Play all audios:
ABSTRACT Competing endogenous RNAs (ceRNAs) have gained attention in cancer research owing to their involvement in microRNA-mediated gene regulation. Previous studies have identified ceRNA
networks of individual cancers. Nevertheless, none of these studies has investigated different cancer stages. We identify stage-specific ceRNAs in breast cancer using the cancer genome atlas
data. Moreover, we investigate the molecular functions and prognostic ability of ceRNAs involved in stage I–IV networks. We identified differentially expressed candidate ceRNAs using edgeR
and limma R packages. A three-step analysis was used to identify statistically significant ceRNAs of each stage. Survival analysis and functional enrichment analysis were conducted to
identify molecular functions and prognostic ability. We found five genes and one long non-coding RNA unique to the stage IV ceRNA network. These genes have been described in previous breast
cancer studies. Genes acted as ceRNAs are enriched in cancer-associated pathways. Two, three, and three microRNAs from stages I, II, and III were prognostic from the Kaplan–Meier survival
analysis. Our results reveal a set of unique ceRNAs in metastatic breast cancer. Further experimental work is required to evaluate their role in metastasis. Moreover, identifying
stage-specific ceRNAs will improve the understanding of personalised therapeutics in breast cancer. SIMILAR CONTENT BEING VIEWED BY OTHERS INTEGRATED WHOLE TRANSCRIPTOME PROFILING REVEALED A
CONVOLUTED CIRCULAR RNA-BASED COMPETING ENDOGENOUS RNAS REGULATORY NETWORK IN COLORECTAL CANCER Article Open access 02 January 2024 CONSTRUCTION AND ANALYSIS OF PSEUDOGENE-RELATED CERNA
NETWORK IN BREAST CANCER Article Open access 10 December 2023 THE PROGNOSTIC VALUE OF CIRCULAR RNA REGULATORY GENES IN COMPETITIVE ENDOGENOUS RNA NETWORK IN GASTRIC CANCER Article 29 January
2021 INTRODUCTION Breast cancer (BC) is currently diagnosed in 1 in 8 Australian women over their lifetime, making it as the primary cause of female cancer-associated deaths in Australia.
Decades of studies have identified candidate prognostic biomarkers for BC. Recent bioinformatic and experimental studies have found that microRNAs (miRNAs) can act as biomarkers in BC as
they play a crucial role in transcriptional and post-transcriptional gene regulation. miRNAs belong to a group of small non-coding RNAs with 19–25 nucleotides in length. According to
conventional RNA logic, miRNAs inhibit/degrade gene expression binding with miRNA response elements (MREs) of messenger RNAs (mRNAs)1. Salmena, Poliseno2 introduced the competing endogenous
RNA (ceRNA) hypothesis revealing the bi-directional regulation mechanism of miRNAs. The ceRNA logic explains that non-coding RNA transcripts such as long non-coding RNAs (lncRNAs) with
similar MREs can also bind with the relevant miRNAs modulating gene regulation and protein networks. Due to these reasons, ceRNAs have gained considerable attention in cancer studies.
Previous bioinformatics and experimental studies have identified many ceRNAs associated with BC risk3. Apart from generalising results for BC incidents, different cancer stages can also
severely impact response to therapy and mortality. In BC, stage I refers to a tumour or small size confined to the breast; stage II explains the disease that has locally advanced beyond the
breast; stage III describes BC has spread to the neighbouring organs, and stage IV refers to distant metastatic disease4. The early stages, I and II, are considered treatable compared to
advanced stages, III and IV, that require more radical and active treatment strategies5. Therefore, identifying stage-specific biomarkers in BC will significantly contribute to understanding
BC biology under different pathological states. In this study, we performed a ceRNA network analysis to identify stage-specific ceRNAs in BC, and this approach can be applied for any
disease of interest in the future. The identified ceRNAs were further studied using two downstream analyses to identify their molecular functions and prognostic ability. MATERIALS AND
METHODS PATIENTS AND SAMPLES COLLECTION The expression data (RNA-seq and miRNA-seq) and clinical data of BC were collected from the cancer genome atlas (TCGA) that contains 1091 cases and
113 controls. The HTSeq-counts data of RNA-seq (including protein-coding and long non-coding) and isoform quantification data of miRNA-seq (in BC) were downloaded to a local computing server
using the genomics data commons (GDC) data portal6. For selecting miRNA-seq data for individuals, a manifest file was generated using the GDC Data Portal. Then, the GDC data transfer tool
was used to transfer data files listed in the manifest file. The differential expression analysis produced the "group" variable to identify differentially expressed protein-coding
genes and long non-coding RNAs. DIFFERENTIAL EXPRESSION ANALYSIS Firstly, we removed TCGA BC samples with duplicated sample IDs. Then samples that are neither solid tissue normal nor primary
tumour were removed as we compared primary tumour and healthy samples in the differential expression analysis. First, we performed the counts per million (CPM) normalisation to correct
sample library size differences. The low-expressed genes that log (CPM) < 1 in more than 50% of the samples were removed before the differential expression analysis7. Ignoring
low-expressed genes improves the total count of differentially expressed genes and enhances sensitivity and precision. Raw counts expression data were re-normalised using the TMM (trimmed
mean of M values) method implemented in the edgeR (3.40.0) R package (https://bioconductor.org/packages/release/bioc/html/edgeR.html) to compare expression levels between samples (excluding
low-expressed genes)8. The normalised data were transformed into a standard scale using the voom function in the limma (3.54.0,
https://bioconductor.org/packages/release/bioc/html/limma.html) (linear modelling for microarrays) R package9. Previous RNA-seq data analysis-related works have recommended this hybrid
technique, TMM normalisation with voom transformation, due to its better performance in data preprocessing10,11. Moreover, Oshlack et al. have shown TMM normalisation is robust and
outperforms library size normalisation12. In differential expression analysis, linear models were fitted for each gene using the "lmFit" function implemented in the limma (3.54.0,
https://bioconductor.org/packages/release/bioc/html/limma.html) R package9. Then eBayes moderation was applied using information across all the genes to obtain more precise estimates of
gene-wise variability. Four differential expression analyses were conducted for stage I to IV-control comparisons. The cancer stage was determined using the "pathologic stage" as
it provides more accurate information combining results from clinical examinations and surgeries. We gathered 181 (19% basal-like, 5% HER2+, 62% luminal A, 13% luminal B, and 1%
normal-like), 619 (21% basal-like, 13% HER2+, 40% luminal A, 25% luminal B, and 1% normal-like), 247 (12% basal-like, 16% HER2+, 40% luminal A, 29% luminal B, and 3% normal-like), and 20
(17% basal-like, 16% HER2+, 25% luminal A, and 42% luminal B) samples for stages I, II, III, and IV, respectively. In each stage-specific expression analysis, differentially expressed mRNAs,
lncRNAs, and miRNAs were defined at |log2-fold change (FC)|> 1 and Benjamini–Hochberg (BH)-adjusted p value (default in limma package) < 0.0513. COMPETING ENDOGENOUS RNA NETWORK
ANALYSIS The differentially expressed mRNAs, lncRNAs, and miRNAs in each cancer stage were applied in the stage-specific ceRNA network analysis. The ceRNA network analysis consists of three
main steps: (1) identifying lncRNA-mRNA pairs that share the significant number of miRNAs, (2) selecting positively correlated lncRNA-mRNA pairs, and (3) jointly estimating the significance
of multiple miRNAs in lncRNA-mRNA pairs. These three steps are described in detail in previous ceRNA papers14,15. The mRNA-miRNA and lncRNA-miRNA interactions are required to perform steps i
and iii. We used miRcode and starBase databases for miRNA-target predictions16,17. The miRcode database facilitates mRNA-miRNA and lncRNA-miRNA target predictions using a broad searchable
map that contains 10,419 lncRNAs. The starBase includes miRNA-mRNA interactions predicted by analysing 108 CLIP-seq datasets. Steps i and ii were performed using the hypergeometric test and
the Pearson correlation test, respectively. These two testing methods have been implemented in the GDCRNATools (1.18.0, https://bioconductor.org/packages/release/bioc/html/GDCRNATools.html)
R/Bioconductor package18. The third step, multiple sensitivity correlation (mscor) analysis, was executed using the SPONGE (1.20.0,
https://bioconductor.org/packages/release/bioc/html/SPONGE.html) (sparse partial correlation on gene expression) R/Bioconductor package19. The significant ceRNA interactions were filtered by
three user-defined thresholds, (1) false discovery rate (FDR) < 0.01 in hypergeometric test, (2) Pearson correlation coefficient > 0.40, and (3) the adjusted p value of mscor in
SPONGE method < 0.05. The resulting lncRNA-miRNA-mRNA associations in each BC stage were combined into a single column, as "<lncRNA gene ensemble ID>_<gene ensemble
ID>_<miRNA name>". Then set of values in each BC stage were applied into a four-sets (for four stages) Venn diagram representation. FUNCTIONAL ENRICHMENT ANALYSIS A total of
unique 47 aberrantly expressed genes (25 from stage I, 42 of stage II, 40 of stage III, and 47 of stage IV) were analysed to understand the biological functions of identified ceRNAs in this
study. The Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analyses were conducted using the R/Bioconductor clusterProfiler (4.6.0,
https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html) R package20. SURVIVAL ANALYSIS Survival analysis was performed using the Kaplan–Meier (K–M) survival curves,
implemented in the survival (3.4.0, https://cran.r-project.org/web/packages/survival/index.html) R package to explore the impact of the expression level of RNAs/miRNAs on the prognostic
survival of patients21. For each gene/lncRNA/miRNA, the tumour samples were divided into two groups (low-expressed and high-expressed) according to the median expression level. The log-rank
test (Mantel–Haenszel test) was used as the statistical method for the K–M curves. The log-rank test statistic has a chi-square (χ2) distribution with one degree of freedom. Therefore,
significant genes, lncRNAs and miRNAs were chosen under the χ2 test statistic p value < 0.05. The survival-significant mRNAs were checked for tumour-suppressive/oncogenic/cancer-driven
roles using the CancerMine database22. ETHICAL APPROVAL The study was approved by the Human Research Ethics Committees of the Queensland University of Technology (protocol code: 1900001147,
date of approval: 19 December 2019) and the QIMR Berghofer Medical Research Institute (protocol code: P1051, date of approval: 23 August 2019). RESULTS DIFFERENTIAL EXPRESSION ANALYSIS
RESULTS First, we conducted stagewise differential expression analysis to determine which genes, lncRNAs, and miRNAs are expressed at different levels between tumour and healthy groups. The
number of up/downregulated lncRNAs, genes and miRNAs are available in Table 1. The above-listed mRNAs, lncRNAs, and miRNAs were involved in the ceRNA network analysis. COMPETING ENDOGENOUS
RNA NETWORKS OF BC STAGES We constructed four ceRNA networks for BC stages I–IV. In Table 2, we have included the count of significant ceRNA associations in each BC stage. The number of
lncRNAs, genes, and miRNAs involved in stage-specific ceRNA networks is given within brackets. According to Table 2, stage II and IV analyses have resulted in a considerably large set of
ceRNA networks compared to stages I and III. The detailed list of significant lncRNA-mRNA-miRNA associations of each BC stage is available in Supplementary Information. The Cytoscape tool
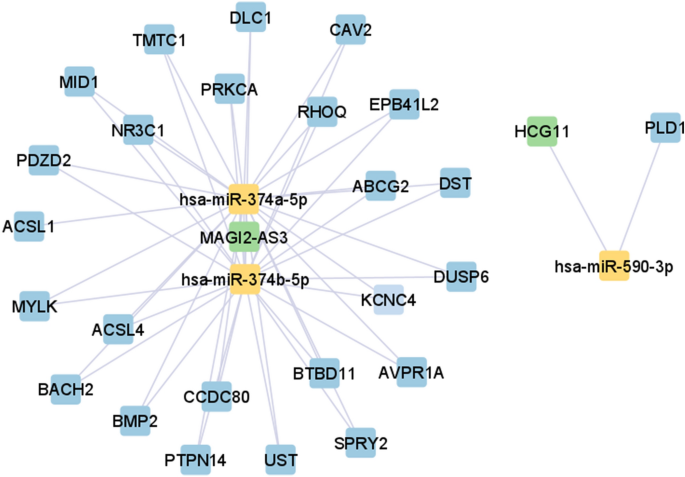
version 3.9.1 (https://cytoscape.org/download.html)23 was used to visualise ceRNA networks in each BC stage. Figures 1, 2, 3 and 4 illustrates ceRNA networks for BC stage I, II, II, and IV,
respectively. In Figs. 1, 2, 3 and 4, blue-, green-, and yellow-coloured squares represent genes, lncRNAs, and miRNAs, respectively. According to Figs. 1, 2, 3 and 4, hsa-miR-374a-5p and
374b-5p tend to build up separated ceRNA clusters in each BC stage. As shown in Fig. 4, the KLF5 gene and TRAF3IP2-AS1 lncRNA, unique to stage IV, create a unique triplet with
hsa-miR-153-3p. The lncRNAs, genes and miRNAs list of significant ceRNA associations were combined for a single variable as "<lncRNA gene ensemble ID>_<gene ensemble
ID>_<miRNA name>" (Ex: ENSG00000234456_ENSG00000125845_hsa-miR-374b-5p). The values of gene ensemble ID, lncRNA ensemble ID, and the derived variable columns were inserted into
a four-set Venn diagram representation (indicating four BC stages) as Fig. 5a–c, respectively. According to Fig. 5, most lncRNAs, mRNAs, and lncRNA-mRNA-miRNA associations found in each BC
stage ceRNA network have been shared among more than one stage. One lncRNA (TRAF3IP2-AS1) and five mRNAs (_KDR_, _SGCB_, _PRTG_, _KLF5_, and _PCNX1_) have been observed only in the
stage-IV-specific ceRNA network. We identified 29 and 15 unique ceRNA associations in stage-III and stage-IV BC, respectively. FUNCTIONAL ENRICHMENT ANALYSIS FOR STAGE-SPECIFIC COMPETING
ENDOGENOUS RNA NETWORKS We conducted functional enrichment analyses on 25, 42, 40, and 47 genes obtained from the stage I, stage II, stage III, and stage IV ceRNA networks. None of the
stage-specific genes was enriched in KEGG pathways24. Two genes resulting from stage I analysis were enriched in four CoA ligase activity-associated GO-molecular function (GO-MF) pathways.
The GO-MF hormone-binding pathway was significant across stages II, III, and IV. In stage IV, six genes were enriched in three GO-cellular components (CC) pathways, membrane raft, membrane
microdomain, and membrane region. Figure 6 illustrates GO pathway results for each BC stage. SURVIVAL ANALYSIS FOR STAGE-SPECIFIC COMPETING ENDOGENOUS RNA NETWORKS K-M survival analyses and
log-rank tests were performed to identify the potential stage-specific differentially expressed genes, lncRNAs and miRNAs strongly correlated with BC patients' prognostic
characteristics. The significant genes, lncRNAs and miRNAs, were chosen under p value < 0.05. We found two, three, three, and one gene(s) as prognostic biomarkers in stage I, stage II,
stage III, and stage IV, respectively and described in Table 3. None of the lncRNAs was identified as biomarkers in stage-specific BC survival analysis. In miRNA-based survival analyses,
two, three, and three miRNAs were statistically significant in stage I, stage II, and III, respectively. Figure 7 illustrates K–M curves for the top significant miRNA of each stage,
hsa-miR-106b-5p in stage I, hsa-miR-31-5p in stage II, and hsa-miR-551b-3p in stage III. DISCUSSION This study identified stage-specific lncRNA-mRNA-miRNA ceRNA associations in BC. According
to ceRNA network analysis results, most ceRNA associations were shared across all four stages. In contrast, one lncRNA (TRAF3IP2-AS1) and five genes (_KDR_, _PRTG_, _KLF5_, _SGCB_, and
_PCNX1_) were statistically significant only in metastatic BC, i.e., stage IV. The lncRNA TRAF3IP2-AS1 has been previously reported in renal cell carcinoma and glioblastoma but not in BC25.
According to AACR (American Association for Cancer Research) project GENIE (Genomics Evidence Neoplasia Information Exchange), the _KDR_ gene is altered in 1.54% of BC patients. It can play
an essential role in mediating endothelial cells proliferation, migration, and permeability26. Endothelial cells are actively involved in cancer metastasis27. The _PRTG_ gene has not been
previously described in BC. Nevertheless, _PRTG_ has been identified as an oncogenic protein in gastric carcinogenesis by activating the downstream cGMP/PKG signalling pathway28. The _KLF5_
gene is found to play a role in BC, but its precise function remains determined. On the one hand, the _KLF5_ locus at chromosome 13 is frequently deleted in human BC, and its protein is
degraded by the WWP1 oncogenic ubiquitin E3 ligase, which suggests a tumour-suppressor function29. On the other hand, increased expression of _KLF5_ is associated with expression of the HER2
oncoprotein and shorter survival in BC patients suggesting an oncogenic function of _KLF5_ in BC30. A recent bioinformatic study has found that SGCB protein is specific in basal A subtyped
BC gene regulatory networks31. The _PCNX1_ has been a potential marker of response to chemotherapy in BC, and therapeutic modulation of its activities could enhance chemotherapy responses32.
The BC studies mentioned above have described four out of five genes found from our stage IV-specific ceRNA network, and none of these studies explains their contribution to metastatic BC.
Therefore, wet-lab experiments will be carried out in the future to investigate their role in BC metastatic nature. Two downstream analyses, functional enrichment analysis and survival
analysis, have ensured stage-specific ceRNA components found in our study. All statistically significant genes resulting from stagewise survival analyses have been previously reported in
cancer studies22. _BMP2_ from the stage I survival analysis has shown oncogenic function in BC33. Only _DUSP6_ and _ACSL1_ in stage II have acted as an oncogene and a tumour suppressor in
BC, respectively34, 35. _DST_ and _AHNAK_ genes that were significant from the stage III survival analysis have shown tumour suppressive characteristics in both experimental and
computational BC studies36,37,38,39,40. In stage IV, we identified _MID1_ as the only survival significant gene and it has been linked with invasive lobular carcinoma22. The invasive lobular
carcinoma is the second most common type of BC. It originates in the milk-producing gland (lobules) of the breast. Invasive cancer is recognised as the cancer cells have broken out of the
lobules where they initiated and are potential to expand to the lymph nodes and other areas of the body, leading to metastasis41. Therefore, _MID1_ gene should be further investigated to
identify its role in metastatic BC (stage IV). We found eight miRNAs from the stage-specific survival analyses, and these miRNAs have shown a tumour-suppressive/oncogenic role in previous BC
experimental studies. The hsa-miR-106b-5p and hsa-miR-374b-5p were associated with the prognosis of stage I BC patients. The hsa-miR-106b-5p promoted cell migration, invasion, and
proliferation by targeting FUT642. Abnormal hsa-miR-374b-5p expression in luminal-HER2-positive BC cells can be used for classifying clinicopathologic subtypes of BC43. Three miRNAs,
hsa-miR-150-5p and hsa-miR-31-5p, are significant in stage II BC survival analysis, and hsa-miR-374b-5p was significant in both stages I and II. The other two miRNAs, hsa-miR-150-5p and
hsa-miR-31-5p, have shown an oncogenic and tumour-suppressive role in BC, respectively44,45. We identified three miRNA signatures (hsa-miR-551b-3p, hsa-miR-101-3p, and hsa-miR-26a-5p) as
prognostics in stage III BC survival. Among them, hsa-miR-551b-3p have promoted oncogenic features in BC cells46. In previous BC experimental studies, both hsa-miR-101-3p and hsa-miR-26a-5p
have shown a tumour-suppressive role47,48. We did not find statistically significant prognostic miRNAs from the stage IV BC survival analysis. Functional enrichment analyses found molecular
pathways associated with protein-coding genes in stage-specific ceRNA networks. Two genes in Acyl-CoA Synthetase Long (ACSL) Chain family, _ACSL1_ and _ACSL4_, included only in stage I ceRNA
networks, are enriched in four CoA ligase activity-associated pathways. Therefore, wet-lab experiments are required to understand the tumour-suppressive/oncogenic/cancer-driven role of ACSL
Chain family members among early-stage BC patients. Four genes found in stages II, III, and IV (_NR3C1_, _AVPR1A_, _LEPR_, and _THRB_) are associated with hormone binding, which plays a
role in BC pathophysiology and defining risk. Six genes in the stage IV ceRNA network are enriched in three components in the GO-CC pathway: membrane raft, membrane microdomain, and membrane
region. These membrane domains have shown an important role in cancer metastasis49. Our study has shared a limited set of ceRNAs with the previous ceRNA network study for overall BC cases
by Tuersong et al.3. Four miRNAs (hsa-miR-141, hsa-miR-200a, hsa-miR-204, and hsa-miR-301b) have been identified in ceRNA networks in both studies. Among these four miRNAs, Tuersong et al.3
have demonstrated that hsa-miR-204 was downregulated and hsa-miR-301b was upregulated in patients with BRCA compared with healthy controls and were associated with overall survival. Our
previous transcriptome-wide association study also demonstrated hsa-miR-204 as a tumour-suppressive miRNA in prostate cancer with statistically significant low expressed levels in prostate
cancer cell lines50. Moreover, we found two genes, _SPRY2_ and _CHL1_, involved in Tuersong et al.3 and our works (_SPRY2_ in stages I–IV and _CHL1_ in stages II, III, and IV). Observing a
smaller number of shared ceRNAs between studies can be occurred due to higher heterogeneity between breast cancer stages, and it will lead to different RNA/gene expression levels. Zhou et
al. conducted a ceRNA network analysis on BRCA subtypes, basal-like, HER2+, luminal A, and luminal B51. The authors have identified three lncRNAs, NEAT1, OPI5-AS1, and AC008124.1, among all
four subtype-related ceRNA networks. Moreover, three lncRNAs, NEAT1, FAM83H-AS1, and XIST1, were significantly differentially expressed in the basal-like subtype-related network.
Nevertheless, we could not find a shared outcome between our study and subtype-related networks. This can be due to our stage-based analyses containing RNA/gene expression levels from
multiple subtypes. This study is limited to ceRNA networks mediated by microRNA expression levels. Other genomic (copy number alteration), transcriptomic (transcription factors), and
epigenetic (DNA methylation) factors were not considered in the ceRNA network analysis52. Moreover, other possible ceRNA components such as pseudogenes and lincRNAs were not considered.
Therefore, future studies should be extended to address these concerns. Nevertheless, this study elucidates a new level of ceRNA network analysis, stage-specific ceRNA networks, to
understand better common/unique ceRNA(s) among/within the stage(s) of a given cancer. Identifying novel stage-level cancer biomarkers will significantly contribute to the knowledge of
personalised therapeutics and determining risk. CONCLUSIONS We conducted ceRNA networks analyses in four stages of BC. Only one lncRNA and five genes were significant in the stage IV BC
ceRNA network. Further validation experiments are required to characterise their role in BC metastatic nature. Identifying ceRNA components across cancer stages will advance the diagnosis,
risk identification, and therapeutics. DATA AVAILABILITY Publicly available TCGA BC RNA-seq and miRNA-seq expression data were downloaded through the GDC Data Portal
(https://portal.gdc.cancer.gov/repository). All statistical analyses and graph preparations were performed using the R statistical software, freely available at https://cran.r-project.org/.
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request. REFERENCES * Bhaskaran, M. & Mohan, M. MicroRNAs: History,
biogenesis, and their evolving role in animal development and disease. _Vet. Pathol._ 51(4), 759–774 (2014). Article CAS PubMed Google Scholar * Salmena, L. _et al._ A ceRNA hypothesis:
The Rosetta Stone of a hidden RNA language?. _Cell_ 146(3), 353–358 (2011). Article CAS PubMed PubMed Central Google Scholar * Tuersong, T. _et al._ Comprehensive analysis of the
aberrantly expressed lncRNA-associated ceRNA network in breast cancer. _Mol. Med. Rep._ 19(6), 4697–4710 (2019). CAS PubMed PubMed Central Google Scholar * Sharma, G. N. _et al._ Various
types and management of breast cancer: An overview. _J. Adv. Pharm. Technol. Res._ 1(2), 109–126 (2010). PubMed PubMed Central Google Scholar * Kwapisz, D. Oligometastatic breast cancer.
_Breast Cancer_ 26(2), 138–146 (2019). Article PubMed Google Scholar * Grossman, R. L. _et al._ Toward a shared vision for cancer genomic data. _N. Engl. J. Med._ 375(12), 1109–1112
(2016). Article PubMed PubMed Central Google Scholar * Liu, J. _et al._ Regulation of long non-coding RNA KCNQ1OT1 network in colorectal cancer immunity. _Front. Genet._ 12, 684002
(2021). Article CAS PubMed PubMed Central Google Scholar * Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A bioconductor package for differential expression analysis of
digital gene expression data. _Bioinformatics_ 26(1), 139–140 (2010). Article CAS PubMed Google Scholar * Ritchie, M. E. _et al._ limma powers differential expression analyses for
RNA-sequencing and microarray studies. _Nucleic Acids Res._ 43(7), e47 (2015). Article PubMed PubMed Central Google Scholar * Morovat, _et al._ Identification of potentially functional
circular RNAs hsa_circ_0070934 and hsa_circ_0004315 as prognostic factors of hepatocellular carcinoma by integrated bioinformatics analysis. _Sci. Rep._ 12, 4933 (2022). Article ADS CAS
PubMed PubMed Central Google Scholar * Berti, F. C. B. _et al._ Comprehensive analysis of ceRNA networks in HPV16- and HPV18-mediated cervical cancers reveals XIST as a pivotal competing
endogenous RNA. _Biochim. Biophys. Acta Mol. Basis Dis._ 1867(10), 166172 (2021). Article CAS PubMed Google Scholar * Robinson, M. D. & Oshlack, A. A scaling normalisation method for
differential expression analysis of RNA-seq data. _Genome Biol._ 254, 11 (2010). Google Scholar * Chen, F. _et al._ RNA-seq analysis identified hormone-related genes associated with
prognosis of triple negative breast cancer. _J. Biomed. Res._ 34(2), 129–138 (2020). Article PubMed PubMed Central Google Scholar * Fiannaca, A. _et al._ miRTissue (ce): Extending
miRTissue web service with the analysis of ceRNA-ceRNA interactions. _BMC Bioinform._ 21(Suppl 8), 199 (2020). Article CAS Google Scholar * Jayarathna, D. K. _et al._ Identifying complex
lncRNA/pseudogene–miRNA–mRNA crosstalk in hormone-dependent cancers. _Biology_ 10, 10 (2021). Article Google Scholar * Jeggari, A., Marks, D. S. & Larsson, E. miRcode: A map of
putative microRNA target sites in the long non-coding transcriptome. _Bioinformatics_ 28(15), 2062–2063 (2012). Article CAS PubMed PubMed Central Google Scholar * Li, J. H. _et al._
starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. _Nucleic Acids Res._ 42(Database issue), 92–97 (2014). Article ADS
Google Scholar * Li, R. _et al._ GDCRNATools: An R/bioconductor package for integrative analysis of lncRNA, miRNA and mRNA data in GDC. _Bioinformatics_ 34(14), 2515–2517 (2018). Article
PubMed Google Scholar * List, M. _et al._ Large-scale inference of competing endogenous RNA networks with sparse partial correlation. _Bioinformatics_ 35(14), i596–i604 (2019). Article
CAS PubMed PubMed Central Google Scholar * Yu, G. _et al._ clusterProfiler: An R package for comparing biological themes among gene clusters. _OMICS_ 16(5), 284–287 (2012). Article CAS
PubMed PubMed Central Google Scholar * Therneau, T. M. & Grambsch, P. M. _Modeling Survival Data: Extending the Cox Model_ (Springer, 2000). Book MATH Google Scholar * Lever, J.
_et al._ CancerMine: A literature-mined resource for drivers, oncogenes and tumor suppressors in cancer. _Nat. Methods_ 16(6), 505–507 (2019). Article CAS PubMed Google Scholar * Ideker,
T. _et al._ Cytoscape: A software environment for integrated models of biomolecular interaction networks. _Genome Res._ 13(11), 2498–2504 (2003). Article PubMed PubMed Central Google
Scholar * Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. _Nucleic Acids Res._ 28(1), 27–30 (2000). Article CAS PubMed PubMed Central Google Scholar * Yang,
L. _et al._ Low expression of TRAF3IP2-AS1 promotes progression of NONO-TFE3 translocation renal cell carcinoma by stimulating N(6)-methyladenosine of PARP1 mRNA and downregulating PTEN. _J.
Hematol. Oncol._ 14(1), 46 (2021). Article CAS PubMed PubMed Central Google Scholar * Zhang, X. _et al._ Downregulation of KDR expression induces apoptosis in breast cancer cells.
_Cell Mol. Biol. Lett._ 19(4), 527–541 (2014). Article CAS PubMed PubMed Central Google Scholar * Jahroudi, N. & Greenberger, J. S. The role of endothelial cells in tumor invasion
and metastasis. _J. Neurooncol._ 23, 99–108 (1995). Article CAS PubMed Google Scholar * Xiang, T. _et al._ The novel ZEB1-upregulated protein PRTG induced by _Helicobacter pylori_
infection promotes gastric carcinogenesis through the cGMP/PKG signaling pathway. _Cell Death Dis._ 12(2), 150 (2021). Article ADS CAS PubMed PubMed Central Google Scholar * Chen, C.
_et al._ A possible tumor suppressor role of the KLF5 transcription factor in human breast cancer. _Oncogene_ 21(43), 6567–6572 (2002). Article CAS PubMed Google Scholar * Tong, D. _et
al._ Expression of KLF5 is a prognostic factor for disease-free survival and overall survival in patients with breast cancer. _Clin. Cancer Res._ 12(8), 2442–2448 (2006). Article CAS
PubMed Google Scholar * Qin, S., Ma, F. & Chen, L. Gene regulatory networks by transcription factors and microRNAs in breast cancer. _Bioinformatics_ 31(1), 76–83 (2015). Article CAS
PubMed Google Scholar * Amri, W. A., _et a_l. MUC17 and PCNX1 as mediators of chemotherapy response in breast cancer. In San Antonio Breast Cancer Symposium. 2019. Texas. * Jung, J. W.
_et al._ An Activin A/BMP2 chimera, AB215, blocks estrogen signaling via induction of ID proteins in breast cancer cells. _BMC Cancer_ 14, 549 (2014). Article PubMed PubMed Central Google
Scholar * Kui, L. _et al._ High-throughput in vitro gene expression profile to screen of natural herbals for breast cancer treatment. _Front. Oncol._ 11, 684351 (2021). Article PubMed
PubMed Central Google Scholar * Chen, W. C. _et al._ Systematic analysis of gene expression alterations and clinical outcomes for long-chain acyl-coenzyme a synthetase family in cancer.
_PLoS One_ 11(5), e0155660 (2016). Article PubMed PubMed Central Google Scholar * Jain, P. B. _et al._ The spectraplakin Dystonin antagonises YAP activity and suppresses tumourigenesis.
_Sci. Rep._ 9(1), 19843 (2019). Article ADS CAS PubMed PubMed Central Google Scholar * Nair, V. A. _et al._ Oncogenic potential of bisphenol A and common environmental contaminants in
human mammary epithelial cells. _Int. J. Mol. Sci._ 21, 10 (2020). Article Google Scholar * Ghodke, I. _et al._ AHNAK controls 53BP1-mediated p53 response by restraining 53BP1
oligomerisation and phase separation. _Mol. Cell_ 81(12), 2596-2610.e7 (2021). Article CAS PubMed PubMed Central Google Scholar * Mehdi, S. _et al._ LY75 ablation mediates
mesenchymal-epithelial transition (MET) in epithelial ovarian cancer (EOC) cells associated with dna methylation alterations and suppression of the Wnt/β-catenin pathway. _Int. J. Mol. Sci._
21, 5 (2020). Article Google Scholar * Park, J. W. _et al._ AHNAK loss in mice promotes type II pneumocyte hyperplasia and lung tumor development. _Mol. Cancer Res._ 16(8), 1287–1298
(2018). Article CAS PubMed Google Scholar * Thomas, M. _et al._ Invasive lobular breast cancer: A review of pathogenesis, diagnosis, management, and future directions of early stage
disease. _Semin. Oncol._ 46(2), 121–132 (2019). Article PubMed Google Scholar * Li, N. _et al._ MicroRNA-106b targets FUT6 to promote cell migration, invasion, and proliferation in human
breast cancer. _IUBMB Life_ 68(9), 764–775 (2016). Article CAS PubMed Google Scholar * Li, J. Y. _et al._ Differential distribution of microRNAs in breast cancer grouped by
clinicopathological subtypes. _Asian Pac. J. Cancer Prev._ 14(5), 3197–3203 (2013). Article PubMed Google Scholar * Augoff, K. _et al._ miR-31 is a broad regulator of β1-integrin
expression and function in cancer cells. _Mol. Cancer Res._ 9(11), 1500–1508 (2011). Article CAS PubMed PubMed Central Google Scholar * Lu, Q., Guo, Z. & Qian, H. Role of
microRNA-150-5p/SRCIN1 axis in the progression of breast cancer. _Exp. Ther. Med._ 17(3), 2221–2229 (2019). CAS PubMed PubMed Central Google Scholar * Parashar, D. _et al._ miRNA551b-3p
activates an oncostatin signaling module for the progression of triple-negative breast cancer. _Cell Rep._ 29(13), 4389-4406.e10 (2019). Article CAS PubMed PubMed Central Google Scholar
* Huang, Z. M. _et al._ MicroRNA-26a-5p inhibits breast cancer cell growth by suppressing RNF6 expression. _Kaohsiung J. Med. Sci._ 35(8), 467–473 (2019). CAS PubMed Google Scholar *
Jiang, H. _et al._ MiR-101–3p and Syn-Cal14.1a synergy in suppressing EZH2-induced progression of breast cancer. _Oncol. Targets Ther._ 13, 9599–9609 (2020). Article CAS Google Scholar *
Greenlee, J. D. _et al._ Rafting down the metastatic cascade: The role of lipid rafts in cancer metastasis, cell death, and clinical outcomes. _Cancer Res._ 81(1), 5–17 (2021). Article CAS
PubMed Google Scholar * Jayarathna, D. K. _et al._ Integrative transcriptome-wide analyses uncover novel risk-associated microRNAs in hormone-dependent cancers. _Front. Genet._ 20, 26
(2021). Google Scholar * Zhou, S. _et al._ Systematical analysis of lncRNA-mRNA competing endogenous RNA network in breast cancer subtypes. _Breast Cancer Res. Treat._ 169(2), 267–275
(2018). Article CAS PubMed Google Scholar * Jayarathna, D. K. _et al._ A supervised machine learning approach identifies gene-regulating factor-mediated competing endogenous RNA networks
in hormone-dependent cancers. _J. Cell. Biochem._ 20, 20 (2022). Google Scholar Download references ACKNOWLEDGEMENTS The datasets reported in this work originated from the TCGA Research
Network: https://www.cancer.gov/tcga. The computational resources and services used in this work were provided by the eResearch Office, Queensland University of Technology, Brisbane,
Australia. FUNDING This research was supported by QUT postgraduate research allowance (QUTPRA); QUT HDR tuition fee sponsorship; Advance Queensland Industry Research Fellowship; the NHMRC
Career Development Fellowship; and the Cancer Council Queensland Grant. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Centre for Genomics and Personalised Health, School of Chemistry and
Physics, Queensland University of Technology, 2 George Street, Brisbane, QLD, 4000, Australia Dulari K. Jayarathna, Jyotsna Batra & Neha S. Gandhi * Department of Genetics and
Computational Biology, QIMR Berghofer Medical Research Institute, Brisbane, QLD, 4006, Australia Dulari K. Jayarathna & Miguel E. Rentería * School of Biomedical Sciences, Faculty of
Health, Queensland University of Technology, Kelvin Grove, Brisbane, QLD, 4059, Australia Miguel E. Rentería & Jyotsna Batra * Translational Research Institute, 37 Kent Street,
Woolloongabba, QLD, 4102, Australia Jyotsna Batra & Neha S. Gandhi Authors * Dulari K. Jayarathna View author publications You can also search for this author inPubMed Google Scholar *
Miguel E. Rentería View author publications You can also search for this author inPubMed Google Scholar * Jyotsna Batra View author publications You can also search for this author inPubMed
Google Scholar * Neha S. Gandhi View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS D.K.J., N.S.G., and M.E.R. designed the study. D.K.J.
conducted statistical analysis and data interpretation. D.K.J. prepared the manuscript. N.S.G., M.E.R, and J.B. supervised the study. All authors reviewed the manuscript. CORRESPONDING
AUTHOR Correspondence to Neha S. Gandhi. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER'S NOTE Springer Nature
remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION. RIGHTS AND PERMISSIONS OPEN ACCESS
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as
long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third
party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the
article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the
copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Jayarathna, D.K., Rentería,
M.E., Batra, J. _et al._ Integrative competing endogenous RNA network analyses identify novel lncRNA and genes implicated in metastatic breast cancer. _Sci Rep_ 13, 2423 (2023).
https://doi.org/10.1038/s41598-023-29585-x Download citation * Received: 04 October 2022 * Accepted: 07 February 2023 * Published: 10 February 2023 * DOI:
https://doi.org/10.1038/s41598-023-29585-x SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative