Play all audios:
ABSTRACT Delivery mode and perinatal antibiotics influence gut microbiome composition in children. Most microbiome studies have used the sequencing of the bacterial 16S marker gene but have
not reported the metabolic function of the gut microbiome, which may mediate biological effects on the host. Here, we used the PICRUSt2 bioinformatics tool to predict the functional profiles
of the gut microbiome based on 16S sequencing in two child cohorts. Both Caesarean section and perinatal antibiotics markedly influenced the functional profiles of the gut microbiome at the
age of 1 year. In machine learning analysis, bacterial fatty acid, phospholipid, and biotin biosynthesis were the most important pathways that differed according to delivery mode.
Proteinogenic amino acid biosynthesis, carbohydrate degradation, pyrimidine deoxyribonucleotide and biotin biosynthesis were the most important pathways differing according to antibiotic
exposure. Our study shows that both Caesarean section and perinatal antibiotics markedly influence the predicted metabolic profiles of the gut microbiome at the age of 1 year. SIMILAR
CONTENT BEING VIEWED BY OTHERS PERSONALIZED MODELING OF GUT MICROBIOME METABOLISM THROUGHOUT THE FIRST YEAR OF LIFE Article Open access 30 December 2024 THE RELATIONSHIP BETWEEN THE GUT
MICROBIOME AND THE RISK OF RESPIRATORY INFECTIONS AMONG NEWBORNS Article Open access 14 July 2022 STRAIN-SPECIFIC IMPACTS OF PROBIOTICS ARE A SIGNIFICANT DRIVER OF GUT MICROBIOME DEVELOPMENT
IN VERY PRETERM INFANTS Article Open access 26 September 2022 INTRODUCTION The perinatal period plays a critical role in gut microbiome development. Several studies have shown that the gut
microbiome composition of infants delivered by Caesarean section (C-section) shows a reduced diversity1,2, lower relative abundance of _Bacteroides_1,2,3,4,5 and a higher relative abundance
of _Enterococcus_2,3,5_, Klebsiella_3,5 and _Clostridium_3,4 as compared to that of vaginally delivered infants1,3. Currently, perinatal antibiotics are frequently used during both vaginal
deliveries and C-sections to prevent early-onset group B streptococcal (GBS) sepsis and after birth to treat suspected neonatal infections6. Intrapartum antibiotic prophylaxis (IAP),
administered to mothers during delivery to prevent GBS transmission to newborn infants, has been associated with reduced gut microbiome richness7 and diversity8,9,10, lower relative
abundance of Bacteroidetes7,8,11 and _Bacteroides_3,7,10,11 and a higher relative abundance of Proteobacteria8,9 and Firmicutes9,11 in term vaginally delivered infants. The human gut
microbiome is highly functionally redundant and different taxonomic compositions can share similar metabolic functions12,13. In most earlier paediatric cohort studies2,4,5, 7,8,9,11, gut
microbiome composition has been presented based on the sequencing of bacterial 16S gene, commonly used as a marker gene in microbiome studies. Yet, the metabolic function of the gut
microbiome has seldom been investigated in paediatric cohorts. Whole genome sequencing (WGS) of all bacteria in gut microbiome would be of great benefit, as WGS identifies the taxa and gene
composition with a higher resolution compared to 16S method14. WGS, however, has rarely been used for large datasets because it is expensive and laborious. In recent years, multiple advanced
bioinformatics tools have been developed to overcome these problems, such as PICRUSt215, Tax4Fun216 and Piphillin17. These tools predict the functional pathways of the microbiome based on
16S rRNA sequences and produce results resembling bacterial whole genome sequencing metabolic pathway data. Our hypothesis was that early perinatal events may markedly change the metabolic
pathways of the gut microbiome and influence the later health of children. In the present study, we characterized and compared the effects of delivery mode and perinatal antimicrobial
exposure on the predicted metabolic pathways of the gut microbiome in infants at 1 year of age. METHODS STUDY DESIGN AND POPULATION We used predicted functional profiles of gut microbiomes
based on 16S data from two prospective cohorts from our laboratory. The delivery mode cohort (DM cohort) consisted of 212 consecutive newborn infants born at the Central Finland Central
Hospital in Jyväskylä, Finland, between February 2014 and March 2014, recruited in the delivery room. The patients of the cohort were term or near-term infants. For the analysis, they were
grouped into two groups based on the mode of delivery: (1) vaginally delivered (N = 60) and (2) born by C-section (N = 23). Reported sample sizes above were the number of samples that did
not get removed in various pre-processing phases. The detailed information on the study population and sample collection has been reported previously18. The Ethics Committee of the Central
Finland Hospital District found the study plan ethically acceptable (decision number 1E/2014). The perinatal antibiotic cohort (PA Cohort) consisted of 149 vaginally delivered term infants
born at the Oulu University Hospital in Oulu, Finland, between February 2014 and June 2015. The participants were recruited the first day after birth. The participants were recruited
according to their perinatal antibiotic exposure and classified into two groups: (1) the control group and (2) those with any perinatal antibiotic exposure. In the control group (N = 27),
the infants were not exposed to any perinatal antibiotics. In the antibiotic exposure group (N = 70), either the mother received antibiotics during delivery, the infant received antibiotics
during the first days of life, or both received antibiotics. Sample sizes above were the number of samples that were not removed in pre-processing phases. The background characteristics and
antibiotic exposures of the participants have been previously reported in detail11,19. The research plan was reviewed and approved by the Regional Ethics Committee of the Northern
Ostrobothnia Hospital District, Oulu University Hospital, Oulu, Finland (decision number EETTMK 76/2013). Parents or caregivers of children in both study cohorts gave written informed
consent before the study. The study was conducted in accordance with the relevant guidelines, regulations and legislation regarding clinical studies and data protection. In both cohorts, at
the age of 1 year, families collected faecal samples from the infant’s potty or diaper and sent them to the laboratory. The faecal samples were processed similarly and were frozen at
temperatures lower than − 22 ℃. All samples were analysed with a similar methodology by 16S rRNA gene sequencing at the University of Oulu, Finland. Details about DNA extraction, primers,
and sequencing protocol has been previously published for DM11 and PA19 data. All the raw sequences were submitted to the GenBank Sequence Read Archive (SRA) with accession numbers SRP152384
and PRJNA605735. We have previously published the results concerning impact of antibiotic exposure on the gut microbiome 16S composition from birth to the age of 6 months11, and compared
the impact or oral antibiotic courses and perinatal antibiotics on gut microbiome 16S composition at 12 months19 in the PA cohort. The impact of delivery mode on the gut microbiome 16S
composition at 12 months has not earlier been reported in DM cohort. Predicted metabolic pathway data, presented in this manuscript, have not earlier been published or submitted for either
cohort. Because microbiome data are high-dimensional, complex, noisy and compositional in nature20,21, increasing the false discovery rate of conventional hypothesis testing methods22, we
used a machine learning (ML) approach in this analysis21,23,24. Sequence pre-processing and strict quality filtering settings in this study were designed to decrease data dimensionality and
sparsity. These upstream choices increase machine learning model performance and interpretability, and as such, differ from previously published work for perinatal antibiotics11,19. As a
result, features that were rare and found in low prevalence were filtered out. We now present side-by-side results from ANCOM2, ALDEx2, and beta diversity for both cohorts to increase the
interpretability of the machine learning models, and effectively showcase the differences in the PA and DM results. SEQUENCE PRE-PROCESSING Raw sequences from both datasets were imported
into Qiime225 (version 2019.10), where they were processed independently from each other. Sequencing primers were trimmed before denoising with the q2-dada2 -plugin. Reads shorter than 270
bp were truncated in the PA cohort and reads shorter than 385 bp were truncated in the DM cohort with an additional 15 bp trimmed from the left side during DADA226. Any quality filtering was
avoided before using DADA2. Taxonomic classifiers were trained using the 132 SILVA27 database trimmed to the study primers and truncated using the DADA2 parameter values for truncation and
filtering. DADA2 outputs an ASV-table (Amplicon Sequence Variant), which represents the abundances of biological features found from the raw sequences. Features from the ASV-tables were
assigned taxonomies and those features were then classified into domains. Features classified as Bacteria and those found in more than one sample were kept. Chimeric features were removed
with the q2-vsearch-plugin using the uchime-denovo tool. Samples with a combined feature frequency of less than 1000 were removed. The PA cohort had 1037 minimum depths, while the DM cohort
had 1290. We chose 1000 as the depth because it was near to the minimum and fit our analysis methods. Unstratified MetaCyc28 pathway abundances were predicted using the q2-picrust2-plugin
using the “mp” hidden-state prediction method with other parameters set to default values. Stratified results, which map each predicted pathway back to the input ASV’s, were produced using
the original python implementation of PICRUSt215 with the same parameters. ASV and metabolic pathway feature tables from both cohorts were independently analysed. ALPHA AND BETA DIVERSITY
ASV-tables and predicted pathway tables were used in both alpha and beta diversity analyses in both cohorts. Bray–Curtis dissimilarity was chosen for both types of feature tables for beta
diversity, while Shannon index was chosen for alpha diversity. Feature tables were rarefied to depths of 1000 and 10,000 in ASV-tables and predicted pathway tables, respectively. Principal
coordinates analysis (PCoA) was done for Bray–Curtis dissimilarity distance matrices. Alpha and beta diversity analyses were done using the q2-diversity-plugin and visualized with
Matplotlib29. The Kruskal–Wallis H test was performed to test within-sample group differences for alpha diversity. Adonis PERMANOVA analyses were done using the pairwise
beta-diversity-significance command in q2-diversity to test group differences. Adonis PERMANOVA was used for multivariate analysis using both perinatal antibiotic exposure and delivery mode
in DM cohort, while only perinatal antibiotics was used with the PA cohort. PCoA analysis was performed with the beta diversity distance matrix and visualized with ellipses drawn onto the
two-dimensional space using the Pearson correlation coefficient of the two principal coordinates with the highest explained variance. DIFFERENTIAL ABUNDANCE ANALYSIS ALDEx230 and ANCOM22
were used for differential abundance analysis to examine group differences in both genus level and predicted pathway data. Features that were found in 10% or more samples were kept. ALDEx2
analyses were done using the q2-aldex2-plugin with a Q-score significance threshold of 0.05. ANCOM2 (further developed based on ANCOM22) analyses were done using the R-package31 with default
parameters. For DM cohort the ANCOM2 analyses were adjusted according to perinatal antibiotic treatments. ANCOM2 calculates a threshold value for the proportion of feature ratios that show
significant differences. Prior to testing, we chose the value of 0.7 or higher as the significant value, meaning that when output was 0.7 or higher, ANCOM2 found the feature to be
significantly different between study groups. Values below 0.7 were considered non-significant. A significance threshold of 0.7 was recommended by the author of ANCOM231 as a common choice.
ALDEx2 uses p-values from Benjamini–Hochberg adjusted (to control false discovery rate) Welch t-tests (p-value of < 0.05 was considered significant prior to testing). Additionally, ALDEx2
outputs the effect size, which indicates the direction and volume of change of the centred log-ratios. In our study, a positive sign indicated that the feature was more abundant in the
C-section or perinatal antibiotics treatment group whereas a negative sign indicated greater abundance in their respective control groups (i.e., vaginal birth and no-perinatal antibiotics
treatments). MACHINE LEARNING ANALYSIS Machine learning models were trained to predict the target variables of delivery mode and perinatal antibiotic treatments in DM and PA cohorts,
respectively. Models were created using a nested cross-validation (CV) setting where parameters were only tuned using the inner cross-validation loop. Random Forest (RF)32, Extremely
Randomized Trees (EXTRA)33 and Adaptive Boosting (Adaboost)34 models were tuned and trained independently of each other. Model performance was tested against the validation fold that was
unseen to the models. Performance was evaluated using the Receiver Operating Characteristic (ROC) of the Area-Under-the-Curve (AUC). Model selection was only done in the inner CV folds using
the ROC AUC metric. Machine learning analyses were implemented using the scikit-learn package35. Rank aggregation analysis was used to highlight the key shared features for Random Forest,
ExtraTrees and Adaboost models during model training. The importance of each taxon or pathway feature was recorded and compiled in a rank aggregation analysis. CROSS-STUDY PREDICTIONS We
aimed to find out whether similar machine learning models were able to differentiate the impact of both perinatal antibiotics and C-section on the predicted metabolic pathway composition of
the gut microbiome using a cross-study prediction between two available cohorts. Machine learning models were trained using prevalence filtered feature tables from both taxon and pathway
data. Models were given the task to differentiate positive (C-section and perinatal antibiotics treatment) and negative (vaginal delivery and no antibiotic treatments) classes from each
other. In this analysis, we chose a prevalence level such that each feature needed to be found in a percentage of all samples in both cohorts to be included. Prior to model building, we
tried prevalence cut-offs in the range of 10–50% in both taxa and pathway data. In previous studies, researchers have experimented with prevalence thresholds varying from 1 to 10% in one
study36, while selecting as high as 45% in another37. We chose as high prevalence cut-offs as possible while leaving as many features as possible in both types of data. For metabolic pathway
data, the percentage chosen for prevalence cut-off was 50% (240 features) and for taxa data, 30% (12 features). Prevalence thresholds and other data filtering choices were not readjusted
after initial model building to prevent leaking information between training and testing folds. RF, EXTRA and Adaboost models were trained using only one cohort’s data using the same scheme
described earlier. After each iteration of the nested cross-validation loop, the best RF, EXTRA and Adaboost models were combined into an ensemble classifier. This classifier was then used
to predict a randomly sampled subset from the other cohort that was completely unseen by the models. Feature importances were estimated using the prefitted ensemble classifier with
permutation_importance function from scikit-learn35. Next, we examined if models trained on the PA cohort were biased towards samples that had been exposed to perinatal antibiotics in the DM
cohort. We pooled together all the test predictions from C-section and vaginal samples into two groups. Samples that had been exposed to perinatal antibiotic treatments and those that had
not been exposed and produced the pooled AUC of both groups. There were no marked differences in the AUC when predicting DM cohort samples that had been exposed to perinatal antibiotics
(0.71 AUC) to those that had not been (0.7 AUC). RESULTS WITHIN AND BETWEEN-SAMPLE DIVERSITY ACCORDING TO DELIVERY MODE AND PERINATAL ANTIBIOTICS First we analysed beta diversity, i.e.,
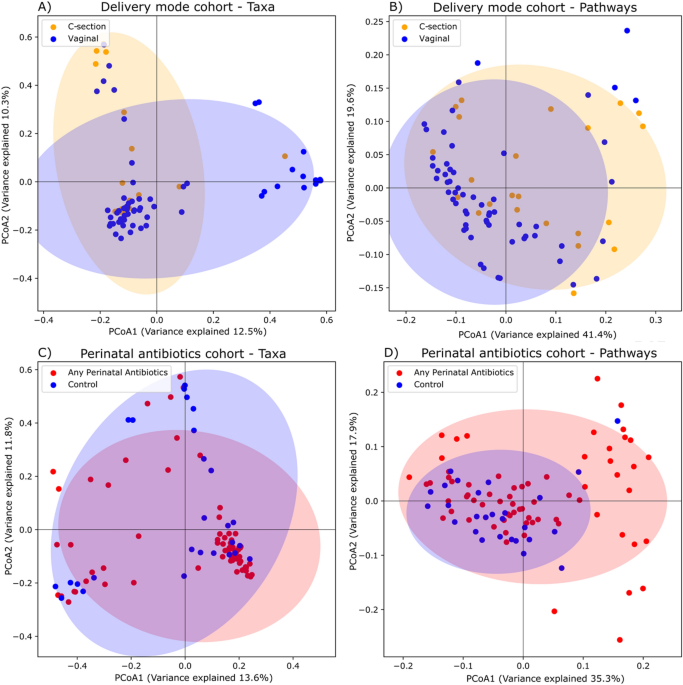
between-sample diversity, to examine differences in the taxonomic and metabolic pathway composition of the gut microbiome according to delivery mode (Fig. 1A,B, delivery mode cohort) and
perinatal antibiotic exposure (Fig. 1C,D, perinatal antibiotics cohort) at the age of 1 year using principal coordinate analysis (PCoA). Delivery mode was not associated according to
PERMANOVA analyses for taxa or predicted metabolic pathways (Supplementary Table 1). Perinatal antibiotics were associated for taxa (_R2_ = 0.0196, _Pr_(> _F_) = 0.008), but not for
predicted metabolic pathways (Supplementary Table 1). Alpha diversity metrics, i.e., within-sample diversity, showed no significant differences for taxonomic or metabolic pathway data
according to delivery mode or perinatal antibiotic exposure (Supplementary Fig. 1). EFFECTS ON FATTY ACID BIOSYNTHESIS, LIPID BIOSYNTHESIS AND BIOTIN METABOLISM PATHWAYS We then investigated
which taxa and metabolic pathways of the gut microbiome were differentially abundant in the gut microbiomes of children depending on the delivery mode (C-section vs vaginal) or perinatal
antibiotic exposure (any vs none) (Table 1, Supplementary Table 2). The full output of ANCOM2 and ALDEx2 can be found as “Supplementary Tables S1” (Supplementary Tables 3–10). Using ANCOM2
analysis for taxa, the abundance of Bacteroides and Erysipelatoclostridium were significantly different in the gut microbiomes of infants born via C-section as compared to that of vaginally
born infants (Table 1). When comparing the metabolic pathway data of the gut microbiome, we found several metabolic pathways that were significantly different between functional profiles of
gut microbiomes of infants at 1 year of age depending on whether they were born via C-section or vaginal route (Table 1). Five of these pathways were linked to fatty acid and lipid
biosynthesis. In ALDEx2 analysis, none of the taxa differed in a statistically significant manner depending on delivery mode, while one pathway related to fatty acid biosynthesis (PWYG-321)
was differentially abundant (Supplementary Table 2). Bacteroides was significantly different depending on exposure to perinatal antibiotics (Table 1). When comparing metabolic pathways
according to perinatal antibiotic exposure, we identified seven pathways as significantly different (Table 1), two of which were linked to vitamin B6 metabolism and two to carbohydrate
degradation. Three pathways (biotin metabolism, biotin synthesis and glycosaminoglycan degradation) were significantly different according to both the delivery mode and perinatal exposure
(Table 1). When we used ALDEx2 for comparisons, we found one genus and 25 differentially abundant metabolic pathways depending on the perinatal antibiotic exposure (Supplementary Table 2).
INFLUENCE OF PERINATAL EVENTS ON PREDICTED METABOLIC PATHWAYS IN GUT MICROBIOME We used machine learning models on predicted metabolic pathways in gut microbiome to differentiate whether the
child was born via C-section or vaginal route (Fig. 2A,B), or whether the child was exposed or unexposed to perinatal antibiotics (Fig. 2C,D). Three algorithms (RF, ExtraTrees, Adaboost)
were used to differentiate both target variables using the 16S sequencing derived genera and predicted metabolic pathway data. Models were able to differentiate their respective test samples
more precisely when using metabolic pathway data instead of taxa data (Fig. 2). Children born via C-section were well differentiated from those born vaginally by using machine learning
models on gut microbiome data at the age of 12 months (Fig. 2A,B) Using RF for taxa by delivery mode, the highest AUC, representing the measure of separability under ROC, was 0.69 (_SD_ =
0.05) (Fig. 2A), while ExtraTrees achieved the highest performance for metabolic pathways of the gut microbiome with an AUC = 0.71 (SD = 0.04) (Fig. 2B), differentiating data by delivery
mode. Similarly, children exposed to perinatal antibiotics were well differentiated from those unexposed by using machine learning models on gut microbiome data at the age of 12 months. RF
models trained using taxa to differentiate data by perinatal antibiotic exposure performed better (AUC of 0.69, SD = 0.04) than ExtraTrees or Adaboost (Fig. 2C). Additionally, RF was again
the top performer with metabolic pathways in differentiating data by perinatal antibiotic exposure with an AUC of 0.74 (_SD_ = 0.04) (Fig. 2D). MOST IMPORTANT GENERA AND PREDICTED METABOLIC
PATHWAYS FOR MACHINE LEARNING MODELS Machine learning models are often referred to black boxes, as the inner workings are often obscured or complicated to understand. Random forests and
other decision tree-based algorithms have the benefit of providing continuous value for feature importance’s, effectively ranking how well each feature (genera or predicted pathway) affected
the overall result. In rank aggregation analysis, the importance of each taxon or pathway feature in machine learning models was recorded and compiled (Fig. 3). In children born via
C-section or vaginally, using gut microbiome data at the age of 1 year, the most important features in gut microbiome 16S composition, differentiating children in machine learning analysis
for delivery mode, were Erysipelatoclostridium followed by _Bacteroides_, _Roseburia_, _Alistipes_ and _Lactococcus_ (Fig. 3A). At the same time, the most important predicted metabolism
related pathways, differentiating children in machine learning analysis according to delivery mode, were fatty acid biosynthesis, phospholipid biosynthesis, biotin biosynthesis and biotin
metabolism related pathways. (Fig. 3B). In children exposed or unexposed to perinatal antibiotics, using gut microbiome at the age of 12 months, the most important features differentiating
children in machine learning analysis for perinatal antibiotic exposure were _Bacteroides_, _Faecalibacterium_ and _Dorea_ (Fig. 3C). At the same time, the most important predicted
metabolism related pathways, differentiating children in machine learning analysis according to perinatal antibiotic exposure, were proteinogenic amino acid biosynthesis, carbohydrate
degradation, pyrimidine deoxyribonucleotide biosynthesis, vitamin biosynthesis and biotin biosynthesis (Fig. 3D). In both cohorts, there were multiple features with similar median ranks,
indicating a high amount of collinearity in pathway data, where many features were correlated with each other. Rank aggregation values for each variable are reported in the “Supplementary
File S1” (Supplementary Tables 11–14). CROSS-STUDY ANALYSIS: TRAINING MODELS ON ONE COHORT AND TESTING THEM ON THE OTHER COHORT As the direct comparison of taxon or pathway data from the two
datasets using conventional statistics is not optimal, even for datasets from the same laboratory, we then performed a cross-study comparison using machine learning to assess effects
associated with delivery mode or perinatal antibiotic exposure (presented in Table 1 and Figs. 2, 3). We examined whether patterns found by machine learning models in one dataset for
delivery mode could be generalized to the samples from the other cohort for perinatal antibiotic exposure and vice versa, possibly indicating analogous or similarly differentiating changes
in gut microbiome after C-section or perinatal antibiotics as compared to those with undisturbed early gut colonization. We thus trained models on one cohort and tested them on the other
cohort. In the cross-study analysis, gut microbiome 16S composition changes due to C-section or perinatal antibiotic were not analogous or similarly differentiating. Model performance was
low when using taxon data to train and test models in both cohorts. Models trained on the PA cohort and tested on the DM cohort achieved an AUC of 0.64 (Fig. 4A). Training on the DM cohort
and testing on the PA cohort showed an AUC of 0.54. When using predicted metabolic pathway features, the cross-study models were able to predict cross-study sample classes as positive
(C-section or any perinatal antibiotics treatment) or negative (vaginal delivery or no perinatal antibiotics treatments) (Fig. 4B). Models trained on differentiating perinatal antibiotic
exposure (any vs none) were able to differentiate the delivery mode (C-section vs vaginal) with an AUC of 0.72, while models trained on delivery mode cohort achieved an AUC of 0.70 in
differentiating perinatal antibiotic exposure (Fig. 5B). THE MOST IMPORTANT METABOLIC PATHWAYS IN THE CROSS-STUDY PREDICTIVE MODELS We then analysed which features were most important for
the performance of the models by using permutation importance analysis. After “shuffling”, i.e., deliberately removing the true grouping data for the delivery mode or perinatal antibiotic
exposure and replacing it with a random grouping variable, Bacteroides resulted in the largest reduction in the AUC of models in differentiating both delivery modes (mean reduction = 0.14,
_SD_ = 0.06) (Fig. 5A) and perinatal antibiotic exposure (mean reduction = 0.06, _SD_ = 0.04, Fig. 5C) using taxon data from the samples. After shuffling, using metabolic pathway data, we
found several features decreasing only slightly the performance of the model, with the largest average change of 0.03 to AUC for proteinogenic amino acid biosynthesis according to delivery
mode (Fig. 5B) and 0.005 for biotin biosynthesis according to perinatal antibiotic exposure (Fig. 5D). In the DM cohort (C-section vs vaginal delivery), pathways related to proteinogenic
amino acid biosynthesis, biotin biosynthesis and fermentation were the most important (Fig. 5B). In the PA cohort (any antibiotics vs none in vaginally delivered infants), pathways related
to biotin biosynthesis, polysaccharide degradation, proteinogenic amino acid biosynthesis and pyrimidine nucleotide biosynthesis were the most important (Fig. 5D). Values for each tested
variable are reported in the “Supplementary File S1” (Supplementary Tables 15–19). RELATIVE ABUNDANCES OF IMPORTANT TAXA AND PATHWAYS Lastly, we present the relative abundances between study
groups of genera and pathways that were found significantly different by ANCOM2 (Table 1) or important by machine learning models (Figs. 2 and 4). Additionally, the stratified relative
abundance was gathered from PICRUSt2, which allows for a direct link from each predicted pathway back to the ASV’s. Bacteroides was differentially abundant in both DM and PA and was the most
important feature of cross-study machine learning models. We additionally stratified the predicted pathway data according to Bacteroides to gain further insight. In the DM cohort,
Bacteroides (Supplementary Table 19) and Erysipelatoclostridium were highlighted by ANCOM2 (Table 1) as differentially abundant, while also being important for machine learning models (Fig.
2A). Bacteroides (36% in C-section and 58% in vaginal) had lower relative abundance in C-section, while Erysipelatoclostridium (6% in C-section and 2.2% in vaginal) was enriched in children
exposed to C-section. Several predicted metabolic pathways related to biotin metabolism, biotin biosynthesis, glycosaminoglycan degradation, and fatty acid biosynthesis had decreased
relative abundance in C-section children (Supplementary Table 19). Based on Bacteroides stratified data, all the above pathways had 44–99% of their relative abundances come from Bacteroides
sequences in C-section samples, while 73–99% of their relative abundances in vaginal group (Supplementary Table 20). One pathway related to fatty acid biosynthesis (PHOSLIPSYN-PWY) and
fermentation (P161-PWY) were enriched in C-section samples. Bacteroides was the only significantly different genera in PA cohort while also being important to machine learning models.
Bacteroides had lower relative abundance (38% in perinatal antibiotics and 66% in no antibiotics) samples (Supplementary Table 19). Predicted pathways related to biotin metabolism, biotin
biosynthesis, glycosaminoglycan degradation, vitamin B6 metabolism, and fatty acid biosynthesis pathways were decreased according to perinatal antibiotics usage. According to Bacteroides
stratified data, a large portion of the above pathways relative abundances were derived from Bacteroides in both PA (37–97%) and non-PA groups (64–99%) (Supplementary Table 20). Pathways
related to sugar degradation (GLUCOSE1PMETAB-PWY), pyrimidine deoxyribonucleotide biosynthesis (PWY-6545), and vitamin biosynthesis (PWY-6891) were enriched according to perinatal
antibiotics usage. DISCUSSION Delivery mode and perinatal antibiotics have been shown to influence gut microbiome composition in children1,2,3,4,5,7,8,9,10,11. Most microbiome
studies2,4,5,7,8,9,11 have used the sequencing of the bacterial 16S marker gene but have not reported the metabolic function of gut microbiome, which may mediate biological effects on the
host. Whole genome sequencing of the gut microbiome would be an ideal solution but is still not feasible for every study or dataset14,38,39. Here, we used the PICRUSt215 bioinformatics tool
to predict the functional profiles of the gut microbiome based on 16S sequencing in two prospective cohorts from our laboratory. Our study shows that both Caesarean section and perinatal
antibiotics markedly influence the predicted metabolic profiles of the gut microbiome at the age of 1 year. Earlier, in a study of 60 infants using metagenome sequencing of faecal samples,
perinatal antibiotics predicted microbiome alterations, but delivery mode had no enduring effects40. In accordance with our results, a metagenomic study by Chu et al.41 found several similar
changes in metabolic pathways; intrapartum antibiotic exposure enriched pathways associated with glycolysis and pyrimidine metabolism whereas pathways associated with folate and biotin
metabolism were decreased. Similarly, we found that predicted pathways related to sugar degradation and pyrimidine deoxyribonucleotide biosynthesis were enriched with perinatal antibiotics
exposure, while biotin biosynthesis and metabolism pathways decreased. Perinatal events affected pathways related to biotin metabolism, vitamin metabolism, and vitamin biosynthesis in the
present study. Several microbial species in gut microbiome can synthesize vitamin K2 and water-soluble B-vitamins, such as biotin42. Vitamin metabolism genes are found across different phyla
suggesting that they have a core function in gut microbiome metabolism42,43. In a previous metagenomic analysis of fecal samples in four countries, some of vitamin metabolism related
pathways differed between study participants with type 2 diabetes or inflammatory bowel disease42. Overall, the clinical relevance of gut vitamin metabolism is still poorly understood but
according to the present study, it appears that perinatal events, delivery mode or perinatal antibiotics, may change the microbiome-mediated vitamin metabolism in human gut of children. In
the present study, perinatal events also influenced predicted metabolic pathways of fatty acid metabolism. Short-chain fatty acids, especially butyrate, likely play an important role in the
maintenance of gut health44. Butyrate modulates inflammatory responses and intestinal barrier function44. Decreased fatty acid synthesis has been reported in inflammatory bowel disease using
shotgun metagenomics of faecal samples45 and in asthma46 using mass spectrometry of faecal samples for metabolome analysis. Furthermore, changes in butyrate-producing bacteria may modulate
the function of nervous systems44,47. In an animal model, mice treated with butyrate after a high-fat diet had reduced glucose intolerance and insulin resistance and improved cardiovascular
disease related metabolic disorder48. Thus, the observed changes in fatty acid metabolism may associate with the long-term health of children if the observed changes in the predicted
metabolic pathways of gut microbiome, observed here at the age of 1 year, persist in adolescence and adulthood. We found that genera Bacteroides to be differentially abundant according to
both delivery mode and antibiotic exposure. The mean relative abundance of Bacteroides was decreased in children born by C-section and those exposed to perinatal antibiotics. According to
our results, Bacteroides is a major contributor to several predicted pathways related to biotin metabolism, biotin biosynthesis, glycosaminoglycan degradation, and fatty acid biosynthesis.
These predicted pathways were found to be differentially abundant or important to machine learning models according to delivery mode and perinatal antibiotics. Bacteroides contributed 64–99%
of the total copy number of these pathways found in children vaginally delivered or not exposed to perinatal antibiotics. Subsequently, as relative abundance of Bacteroides decreased when
exposed to C-section or perinatal antibiotics, so did the relative abundances of these pathways. The gut microbiome is highly redundant; many different bacteria can perform the same
metabolic function12,13. Our results suggest that not all metabolic pathways can be replenished by redundant bacteria, as several predicted pathways linked to Bacteroides were decreased in
children exposed C-section or perinatal antibiotics. Still, several predicted pathways, related to proteinogenic amino acid biosynthesis and nucleotide biosynthesis were found to be
important for machine learning models according to exposure to perinatal antibiotics, but not clearly decreased or increased based on mean relative abundances. These results suggest that
some metabolic pathways interact with C-section and exposure to perinatal antibiotics in a complex way. Thus, mere 16S sequencing and presenting taxonomic data is not the most optimal method
for microbiome research. Whole genome sequencing would be an ideal method but is still prohibitively expensive for most research groups investigating clinical associations in large study
cohorts. Furthermore, currently available WGS datasets from microbiome research groups are too small for modern “data hungry” statistical methods, such as deep learning. Several tools, such
as PICRUSt215, Tax4Fun216 and Piphillin17, have been developed to overcome this issue. These tools can estimate the metabolic pathway, gene or metabolite compositions of samples based on
their 16S sequences. In the present study, we used PICRUSt2 to predict functional metabolic profiles of the gut microbiome in two child cohorts. Previously, PICRUSt2 was used to investigate
the gut microbiome in animal models of multiple sclerosis49 and the gut microbiome of Crohn’s disease50 but rarely in prospective paediatric cohorts investigating the influence of perinatal
events on subsequent microbiome composition, its function and later health. In the present study, we analysed the gut microbiome from samples obtained at the age of 1 year in two prospective
child cohorts from our laboratory11,18,51,52. Our predictive machine learning models confirmed the results from previous studies that both delivery mode (C-section vs vaginal delivery) and
antimicrobial exposure at birth influence the abundance of Bacteroides in the gut microbiome in children1,2,3,4,7,10,11. In this study, we also report the novel finding that several
predicted metabolic pathways linked to amino acid biosynthesis and biotin biosynthesis were identified as important when predicting unseen validation samples across the two study cohorts.
However, both cohorts still showed unique differences not found in the other cohorts when analysed independently with machine learning and a traditional approach. Recently, the machine
learning approach has become popular in translational research due to its several advantages as compared to conventional statistical analysis. For instance, machine learning has been used to
predict skin cancer from images53, type 2 diabetes from health records54, circumstances of death with the microbiome55 and exercise response to metabolic homeostasis with the microbiome56.
Random forest, a type of machine learning algorithm, has been used to differentiate microbiome samples with taxonomic data between C-section and vaginally delivered infants57. Le Goellec et
al. used a variety of algorithms, including random forest and other decision-tree based algorithms, to predict antibiotic usage and delivery mode from infant gut microbiome samples58.
Stewart et al. used random forest models to examine important operational taxonomic units in predicting the age of infants in the first 40 months of life59. In the present study, we report
the same taxonomic differences found previously. In addition, we report the predicted functional profiles of the gut microbiome according to delivery mode and perinatal antibiotic exposure
using a machine learning approach. Interestingly, transforming the 16S taxonomic data to predicted metabolic pathways using the bioinformatics tool PICRUSt2 increased model performance in
both cross-cohort and inner-cohort predictions. Previously published studies showed poor generalization performance when used to predict samples in a cross-study way24,36. Similarly, our
models were unable to predict the C-section or exposure to perinatal antibiotics using only taxa as input. Using a tool to predict the metabolic pathway composition of a sample is a form of
feature engineering, the process of extracting or augmenting the feature space to form new features, which is a commonly used technique in machine learning60,61. Contrary to most feature
engineering methods, PICRUSt2 pulls new information from an outside source, such as MetaCyc28 dabase, to transform each feature to many new informative ones. This might be one of the reasons
why our models had increased performance when using PICRUSt2 enhanced data. As such, our results suggest that predicted metabolic pathway composition may be more informative for host trait
classification problems than taxonomic features alone. In the present study, we found that mode of delivery and perinatal antibiotic exposure influenced several metabolic pathways of the gut
microbiome, such as pathways for vitamin B metabolism and fatty acid synthesis. Short-chain fatty acids, especially butyrate, likely play an important role in the maintenance of gut
health44. Butyrate modulates inflammatory responses and intestinal barrier function44. Decreased fatty acid synthesis has been reported in inflammatory bowel disease using shotgun
metagenomics of faecal samples45 and in asthma46 using mass spectrometry of faecal samples for metabolome analysis. Furthermore, changes in butyrate-producing bacteria may modulate the
function of nervous systems44,47. Earlier, in a study of 60 infants using metagenome sequencing of faecal samples, perinatal antibiotics predicted microbiome alterations, but delivery mode
had no enduring effects40. In accordance with our results, a metagenomic study by Chu et al.41 found several similar changes in metabolic pathways; intrapartum antibiotic exposure enriched
pathways associated with glycolysis and pyrimidine metabolism whereas pathways associated with folate and biotin metabolism were decreased. The strength of the present study is that it
demonstrates the successful use of predicted functional metabolic profiles based on 16S data in paediatric cohorts. There are many large clinical and translational paediatric cohorts with
16S data available, but metagenome sequencing has rarely been done due to constraints of cost and availability of whole genome sequencing of the gut microbiome. Metabolic pathways of gut
microbiome data are more stable and more widely shared between individuals than taxonomic level data62,63. Thus, the approach presented in this study is likely to be more reasonable than
mere taxonomic level comparisons. Furthermore, we had two prospective cohorts from the same laboratory for the present study. We analysed data with a machine learning approach, including
cross-cohort comparisons. Our study approach shows possibilities how to utilize gut microbiome 16S datasets in a novel and meaningful way, including cross-study comparisons of functional
profiles using machine learning. The main strength of the study is also the main limitation of the study. We do not present actual metagenome sequencing data, derived from whole genome
sequencing of gut microbiome, or metabolome data, derived from mass spectrometry of faecal samples, but rather use predicted functional profiles of the gut microbiome. The metabolic pathway
genes of the gut microbiome predicted based on 16S data may not express the predicted proteins or the metabolites responsible for biological effects. We did not validate our results against
metagenome sequencing or mass spectrometry, however, high-quality reports of PICRUSt64, PICRUSt215, Piphillin17, and Tax4Fun216 benchmark the methodology against WGS data in varying
microbiomes. These studies show, that the predicted functional profiles strongly correlate to functional profiles from a WGS approach. In conclusion, using two prospective paediatric
cohorts, perinatal events, both Caesarean section and perinatal antibiotics, markedly influenced the functional profiles of the gut microbiome at the age of 1 year. The observed changes in
metabolic pathways of gut microbiome may potentially influence the later health of children. CHANGE HISTORY * _ 23 NOVEMBER 2023 A Correction to this paper has been published:
https://doi.org/10.1038/s41598-023-47520-y _ REFERENCES * Bokulich, N. A. _et al._ Antibiotics, birth mode, and diet shape microbiome maturation during early life. _Sci. Transl. Med._ 8,
343ra82 (2016). Article PubMed PubMed Central Google Scholar * Jakobsson, H. E. _et al._ Decreased gut microbiota diversity, delayed Bacteroidetes colonisation and reduced Th1 responses
in infants delivered by Caesarean section. _Gut_ 63, 559–566 (2014). Article CAS PubMed Google Scholar * Shao, Y. _et al._ Stunted microbiota and opportunistic pathogen colonization in
caesarean-section birth. _Nature_ 574, 117 (2019). Article ADS CAS PubMed PubMed Central Google Scholar * Penders, J. _et al._ Factors influencing the composition of the intestinal
microbiota in early infancy. _Pediatrics_ 118, 511–521 (2006). Article PubMed Google Scholar * Reyman, M. _et al._ Impact of delivery mode-associated gut microbiota dynamics on health in
the first year of life. _Nat. Commun._ 10, 4997 (2019). Article ADS PubMed PubMed Central Google Scholar * Le Doare, K. _et al._ Intrapartum antibiotic chemoprophylaxis policies for the
prevention of group B streptococcal disease worldwide: Systematic review. _Clin. Infect. Dis._ 65, S143–S151 (2017). Article PubMed PubMed Central Google Scholar * Azad, M. B. _et al._
Impact of maternal intrapartum antibiotics, method of birth and breastfeeding on gut microbiota during the first year of life: A prospective cohort study. _BJOG_ 123, 983–993 (2016). Article
CAS PubMed Google Scholar * Nogacka, A. _et al._ Impact of intrapartum antimicrobial prophylaxis upon the intestinal microbiota and the prevalence of antibiotic resistance genes in
vaginally delivered full-term neonates. _Microbiome_ 5, 93 (2017). Article PubMed PubMed Central Google Scholar * Mazzola, G. _et al._ Early gut microbiota perturbations following
intrapartum antibiotic prophylaxis to prevent group B streptococcal disease. _PLoS ONE_ 11, e0157527 (2016). Article MathSciNet PubMed PubMed Central Google Scholar * Coker, M. O. _et
al._ Specific class of intrapartum antibiotics relates to maturation of the infant gut microbiota: A prospective cohort study. _BJOG_ 127, 217–227 (2020). Article CAS PubMed Google
Scholar * Tapiainen, T. _et al._ Impact of intrapartum and postnatal antibiotics on the gut microbiome and emergence of antimicrobial resistance in infants. _Sci. Rep._ 9, 10635 (2019).
Article ADS PubMed PubMed Central Google Scholar * Huttenhower, C. _et al._ Structure, function and diversity of the healthy human microbiome. _Nature_ 486, 207–214 (2012). Article ADS
CAS Google Scholar * Moya, A. & Ferrer, M. Functional redundancy-induced stability of gut microbiota subjected to disturbance. _Trends Microbiol._ 24, 402–413 (2016). Article CAS
PubMed Google Scholar * Ranjan, R., Rani, A., Metwally, A., McGee, H. S. & Perkins, D. L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing.
_Biochem. Biophys. Res. Commun._ 469, 967–977 (2016). Article CAS PubMed Google Scholar * Douglas, G. M. _et al._ PICRUSt2 for prediction of metagenome functions. _Nat. Biotechnol._ 38,
685–688 (2020). Article CAS PubMed PubMed Central Google Scholar * Wemheuer, F. _et al._ Tax4Fun2: Prediction of habitat-specific functional profiles and functional redundancy based on
16S rRNA gene sequences. _Environ. Microbiome_ 15, UNSP 11 (2020). Article Google Scholar * Narayan, N. R. _et al._ Piphillin predicts metagenomic composition and dynamics from
DADA2-corrected 16S rDNA sequences. _BMC Genom._ 21, 56 (2020). Article CAS Google Scholar * Tapiainen, T. _et al_. Maternal influence on the fetal microbiome in a population-based study
of the first-pass meconium. _Pediatr. Res._ 84, 371–379 (2018). Article CAS PubMed Google Scholar * Ainonen, S. _et al_. Antibiotics at birth and later antibiotic courses: Effects on gut
microbiota. _Pediatr. Res._ https://doi.org/10.1038/s41390-021-01494-7 (2021). Article PubMed PubMed Central Google Scholar * Gloor, G. B., Macklaim, J. M., Pawlowsky-Glahn, V. &
Egozcue, J. J. Microbiome datasets are compositional: And this is not optional. _Front. Microbiol._ 8, 2224 (2017). Article PubMed PubMed Central Google Scholar * Zitnik, M. _et al._
Machine learning for integrating data in biology and medicine: Principles, practice, and opportunities. _Inf. Fusion_ 50, 71–91 (2019). Article PubMed Google Scholar * Mandal, S. _et al._
Analysis of composition of microbiomes: A novel method for studying microbial composition. _Microb. Ecol. Health Dis._ 26, 27663 (2015). PubMed Google Scholar * Bokulich, N. _et al._
q2-sample-classifier: Machine-learning tools for microbiome classification and regression. _J. Open Source Softw._ 3, 934 (2018). Article ADS Google Scholar * Pasolli, E., Truong, D. T.,
Malik, F., Waldron, L. & Segata, N. Machine learning meta-analysis of large metagenomic datasets: Tools and biological insights. _PLoS Comput. Biol._ 12, e1004977 (2016). Article ADS
PubMed PubMed Central Google Scholar * Bolyen, E. _et al._ Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. _Nat. Biotechnol._ 37, 852–857 (2019).
Article CAS PubMed PubMed Central Google Scholar * Callahan, B. J. _et al._ DADA2: High-resolution sample inference from Illumina amplicon data. _Nat. Methods_ 13, 581 (2016). Article
CAS PubMed PubMed Central Google Scholar * Quast, C. _et al._ The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. _Nucleic Acids Res._ 41,
D590–D596 (2013). Article CAS PubMed Google Scholar * Caspi, R. _et al._ The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases.
_Nucleic Acids Res._ 42, D459–D471 (2014). Article CAS PubMed Google Scholar * Hunter, J. D. Matplotlib: A 2D graphics environment. _Comput. Sci. Eng._ 9, 90–95 (2007). Article Google
Scholar * Fernandes, A. D., Macklaim, J. M., Linn, T. G., Reid, G. & Gloor, G. B. ANOVA-like differential expression (ALDEx) analysis for mixed population RNA-Seq. _PLoS ONE_ 8, e67019
(2013). Article ADS CAS PubMed PubMed Central Google Scholar * Lin, F.H. HuangLin/ANCOM: Third release of ANCOM. v2.1. https://doi.org/10.5281/zenodo.3577802 (2019). * Breiman, L.
Random forests. _Mach. Learn._ 45, 5–32 (2001). Article MATH Google Scholar * Geurts, P., Ernst, D. & Wehenkel, L. Extremely randomized trees. _Mach. Learning_ 63, 3–42 (2006).
Article MATH Google Scholar * Freund, Y. & Schapire, R. E. A decision-theoretic generalization of on-line learning and an application to boosting. _J. Comput. Syst. Sci._ 55, 119–139
(1997). Article MathSciNet MATH Google Scholar * Pedregosa, F. _et al._ Scikit-learn: Machine learning in python. _J. Mach. Learn. Res._ 12, 2825–2830 (2011). MathSciNet MATH Google
Scholar * Wirbel, J. _et al_. Microbiome meta-analysis and cross-disease comparison enabled by the SIAMCAT machine learning toolbox. _Genome Biol._ 22, 93 (2021). Article PubMed PubMed
Central Google Scholar * Russell, J. T. _et al_. Genetic risk for autoimmunity is associated with distinct changes in the human gut microbiome. _Nat. Commun._ 10, 3621 (2019). Article ADS
PubMed PubMed Central Google Scholar * Knight, R. _et al._ Best practices for analysing microbiomes. _Nat. Rev. Microbiol._ 16, 410–422 (2018). Article CAS PubMed Google Scholar *
Liu, Y. _et al_. A practical guide to amplicon and metagenomic analysis of microbiome data. _Protein Cell_. 12, 315–330 (2021). Article PubMed Google Scholar * Baumann-Dudenhoeffer, A.
M., D’Souza, A. W., Tarr, P. I., Warner, B. B. & Dantas, G. Infant diet and maternal gestational weight gain predict early metabolic maturation of gut microbiomes. _Nat. Med._ 24, 1822
(2018). Article CAS PubMed PubMed Central Google Scholar * Chu, D. M. _et al._ Maturation of the infant microbiome community structure and function across multiple body sites and in
relation to mode of delivery. _Nat. Med._ 23, 314–326 (2017). Article CAS PubMed PubMed Central Google Scholar * Das, P., Babaei, P. & Nielsen, J. Metagenomic analysis of
microbe-mediated vitamin metabolism in the human gut microbiome. _BMC Genom._ 20, 208 (2019). Article Google Scholar * Engevik, M. A. _et al_. Microbial metabolic capacity for intestinal
folate production and modulation of host folate receptors. _Front. Microbiol._ 10, 2305 (2019). Article PubMed PubMed Central Google Scholar * Tan, J. _et al._ The role of short-chain
fatty acids in health and disease. _Adv. Immunol._ 121, 91–119 (2014). Article CAS PubMed Google Scholar * Quince, C. _et al._ Extensive modulation of the fecal metagenome in children
with Crohn’s disease during exclusive enteral nutrition. _Am. J. Gastroenterol._ 110, 1718–1729 (2015). Article ADS CAS PubMed PubMed Central Google Scholar * Lee-Sarwar, K. A. _et
al._ Integrative analysis of the intestinal metabolome of childhood asthma. _J. Allergy Clin. Immunol._ 144, 442–454 (2019). Article CAS PubMed PubMed Central Google Scholar *
Szentirmai, E., Millican, N. S., Massie, A. R. & Kapas, L. Butyrate, a metabolite of intestinal bacteria, enhances sleep. _Sci. Rep._ 9, 7035 (2019). Article ADS PubMed PubMed Central
Google Scholar * Gao, F. _et al_. Butyrate improves the metabolic disorder and gut microbiome dysbiosis in mice induced by a high-fat diet. _Front. Pharmacol._ 10, 1040 (2019). Article
CAS PubMed PubMed Central Google Scholar * Johanson, D. M. II _et al_. Experimental autoimmune encephalomyelitis is associated with changes of the microbiota composition in the
gastrointestinal tract. _Sci. Rep._ 10, 15183 (2020). Article PubMed PubMed Central Google Scholar * Kwak, M. S., Cha, J. M., Shin, H. P., Jeon, J. W. & Yoon, J. Y. Development of a
NOVEL metagenomic biomarker for prediction of upper gastrointestinal tract involvement in patients with Crohn’s disease. _Front. Microbiol._ 11, 1162 (2020). Article PubMed PubMed Central
Google Scholar * Korpela, K. _et al_. Microbiome of the first stool and overweight at age 3 years: A prospective cohort study. _Pediatric Obesity_. 15, e12680 (2020). Article PubMed
Google Scholar * Korpela, K. _et al_. Microbiome of the first stool after birth and infantile colic. _Pediatric Res_. 88, 776–783 (2020). Article Google Scholar * Esteva, A. _et al._
Dermatologist-level classification of skin cancer with deep neural networks. _Nature_ 542, 115 (2017). Article ADS CAS PubMed PubMed Central Google Scholar * Anderson, J. P. _et al._
Reverse Engineering and evaluation of prediction models for progression to type 2 diabetes: An application of machine learning using electronic health records. _J. Diabetes Sci. Technol._
10, 6–18 (2015). Article PubMed PubMed Central Google Scholar * Zhang, Y. _et al._ Machine learning performance in a microbial molecular autopsy context: A cross-sectional postmortem
human population study. _PLoS ONE_ 14, e0213829 (2019). Article CAS PubMed PubMed Central Google Scholar * Liu, Y. _et al._ Gut microbiome fermentation determines the efficacy of
exercise for diabetes prevention. _Cell Metab._ 31, 77-91.e5 (2020). Article CAS PubMed Google Scholar * Dominguez-Bello, M. G. _et al._ Partial restoration of the microbiota of
cesarean-born infants via vaginal microbial transfer. _Nat. Med._ 22, 250–253 (2016). Article CAS PubMed PubMed Central Google Scholar * Le Goallec, A. _et al._ A systematic machine
learning and data type comparison yields metagenomic predictors of infant age, sex, breastfeeding, antibiotic usage, country of origin, and delivery type. _PLoS Comput. Biol._ 16, 1007895
(2020). Article Google Scholar * Stewart, C. J. _et al._ Temporal development of the gut microbiome in early childhood from the TEDDY study. _Nature_ 562, 583 (2018). Article ADS CAS
PubMed PubMed Central Google Scholar * Hira, Z. M. & Gillies, D. F. A review of feature selection and feature extraction methods applied on microarray data. _Adv. Bioinform._ 2015,
198363–198413 (2015). Article Google Scholar * Heaton, J. An empirical analysis of feature engineering for predictive modelling. In _SoutheastCon 2016_.
https://doi.org/10.1109/SECON.2016.7506650 (2016). * Zhou, S. _et al._ Diversity of gut microbiota metabolic pathways in 10 pairs of Chinese infant twins. _PLoS ONE_ 11, e0161627 (2016).
Article PubMed PubMed Central Google Scholar * Visconti, A. _et al._ Interplay between the human gut microbiome and host metabolism. _Nat. Commun._ 10, 4505 (2019). Article ADS PubMed
PubMed Central Google Scholar * Langille, M. G. I. _et al._ Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. _Nat. Biotechnol._ 31, 814–821
(2013). Article CAS PubMed PubMed Central Google Scholar Download references FUNDING Financial support was received from Academy of Finland, Finnish Pediatric Research Foundation,
University of Oulu Graduate School, and Oulu University Hospital, Finland. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * PEDEGO (Pediatrics, Dermatology, Gynecology, Obstetrics) Research
Unit and Medical Research Center Oulu, University of Oulu, P.O. Box 5000, 90014, Oulu, Finland Petri Vänni, Mysore V. Tejesvi, Sofia Ainonen, Katja Korpela, Jarmo Salo, Niko Paalanne &
Terhi Tapiainen * Ecology and Genetics, Faculty of Science, University of Oulu, Oulu, Finland Mysore V. Tejesvi * Department of Paediatrics, University of Eastern Finland and Kuopio
University Hospital, Kuopio, Finland Marjo Renko * Department of Pediatrics and Adolescent Medicine, Oulu University Hospital, Oulu, Finland Jarmo Salo, Niko Paalanne & Terhi Tapiainen *
Biocenter Oulu, University of Oulu, Oulu, Finland Terhi Tapiainen Authors * Petri Vänni View author publications You can also search for this author inPubMed Google Scholar * Mysore V.
Tejesvi View author publications You can also search for this author inPubMed Google Scholar * Sofia Ainonen View author publications You can also search for this author inPubMed Google
Scholar * Marjo Renko View author publications You can also search for this author inPubMed Google Scholar * Katja Korpela View author publications You can also search for this author
inPubMed Google Scholar * Jarmo Salo View author publications You can also search for this author inPubMed Google Scholar * Niko Paalanne View author publications You can also search for
this author inPubMed Google Scholar * Terhi Tapiainen View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS V.P. and M.T. did the bioinformatics
and computer analyses. M.T. did the laboratory work. T.T., R.M., K.K. and S.J. were responsible for cohorts and sample management. V.P., M.T., T.T., A.S. and P.N. wrote the article. All
authors reviewed the manuscript. CORRESPONDING AUTHOR Correspondence to Petri Vänni. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL
INFORMATION PUBLISHER'S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. The original online version of this
Article was revised: The original version of this Article contained errors in the in-text references in the Discussion and Methods sections. Full information regarding the corrections made
can be found in the correction notice for this Article. SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURE 1. SUPPLEMENTARY TABLES. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed
under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article
are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons
licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of
this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Vänni, P., Tejesvi, M.V., Ainonen, S. _et al._ Delivery mode
and perinatal antibiotics influence the predicted metabolic pathways of the gut microbiome. _Sci Rep_ 11, 17483 (2021). https://doi.org/10.1038/s41598-021-97007-x Download citation *
Received: 20 March 2021 * Accepted: 17 August 2021 * Published: 01 September 2021 * DOI: https://doi.org/10.1038/s41598-021-97007-x SHARE THIS ARTICLE Anyone you share the following link
with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative