIMILAR CONTENT BEING VIEWED BY OTHERS
4-PHENYLBUTYRATE AMELIORATES APOPTOTIC NEURAL CELL DEATH IN DOWN SYNDROME BY REDUCING PROTEIN AGGREGATES Article Open access 20 August 2020 TAU DEPLETION IN HUMAN NEURONS MITIGATES AΒ-DRIVEN
TOXICITY Article Open access 15 February 2024 _APOE2_ MITIGATES DISEASE-RELATED PHENOTYPES IN AN ISOGENIC HIPSC-BASED MODEL OF ALZHEIMER’S DISEASE Article Open access 09 April 2021
INTRODUCTION Down syndrome (DS) is the chromosome abnormality defined by an extra copy of chromosome 21. DS develops various complications such as neurological, skeletal, cardiovascular, and
immunological defects1, but is perhaps best known for being the most common genetic cause of mental retardation and intellectual disability2, occurring at a rate of 1 in 800 to 1000
births3,4. Chromosome 21 contains genes related to neurodegenerative diseases and oxidative stress5, and research on the involvement of these genes on the pathophysiology of DS is underway6.
Consistent with the neurological complications and intellectual disabilities is that individuals with DS have a higher susceptibility to early onset Alzheimer's disease (AD)7,8. One of
the hypothesized reasons is the existence of an extra copy of _amyloid precursor protein (APP)_ gene located on chromosome 21. APP is a precursor of amyloid-beta (Aβ), and the extra copy
increases the expression of _APP_ and subsequent production of Aβ9,10. Since AD-like pathological lesions such as Aβ deposition can be recognized in individuals with DS younger than 40 years
old and symptoms of cognitive impairment due to AD exponentially increases from this age, DS has been regarded as a “young model” of AD1,6,11. Connecting AD and DS is Aβ42, which plays an
important role in the pathogenesis of AD and may also affect the cognitive function seen in DS12,13. It has been suggested that the brains of individuals with DS are exposed to oxidative
stress14,15,16. Oxidative stress plays a central role in neurogenic changes in DS4,17. In animal studies, the administration of antioxidants from the fetal period increases the number of
cells in the hippocampus18, indicating that early intervention to circumvent the effect of oxidative stress may be important to improve the neurological prognosis. Oxidative stress in DS is
attributed to several genes on chromosome 21, including an extra copy of _superoxide dismutase 1 (SOD1)_19. Oxidative stress is enhanced because of the accumulation of H2O2. The
overexpression of _SOD1_ produces a large amount of H2O2 that cannot be catalyzed because the copy number of _catalase_ is normal19. However, in experiments using DS model mice, oxidative
stress was increased even when only two copies of _SOD1_ existed20. Therefore, although oxidative stress is caused by the overexpression of _SOD1_, other factors are also involved. The
relationship between oxidative stress and Aβ is complicated. Oxidative stress may be involved in Aβ production21, and Aβ42 oligomers are known to produce reactive oxygen species (ROS) and
have neurotoxicity22,23,24. Accurate evaluation of the regulatory role of oxidative stress on the production of Aβ production is therefore important for establishing an appropriate
therapeutic strategy for DS-related neuronal degeneration. In this study, we differentiated induced pluripotent stem cells (iPSCs) derived from individuals with DS (D-iPSCs) into neurons
(D-iNs) and measured their Aβ production. Secreted Aβ was increased in D-iNs, and the amount of Aβ was reduced by a high-dose administration of the antioxidant N-acetylcysteine (NAC).
Similar results were obtained from a human embryonic stem cell (ESC) line with trisomy 21. Although many animal models of DS have been constructed, no model animal can completely trace the
symptoms occurring in humans. Our study succeeded in establishing an in vitro human model to evaluate the effect of oxidative stress on neurons with trisomy 21. RESULTS CONVERSION OF D-IPSCS
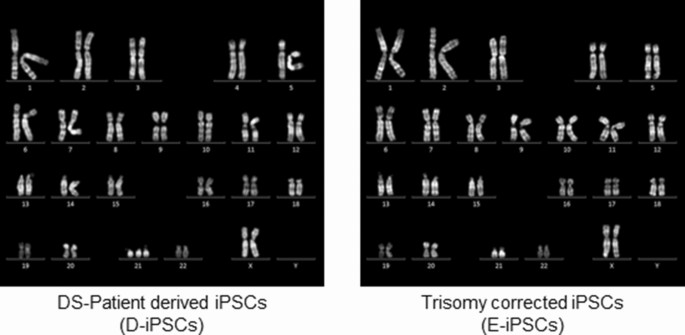
INTO NEURONS To precisely evaluate the phenotype of iNs, we used D-iPSCs and an isogenic euploid control clone established from the D-iPSCs (E-iPSCs)25 (Fig. 1). These iPSCs were then
directly converted into neuronal lineage cells by the transient overexpression of _Neurogenin 2_ (_NGN2_) gene26,27 (Fig. 2a). For this, we incorporated a doxycycline-inducible expression
vector encoding _NGN2_ and mCherry fluorescent protein into the iPSC clones and treated the clones with doxycycline for 5 days (Fig. 2a). At day 5, almost all cells were positive for mCherry
(Fig. 2b) and showed a compatible morphological appearance with neurons, such as elongated neurites (Fig. 2b,c). At day 8, almost all cells expressed an intermediate neuronal progenitor
cell, marker Tbr2, and were positive for a neuron-specific human tubulin protein, Tuj1 (Fig. 2d,e). These observations confirmed the neuronal differentiation of D-iPSCs and E-iPSCs. D-INS
SECRETE A LARGER AMOUNT OF AΒ We next evaluated the secretion of Aβ peptides Aβ40 and Aβ42 from iNs. β-secretase-1 (beta-site APP cleaving enzyme; BACE1) and γ-secretase cleave APP to
produce Aβ peptides, and Aβ peptides exert neurotoxicity through various proposed mechanisms28,29,30,31. In particular, Aβ42 plays an essential and important role in all AD. Aβ42 is highly
hydrophobic and has high aggregation properties. Additionally, a high Aβ42/Aβ40 ratio is associated with the formation of amyloid plaques found in familial AD patients12,32. We therefore
evaluated the amount of Aβ secretion from D-iNs and E-iNs, finding the amount of secreted Aβ40 and Aβ42 was higher in D-iNs at all times observed (Fig. 3a,b). However, the Aβ42/Aβ40 ratio
was not different between the two iN types (Fig. 3c). These data indicated that D-iNs are predisposed to secrete more Aβ protein. OXIDATIVE STRESS AFFECTS THE AMOUNT OF SECRETED AΒ FROM
D-INS The brains of individuals with DS are exposed to high oxidative stress. The deposition of Aβ increases oxidative stress33,34,35, and the existence of an extra copy of _superoxide
dismutase 1 (SOD1)_ causes the overproduction of H2O2. Consistently, antioxidants and catalase show neuroprotective effects in individuals with DS and model mice14,18. Therefore, we
investigated the effect of an antioxidant and an oxidant on the amount of Aβ secretion from iNs. NAC treatment decreased Aβ secretion at the highest dose (Fig. 4a,b). On the contrary, H2O2
treatment significantly increased the secretion of Aβ protein from both D-iNs and E-iNs (Fig. 4d,e). Interestingly, both NAC and H2O2 treatment decreased the Aβ42/Aβ40 ratio (Fig. 4c,f).
These findings show oxidative stress positively correlates with the secretion of Aβ protein from iNs. EFFECT OF OXIDATIVE STRESS ON THE EXPRESSION OF _APP_ We next examined whether oxidative
stress affects the expression of _APP_ in iNs. _APP_ was upregulated during the neuronal differentiation of D-iPSCs, as expected (Fig. 5a). H2O2 treatment upregulated the expression of
_APP_ gene both in D-iNs and E-iNs, but the effect was more prominent in D-iNs (Fig. 5b). NAC treatment also upregulated _APP_ expression (Fig. 5a), indicating that the NAC effect is related
to posttranscriptional modifications. The expression of _SOD1_ was upregulated by treatment with H2O2 or NAC (Fig. 5c,d). VERIFICATION WITH ANOTHER ISOGENIC PSC PAIR To confirm that the
increased Aβ secretion and the inhibitory effect of high-dose NAC are not a clone-specific phenomenon, we tested the reproducibility of the results using another isogenic PSC pair. We used
KhES1, a euploid human ESC line, and a subclone of KhES1 in which chromosome 21 was artificially inserted to make trisomy 2136. We introduced a doxycycline-inducible _NGN2_ expression vector
into these clones and induced neuronal differentiation. Both clones showed good NGN2-dependent differentiation properties and differentiated into neurons expressing Tuj1, MAP2 and Tbr1
(Fig. 6a,b). As in the case with the DS-derived clones, Aβ secretion was increased in a time-dependent manner and higher in the trisomy clone after day 12 (Fig. 6c–e). NAC administration
reduced the production of Aβ42 but had no significant effect on Aβ40 (Fig. 6f–h). These results indicate that the increased secretion of Aβ from trisomy 21 and the inhibitory effect of
high-dose NAC are not clone- dependent. DISCUSSION Here we investigated the effect of trisomy 21 on neuronal Aβ by using trisomy 21 iPSCs and their isogenic euploid control and
differentiating them into neurons. Aβ was produced early after initiating the direct conversion and higher in D-iNs. In addition, the Aβ production was reduced by using an antioxidant, NAC.
The brains of individuals with DS show pathological changes similar to those of AD patients11,37. In familial AD, the proportion of Aβ42 increases from the early stage due to genetic
abnormalities of PSEN1 or PSEN2. A relative increase of Aβ42 to Aβ40 has been considered a risk of synaptic dysfunction12,22,29. On the other hand, in DS, the total amount of both Aβ40 and
Aβ42 increases due to the increased copy number of _APP_ caused by extra chromosome 21. Therefore, the Aβ42/Aβ40 ratio is considered unchanged, especially in the early stage38, which is
consistent with our findings. Treatment with various antioxidants has been tried against the increased oxidative stress in DS. However, the effects were often partial or limited in animal
models39,40,41,42. In the present study, we found that NAC treatment dose-dependently reduced the production of Aβ from iNs, which is consistent with previous studies on DS and AD. NAC is a
precursor of glutathione peroxidase and is known as an antioxidant that can prevent the enhanced death due to oxidative stress of neurons derived from D-iPSCs17. NAC treatment was also seen
to improve the cognitive memory behavior of AD model mice and rats43,44 and to suppress neuroinflammation45. Therefore, NAC itself can reduce oxidative stress in the whole brain and exert a
neuronal cell protective effect. Adding our study gives further argument to NAC improving the prognosis of the cognitive function of DS. Although the addition of NAC suppressed the secretion
of Aβ, it did not down-regulate the expression of _APP_. This finding suggests that the anti-oxidative stress effect of NAC may affect the cleavage of _APP_ protein post-transcriptionally.
Aβ production is known to be affected by oxidative stress, and increased oxidative stress up-regulates _BACE1_ expression46 and _PSEN1_ expression in lipid rafts47,48. Since PSEN1 is an
active center of γ-secretase, the activity of γ-secretase may also be increased by oxidative stress21. In addition, the β and γ secretase-dependent processing of _APP_ is promoted by
α-synuclein49. These reports and our data indicate that, in addition to an extra copy of _APP_, the brains of individuals with DS are exposed to an unfavorable environment where Aβ
production is enhanced due to increased oxidative stress. In anticipation of the antioxidant effect of NAC, clinical trials have been conducted on various neuropsychiatry diseases and
neurodegenerative diseases50,51,52. In this study, we showed the effect of NAC in iNs, suggesting that the neuroprotective effect of NAC can be expanded to diseases associated with Aβ and
oxidative stress. However, our study is based upon a relatively small number of control and DS cases and it will be important for this work to be replicated using larger sample sizes.
MATERIALS AND METHODS ETHICS STATEMENT This study was approved by the Ethics Committee and the recombinant DNA Experiments Safety Committee of Kyoto University. The use of human ESCs was
approved by the Ministry of Education Culture, Sports, Science and Technology of Japan (MEXT). All methods were performed in accordance with the relevant guidelines and regulations. Informed
consent was obtained from the legal guardians of the DS patient. IPSC CLONES AND INTRODUCTION OF THE DOXYCYCLINE-INDUCIBLE NGN2 EXPRESSION VECTOR We used an iPSC clone obtained from a DS
patient and a trisomy-corrected isogenic clone generated from the trisomy clone as previously described25. The source fibroblasts of the iPSCs were obtained from the Coriell Institute for
Medical Research (AG06892). As another isogenic pair, we used an euploid ESC line KhES1 and trisomy KhES1 subclone with chromosome 21 artificially introduced36,53. The original euploid KhES1
clone was kindly provided by Hirofumi Suemori (Institute for Frontier Medical Sciences, Kyoto University, Kyoto, Japan). For the generation of iPSC-derived neurons, we took advantage of the
doxycycline-induced _NGN2_ expression system26,27. We constructed a plasmid vector encoding human NGN2 cDNA under the tetracycline-inducible promoter (_TetO::NGN2-IRES-mCherry_). The
piggyBac backbone vector, PB-TAC-ERN, was a gift from Dr. Knut Woltjen (Addgene plasmid #80475; http://n2t.net/addgene:80475; RRID: Addgene_80475)26. We selected subclones showing high
differentiation ability after the antibiotic selection with G418 disulfate (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan). NEURONAL INDUCTION AND SAMPLE TAKING Neuronal induction by
doxycycline-inducible _NGN2_ expression was performed as previously described27. Briefly, on day 0, iPSCs were dissociated with TrypLE Select (GIBCO, Thermo Fisher Scientific, MA, USA) and
disseminated on a mixed coating of poly-l-lysine (final 0.0002% v/w, Merck, Darmstadt, Germany), and Matrigel (final 2% v/v, Corning, NY, USA). The disseminated iPSCs were cultured in
Neurobasal Medium (GIBCO, Thermo Fisher Scientific, MA, USA) supplemented with 0.5% B27 without vitamin A (GIBCO, Thermo Fisher Scientific, MA, USA), 1 × Glutamax (GIBCO, Thermo Fisher
Scientific, MA, USA), 2 mg/mL doxycycline hydrochloride (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan), and 5 mM Y-27632 (Nacalai-Tesque, Kyoto, Japan). Y-27632 was withdrawn on day
1 or 2. On day 5, we exchanged culture media for new media without doxycycline hydrochloride. To evaluate the amount of Aβ between timepoints, on day 8, all culture media were replaced with
fresh medium. On days 10, 12 and 14, we recovered old media as samples for the ELISA analysis. To check the effect of an oxidant (H2O2) or anti-oxidant (NAC), all culture media were
replaced with fresh medium containing H2O2 or NAC on day 8. The culture media were subjected to analysis on day 10. ENZYME-LINKED IMMUNOSORBENT ASSAY (ELISA) Aβ40 and Aβ42 peptides were
quantified using human Aβ40 and Aβ42 commercially available ELISA kits from Immuno-Biological Laboratories (Gumma, Japan). ELISA measurements were performed according to the
manufacturer's instructions. Biological triplicates were obtained from supernatants derived from separately differentiated neurons. Technical duplicates were obtained by separating
supernatant samples into two wells of primary antibody-conjugated plates. IMMUNOFLUORESCENT STAINING Cells were washed in D-PBS and then fixed in 4% paraformaldehyde at 4 ºC for 15 min.
After washing the cells twice in D-PBS, we incubated the cells for 30 min in 0.025% Triton-10 diluted with blocking reagent (Block Ace, KAC, Kyoto, Japan) at room temperature. Then, primary
antibodies were applied overnight at 4 ºC after washing in D-PBS twice. The next day, cells were washed and incubated with secondary antibodies for 1 h at room temperature. Finally, the
cells were counterstained with DAPI at room temperature. The rate of Tbr2-positive cells was calculated using ImageJ software. Using the "analyze particle" function of Image J, we
set DAPI positive areas as regions of interest (ROIs). For E-iNs and D-iNs, 69–80 and 69–88 ROIs were counted in each experiment, respectively. The ratio of Tbr2-positive cells to
DAPI-positive cells was calculated. QUANTITATIVE PCR RNA samples were prepared with the RNeasy Mini Kit (Qiagen, Hilden, Germany) and subjected to reverse transcription with a PrimeScript RT
Master Mix (Takara Bio, Shiga, Japan). All procedures were performed following the manufacturer's instructions. Quantitative PCR (qPCR) was performed on the StepOnePlus Real-Time PCR
System (Applied Biosystems, Thermo Fisher Scientific, MA, USA). For the detection of transgenes, DNA was subjected to qPCR, and SYBR Premix ExTaqII (Takara Bio, Shiga, Japan) was used for
the detection. Data were processed using the ΔΔ cycle threshold method, converted 2-ΔΔCt values, and the relative quantities are shown in the figures. The primer sequences are shown below.
SOD1 (f) ACAAAGATGGTGTGGCCGAT SOD1 (re) AACGACTTCCAGCGTTTCCT APP (f) GACCACTCGACCAGGTTCTG APP (re) GCCCACCATGAGTCCAATGA GAPDH (f) AATCCCATCACCATCTTCCA GAPDH (re) TGGACTCCACGACGTACTCA
STATISTICAL ANALYSIS GraphPad Prism8 (GraphPad Software, La Jolla, CA, USA) was used for the analysis. All results represent means ± SEM. “n” represents the number of independent cultures.
Statistical analysis was performed using Student’s t-test, two-way ANOVA and post-hoc tests. A p value less than 0.05 was considered significant. REFERENCES * Barca, D. _et al._ Intellectual
disability and epilepsy in Down syndrome. _Maedica_ 9, 344–350 (2014). PubMed PubMed Central Google Scholar * AgarwalGupta, N. & Kabra, M. Diagnosis and management of Down syndrome.
_Indian J. Pediatr._ 81, 560–567. https://doi.org/10.1007/s12098-013-1249-7 (2014). Article Google Scholar * Carmona-Iragui, M., Videla, L., Lleo, A. & Fortea, J. Down syndrome,
Alzheimer disease, and cerebral amyloid angiopathy: The complex triangle of brain amyloidosis. _Dev. Neurobiol._ 79, 716–737. https://doi.org/10.1002/dneu.22709 (2019). Article PubMed
Google Scholar * Haydar, T. F. & Reeves, R. H. Trisomy 21 and early brain development. _Trends Neurosci._ 35, 81–91. https://doi.org/10.1016/j.tins.2011.11.001 (2012). Article CAS
PubMed Google Scholar * Lott, I. T., Head, E., Doran, E. & Busciglio, J. Beta-amyloid, oxidative stress and Down syndrome. _Curr Alzheimer Res._ 3, 521–528.
https://doi.org/10.2174/156720506779025305 (2006). Article CAS PubMed Google Scholar * Vacca, R. A. _et al._ Down syndrome: Neurobiological alterations and therapeutic targets.
_Neurosci. Biobehav. Rev._ 98, 234–255. https://doi.org/10.1016/j.neubiorev.2019.01.001 (2019). Article CAS PubMed Google Scholar * Wiseman, F. K. _et al._ A genetic cause of Alzheimer
disease: mechanistic insights from Down syndrome. _Nat. Rev. Neurosci._ 16, 564–574. https://doi.org/10.1038/nrn3983 (2015). Article CAS PubMed PubMed Central Google Scholar * Hartley,
D. _et al._ Down syndrome and Alzheimer’s disease: Common pathways, common goals. _Alzheimers Dement._ 11, 700–709. https://doi.org/10.1016/j.jalz.2014.10.007 (2015). Article PubMed Google
Scholar * Lott, I. T. & Dierssen, M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. _Lancet Neurol._ 9, 623–633.
https://doi.org/10.1016/s1474-4422(10)70112-5 (2010). Article PubMed Google Scholar * Head, E., Helman, A. M., Powell, D. & Schmitt, F. A. Down syndrome, beta-amyloid and
neuroimaging. _Free Radic. Biol. Med._ 114, 102–109. https://doi.org/10.1016/j.freeradbiomed.2017.09.013 (2018). Article CAS PubMed Google Scholar * Shi, Y. _et al._ A human stem cell
model of early Alzheimer’s disease pathology in Down syndrome. _Sci. Transl. Med._ 4, 124ra129. https://doi.org/10.1126/scitranslmed.3003771 (2012). Article Google Scholar * Citron, M.
Alzheimer’s disease: Strategies for disease modification. _Nat. Rev. Drug Discov._ 9, 387–398. https://doi.org/10.1038/nrd2896 (2010). Article CAS PubMed Google Scholar * Hartley, S. L.
_et al._ Cognitive decline and brain amyloid-beta accumulation across 3 years in adults with Down syndrome. _Neurobiol. Aging._ 58, 68–76.
https://doi.org/10.1016/j.neurobiolaging.2017.05.019 (2017). Article CAS PubMed PubMed Central Google Scholar * Busciglio, J. & Yankner, B. A. Apoptosis and increased generation of
reactive oxygen species in Down’s syndrome neurons in vitro. _Nature_ 378, 776–779. https://doi.org/10.1038/378776a0 (1995). Article ADS CAS PubMed Google Scholar * Tramutola, A. _et
al._ Activation of p53 in Down syndrome and in the Ts65Dn mouse brain is associated with a pro-apoptotic phenotype. _J. Alzheimers Dis._ 52, 359–371. https://doi.org/10.3233/jad-151105
(2016). Article CAS PubMed PubMed Central Google Scholar * Perluigi, M. & Butterfield, D. A. Oxidative stress and Down syndrome: A route toward Alzheimer-like dementia. _Curr.
Gerontol. Geriatr. Res._ 2012, 724904. https://doi.org/10.1155/2012/724904 (2012). Article CAS PubMed Google Scholar * Briggs, J. A. _et al._ Integration-free induced pluripotent stem
cells model genetic and neural developmental features of Down syndrome etiology. _Stem Cells._ 31, 467–478. https://doi.org/10.1002/stem.1297 (2013). Article CAS PubMed Google Scholar *
Shichiri, M. _et al._ Alpha-Tocopherol suppresses lipid peroxidation and behavioral and cognitive impairments in the Ts65Dn mouse model of Down syndrome. _Free Radic. Biol. Med._ 50,
1801–1811. https://doi.org/10.1016/j.freeradbiomed.2011.03.023 (2011). Article CAS PubMed Google Scholar * Barone, E., Arena, A., Head, E., Butterfield, D. A. & Perluigi, M.
Disturbance of redox homeostasis in Down syndrome: Role of iron dysmetabolism. _Free Radic. Biol. Med._ 114, 84–93. https://doi.org/10.1016/j.freeradbiomed.2017.07.009 (2018). Article CAS
PubMed Google Scholar * Ishihara, K. _et al._ Increased lipid peroxidation in Down’s syndrome mouse models. _J. Neurochem._ 110, 1965–1976. https://doi.org/10.1111/j.1471-4159.2009.06294.x
(2009). Article CAS PubMed Google Scholar * Tamagno, E. _et al._ Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid
precursor protein. _J. Neurochem._ 104, 683–695. https://doi.org/10.1111/j.1471-4159.2007.05072.x (2008). Article CAS PubMed Google Scholar * Castellani, R. J., Plascencia-Villa, G.
& Perry, G. The amyloid cascade and Alzheimer’s disease therapeutics: Theory versus observation. _Lab Invest._ 99, 958–970. https://doi.org/10.1038/s41374-019-0231-z (2019). Article
PubMed Google Scholar * Sanmartin, C. D., Adasme, T., Hidalgo, C. & Paula-Lima, A. C. The antioxidant _N_-acetylcysteine prevents the mitochondrial fragmentation induced by soluble
amyloid-beta peptide oligomers. _Neurodegener. Dis._ 10, 34–37. https://doi.org/10.1159/000334901 (2012). Article CAS PubMed Google Scholar * Cenini, G. _et al._ Association between
frontal cortex oxidative damage and beta-amyloid as a function of age in Down syndrome. _Biochim. Biophys. Acta._ 130–138, 2012. https://doi.org/10.1016/j.bbadis.2011.10.001 (1822). Article
CAS Google Scholar * Li, L. B. _et al._ Trisomy correction in Down syndrome induced pluripotent stem cells. _Cell Stem Cell_ 11, 615–619. https://doi.org/10.1016/j.stem.2012.08.004
(2012). Article CAS PubMed PubMed Central Google Scholar * Kim, S. I. _et al._ Inducible transgene expression in human iPS cells using versatile all-in-one piggyBac transposons.
_Methods Mol. Biol. (Clifton, NJ)._ 1357, 111–131. https://doi.org/10.1007/7651_2015_251 (2016). Article CAS Google Scholar * Kondo, T. _et al._ iPSC-based compound screening and in vitro
trials identify a synergistic anti-amyloid beta combination for Alzheimer’s disease. _Cell Rep._ 21, 2304–2312. https://doi.org/10.1016/j.celrep.2017.10.109 (2017). Article CAS PubMed
Google Scholar * Ahmed, M. _et al._ Structural conversion of neurotoxic amyloid-beta(1–42) oligomers to fibrils. _Nat. Struct. Mol. Biol._ 17, 561–567. https://doi.org/10.1038/nsmb.1799
(2010). Article CAS PubMed PubMed Central Google Scholar * Haass, C. & Selkoe, D. J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid
beta-peptide. _Nat. Rev. Mol. Cell Biol._ 8, 101–112. https://doi.org/10.1038/nrm2101 (2007). Article CAS PubMed Google Scholar * Huynh, T. V., Davis, A. A., Ulrich, J. D. &
Holtzman, D. M. Apolipoprotein E and Alzheimer’s disease: The influence of apolipoprotein E on amyloid-beta and other amyloidogenic proteins. _J. Lipid Res._ 58, 824–836.
https://doi.org/10.1194/jlr.R075481 (2017). Article CAS PubMed PubMed Central Google Scholar * Iadanza, M. G., Jackson, M. P., Hewitt, E. W., Ranson, N. A. & Radford, S. E. A new
era for understanding amyloid structures and disease. _Nat. Rev. Mol. Cell Biol._ 19, 755–773. https://doi.org/10.1038/s41580-018-0060-8 (2018). Article CAS PubMed Google Scholar * Page,
R. M. _et al._ Generation of Abeta38 and Abeta42 is independently and differentially affected by familial Alzheimer disease-associated presenilin mutations and gamma-secretase modulation.
_J. Biol. Chem._ 283, 677–683. https://doi.org/10.1074/jbc.M708754200 (2008). Article CAS PubMed Google Scholar * Butterfield, D. A. & Boyd-Kimball, D. Redox proteomics and amyloid
beta-peptide: Insights into Alzheimer disease. _J. Neurochem._ 151, 459–487. https://doi.org/10.1111/jnc.14589 (2018). Article CAS PubMed PubMed Central Google Scholar * Shelat, P. B.
_et al._ Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. _J. Neurochem._ 106, 45–55.
https://doi.org/10.1111/j.1471-4159.2008.05347.x (2008). Article CAS PubMed Google Scholar * Mao, P. & Reddy, P. H. Aging and amyloid beta-induced oxidative DNA damage and
mitochondrial dysfunction in Alzheimer’s disease: Implications for early intervention and therapeutics. _Biochim. Biophys. Acta._ 1359–1370, 2011.
https://doi.org/10.1016/j.bbadis.2011.08.005 (1812). Article CAS Google Scholar * Kazuki, Y. _et al._ Down syndrome-associated haematopoiesis abnormalities created by chromosome transfer
and genome editing technologies. _Sci. Rep._ 4, 6136. https://doi.org/10.1038/srep06136 (2014). Article CAS PubMed PubMed Central Google Scholar * Gonzalez, C. _et al._ Modeling amyloid
beta and tau pathology in human cerebral organoids. _Mol. Psychiatry._ 23, 2363–2374. https://doi.org/10.1038/s41380-018-0229-8 (2018). Article CAS PubMed PubMed Central Google Scholar
* Head, E. _et al._ Plasma amyloid-β as a function of age, level of intellectual disability, and presence of dementia in Down syndrome. _J. Alzheimers Dis._ 23, 399–409.
https://doi.org/10.3233/jad-2010-101335 (2011). Article CAS PubMed PubMed Central Google Scholar * Sano, M. _et al._ Vitamin E in aging persons with Down syndrome: A randomized,
placebo-controlled clinical trial. _Neurology_ 86, 2071–2076. https://doi.org/10.1212/wnl.0000000000002714 (2016). Article CAS PubMed PubMed Central Google Scholar * Shichiri, M. The
role of lipid peroxidation in neurological disorders. _J. Clin. Biochem. Nutr._ 54, 151–160. https://doi.org/10.3164/jcbn.14-10 (2014). Article CAS PubMed PubMed Central Google Scholar
* Ellis, J. M. _et al._ Supplementation with antioxidants and folinic acid for children with Down’s syndrome: Randomised controlled trial. _BMJ_ 336, 594–597.
https://doi.org/10.1136/bmj.39465.544028.AE (2008). Article CAS PubMed PubMed Central Google Scholar * Lott, I. T. Antioxidants in Down syndrome. _Biochim. Biophys. Acta._ 657–663,
2012. https://doi.org/10.1016/j.bbadis.2011.12.010 (1822). Article CAS Google Scholar * Shahidi, S., Zargooshnia, S., Asl, S. S., Komaki, A. & Sarihi, A. Influence of _N_-acetyl
cysteine on beta-amyloid-induced Alzheimer’s disease in a rat model: A behavioral and electrophysiological study. _Brain Res. Bull._ 131, 142–149.
https://doi.org/10.1016/j.brainresbull.2017.04.001 (2017). Article CAS PubMed Google Scholar * Costa, M. _et al._ _N_-acetylcysteine protects memory decline induced by streptozotocin in
mice. _Chem. Biol. Interact._ 253, 10–17. https://doi.org/10.1016/j.cbi.2016.04.026 (2016). Article CAS PubMed Google Scholar * Fernandes, J. & Gupta, G. L. N-acetylcysteine
attenuates neuroinflammation associated depressive behavior induced by chronic unpredictable mild stress in rat. _Behav. Brain Res._ 364, 356–365. https://doi.org/10.1016/j.bbr.2019.02.025
(2019). Article CAS PubMed Google Scholar * Mouton-Liger, F. _et al._ Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2alpha pathway. _Biochim. Biophys.
Acta._ 885–896, 2012. https://doi.org/10.1016/j.bbadis.2012.01.009 (1822). Article CAS Google Scholar * Cordy, J. M., Hooper, N. M. & Turner, A. J. The involvement of lipid rafts in
Alzheimer’s disease. _Mol. Membr. Biol._ 23, 111–122. https://doi.org/10.1080/09687860500496417 (2006). Article CAS PubMed Google Scholar * Oda, A., Tamaoka, A. & Araki, W. Oxidative
stress up-regulates presenilin 1 in lipid rafts in neuronal cells. _J. Neurosci. Res._ 88, 1137–1145. https://doi.org/10.1002/jnr.22271 (2010). Article CAS PubMed Google Scholar *
Roberts, H. L., Schneider, B. L. & Brown, D. R. Alpha-Synuclein increases beta-amyloid secretion by promoting beta-/gamma-secretase processing of APP. _PLoS ONE_ 12, e0171925.
https://doi.org/10.1371/journal.pone.0171925 (2017). Article CAS PubMed PubMed Central Google Scholar * Berk, M., Malhi, G. S., Gray, L. J. & Dean, O. M. The promise of
N-acetylcysteine in neuropsychiatry. _Trends Pharmacol. Sci._ 34, 167–177. https://doi.org/10.1016/j.tips.2013.01.001 (2013). Article CAS PubMed Google Scholar * Tardiolo, G., Bramanti,
P. & Mazzon, E. Overview on the effects of N-acetylcysteine in neurodegenerative diseases. _Molecules_ https://doi.org/10.3390/molecules23123305 (2018). Article PubMed PubMed Central
Google Scholar * Deepmala, _et al._ Clinical trials of N-acetylcysteine in psychiatry and neurology: A systematic review. _Neurosci. Biobehav. Rev._ 55, 294–321.
https://doi.org/10.1016/j.neubiorev.2015.04.015 (2015). Article CAS PubMed Google Scholar * Nishinaka-Arai, Y. _et al._ Down syndrome-related transient abnormal myelopoiesis is
attributed to a specific erythro-megakaryocytic subpopulation with GATA1 mutation. _Haematologica_ 106, 635–640. https://doi.org/10.3324/haematol.2019.242693 (2021). Article CAS PubMed
Google Scholar Download references ACKNOWLEDGEMENTS We thank Dr. Knut Woltjen (CiRA, Kyoto University, Kyoto, Japan) for providing the plasmid vector, Ms. Harumi Watanabe (CiRA, Kyoto
University, Kyoto, Japan) for providing administrative assistance, and Dr. Peter Karagiannis (CiRA, Kyoto University, Kyoto, Japan) for proofreading the paper. FUNDING Funding was provided
to M.K.S. by the Core Center for iPS Cell Research of Research Center Network for Realization of Regenerative Medicine [JP21bm0104001] from the Japan Agency for Medical Research and
Development (AMED) and the Acceleration Program for Intractable Diseases Research utilizing Disease-specific iPS cells from AMED [17935423], and to Y.K. by JST CREST Grant Number JPMJCR18S4.
AUTHOR INFORMATION Author notes * These authors contributed equally: Hiromitsu Toshikawa and Akihiro Ikenaka. AUTHORS AND AFFILIATIONS * Department of Clinical Application, Center for iPS
Cell Research and Application, Kyoto University, 53 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto, 606-8507, Japan Hiromitsu Toshikawa, Akihiro Ikenaka, Yoko Nishinaka-Arai, Akira Niwa, Tatsutoshi
Nakahata & Megumu K. Saito * Osaka Medical and Pharmaceutical University, Takatsuki, 5690801, Japan Hiromitsu Toshikawa, Akira Ashida & Hiroshi Tamai * Social Welfare Organization
“SAISEIKAI” Imperial Gift Foundation Inc., Saiseikai Suita Hospital, Suita, 5640013, Japan Hiromitsu Toshikawa * Division of Hematology, School of Medicine, University of Washington,
Seattle, WA, 98195, USA Li Li & David W. Russell * Department of Human Health Sciences, Graduate School of Medicine, Kyoto University, Kyoto, 6068507, Japan Yoko Nishinaka-Arai *
Chromosome Engineering Research Center, Tottori University, Tottori, Japan Yasuhiro Kazuki * Division of Genome and Cellular Functions, Department of Molecular and Cellular Biology, School
of Life Science, Faculty of Medicine, Tottori University, Tottori, Japan Yasuhiro Kazuki * Institute for Developmental Brain Research, Osaka Medical and Pharmaceutical University, Takatsuki,
5690801, Japan Hiroshi Tamai Authors * Hiromitsu Toshikawa View author publications You can also search for this author inPubMed Google Scholar * Akihiro Ikenaka View author publications
You can also search for this author inPubMed Google Scholar * Li Li View author publications You can also search for this author inPubMed Google Scholar * Yoko Nishinaka-Arai View author
publications You can also search for this author inPubMed Google Scholar * Akira Niwa View author publications You can also search for this author inPubMed Google Scholar * Akira Ashida View
author publications You can also search for this author inPubMed Google Scholar * Yasuhiro Kazuki View author publications You can also search for this author inPubMed Google Scholar *
Tatsutoshi Nakahata View author publications You can also search for this author inPubMed Google Scholar * Hiroshi Tamai View author publications You can also search for this author inPubMed
Google Scholar * David W. Russell View author publications You can also search for this author inPubMed Google Scholar * Megumu K. Saito View author publications You can also search for
this author inPubMed Google Scholar CONTRIBUTIONS H.To. designed the study, performed almost all the experiments, and analyzed the data. A.I. performed revise experiments. Y.N-A. designed
the study, performed some experiments, and analyzed the data. A.N., A.A., T.N., and H.Ta. analyzed and discussed the data. L.L. and D.W.R. constructed the trisomy and disomy iPSCs. Y.K.
constructed the trisomy ESCs. M.K.S. managed the entire research. H.To. and M.K.S. wrote the manuscript. All authors read and accepted the content of the manuscript. CORRESPONDING AUTHOR
Correspondence to Megumu K. Saito. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER'S NOTE Springer Nature remains
neutral with regard to jurisdictional claims in published maps and institutional affiliations. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original
author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the
article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your
intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence,
visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Toshikawa, H., Ikenaka, A., Li, L. _et al._ _N_-Acetylcysteine prevents
amyloid-β secretion in neurons derived from human pluripotent stem cells with trisomy 21. _Sci Rep_ 11, 17377 (2021). https://doi.org/10.1038/s41598-021-96697-7 Download citation * Received:
20 August 2020 * Accepted: 10 August 2021 * Published: 30 August 2021 * DOI: https://doi.org/10.1038/s41598-021-96697-7 SHARE THIS ARTICLE Anyone you share the following link with will be
able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing
initiative