Play all audios:
ABSTRACT Dopaminergic degeneration is a central feature of Parkinson’s disease (PD), but glial dysfunction may accelerate or trigger neuronal death. In fact, astrocytes play a key role in
the maintenance of the blood–brain barrier and detoxification. 6-hydroxydopamine (6OHDA) is used to induce PD in rodent models due to its specific toxicity to dopaminergic neurons, but its
effect on astrocytes has been poorly investigated. Here, we show that 6OHDA dose-dependently impairs autophagy in human U373 cells and primary murine astrocytes in the absence of cell death.
LC3II downregulation was observed 6 to 48 h after treatment. Interestingly, 6OHDA enhanced NRH:quinone oxidoreductase 2 (NQO2) expression and activity in U373 cells, even if 6OHDA turned
out not to be its substrate. Autophagic flux was restored by inhibition of NQO2 with S29434, which correlated with a partial reduction in oxidative stress in response to 6OHDA in human and
murine astrocytes. NQO2 inhibition also increased the neuroprotective capability of U373 cells, since S29434 protected dopaminergic SHSY5Y cells from 6OHDA-induced cell death when cocultured
with astrocytes. The toxic effects of 6OHDA on autophagy were attenuated by silencing NQO2 in human cells and primary astrocytes from _NQO2_−/− mice. Finally, the analysis of Gene
Expression Omnibus datasets showed elevated NQO2 gene expression in the blood cells of early-stage PD patients. These data support a toxifying function of NQO2 in dopaminergic degeneration
via negative regulation of autophagy and neuroprotection in astrocytes, suggesting a potential pharmacological target in PD. SIMILAR CONTENT BEING VIEWED BY OTHERS MITOCHONDRIAL GLUTAMINE
TRANSPORTER SLC1A5_VAR, A POTENTIAL TARGET TO SUPPRESS ASTROCYTE REACTIVITY IN PARKINSON’S DISEASE Article Open access 09 November 2022 METABOLIC ALTERATIONS IN PARKINSON’S DISEASE
ASTROCYTES Article Open access 02 September 2020 CONNEXIN 43 IS DOWNREGULATED IN ADVANCED PARKINSON’S DISEASE IN MULTIPLE BRAIN REGIONS WHICH CORRELATES WITH SYMPTOMS Article Open access 25
March 2025 INTRODUCTION PD is a neurodegenerative disorder mainly characterized by progressive loss of dopaminergic neurons in the _substantia nigra_. The exact mechanisms of dopaminergic
neuronal loss in the SN are not fully understood, but several factors have been implicated in the onset and progression of PD. Among them are mitochondrial damage and oxidative stress (OS)
caused by alterations in iron and redox metabolism and/or excessive production of toxic dopamine metabolites, followed by protein misfolding and aggregation associated with defective
autophagy and “prion-like protein infection”1,2,3,4. Autophagy is a catabolic process responsible for the removal of protein aggregates and damaged organelles via lysosomal digestion5,6.
Autophagy machinery is also involved in the regulation of related processes with an increasingly recognized role in PD, such as vesicular transport, exocytosis and phagocytosis5,7. Numerous
genetic mutations encoding components of the autophagic machinery have been identified as disease risk factors1,8,9. Notably, autophagy dysregulation is also associated with reactive oxygen
species (ROS) in both cellular signaling and damage10. In particular, all chemical agents inducing ROS and dopaminergic damage, known as Parkinsonian toxins, have been shown to dysregulate
or impair autophagy1,11 in both neurons and astrocytes. Astrocytes maintain redox balance in the brain through antioxidant production and detoxification12, contribute to the formation and
maintenance of the blood–brain barrier13 and regulate inflammatory responses in the central nervous system14,15. Increasing evidence highlights the contribution of astrocyte dysfunction to
the pathogenesis of several neurodegenerative conditions, including PD16. DA metabolism is the main source of ROS in the brain17. Functional glial cells protect neurons against OS by
metabolizing DA via monoamine oxidase-B (MAO-B) and catechol-_O_-methyltransferase (COMT) and by a battery of antioxidant enzymes present in astrocytes18. Prolonged dysfunction of
astrocytes, involving autophagy and ROS detoxification processes, could increase the vulnerability of DA neurons and advance their degeneration during aging, leading to PD16,19. 6OHDA is
widely used to investigate the pathogenesis of PD in preclinical studies since it induces PD-like symptoms in rodents20. 6OHDA is a DA analog that can be produced by DA hydroxylation in the
presence of Fe2+ iron and H2O2. 6OHDA is considered to be a purely synthetic toxin, but several observations suggest the possibility of its endogenous origin, likely during DA catabolism.
Indeed, it has been detected in caudate samples as well as in the urine of PD patients21,22 and in mouse brains following long-term L-dopa administration23. It shows specific PD-like
effects24,25 due to its toxic oxidation metabolites. Upon injection in the brain, it causes selective death of dopaminergic neurons and proinflammatory activation of microglia, while its
effects on the astrocytic compartment are a matter of debate26,27. The mechanisms of 6OHDA toxicity are complex and involve rapid auto-oxidation, leading to the generation of hydrogen
peroxide and hydroxyl radicals28 and impairment of mitochondrial energy production29,30. It is also unclear whether any endogenous toxifying enzymes, such as quinone oxidoreductase 2
(NQO2/QR2), may contribute to this process31,32. NQO2 is a cytosolic and ubiquitously expressed flavoprotein that catalyzes the two-electron reduction of quinone to hydroquinone33. NQO2,
structurally related to NQO1, is considered a phase II detoxifying enzyme, but it enhances the production of activated quinones and ROS in the presence of certain substrates33,34. NQO2
emerged as a possible target in PD more than two decades ago. A case–control Japanese study identified a positive association of a nonfamiliar form of PD with a D (deletion) polymorphism in
the NQO2 promoter region. This “gain of function” variant of the promoter lacking the Sp3 transcriptional suppressor binding site was 3.46 times more frequent in PD patients than in healthy
subjects35. Several other data support a potential role of NQO2 in PD and other neurodegenerative diseases36. NQO2 may play a role in dopamine metabolism as a catechol quinone reductase37
and may negatively influence memory formation and learning processes in mice38. Next, chronic OS induced by the parkinsonian toxin paraquat (PQ) is potently inhibited by the specific NQO2
inhibitor S29434/NMDPEF 39 in vitro and in vivo in rats following intranigral and systemic injection of PQ32. PQ has been shown to inhibit basal autophagy, while treatment with S29434
stimulated autophagy in astrocytes31. Recent data indicate that the function of this enzyme is not limited to the generation of OS and inhibition of autophagy, but NQO2 plays a role in
flavone-induced autophagy by acting as a receptor of proautophagic ligands, including flavonoids and S2943440. Here, we investigated the toxic effects of 6OHDA and the related role of NQO2
in human and murine astrocytes. We show that 6OHDA potently and dose-dependently inhibited autophagic flux in U373 and primary mouse astrocytes, but S29434 restored and enhanced autophagy in
astrocytes and reduced neuronal damage in DA neurons cocultured with astrocytes. A possible role of NQO2 in PD pathogenesis is supported by the higher expression of NQO2 in the majority of
PD patients when compared to healthy subjects. These data indicate a toxifying function of NQO2 in dopaminergic degeneration via negative regulation of autophagy and neuroprotection in
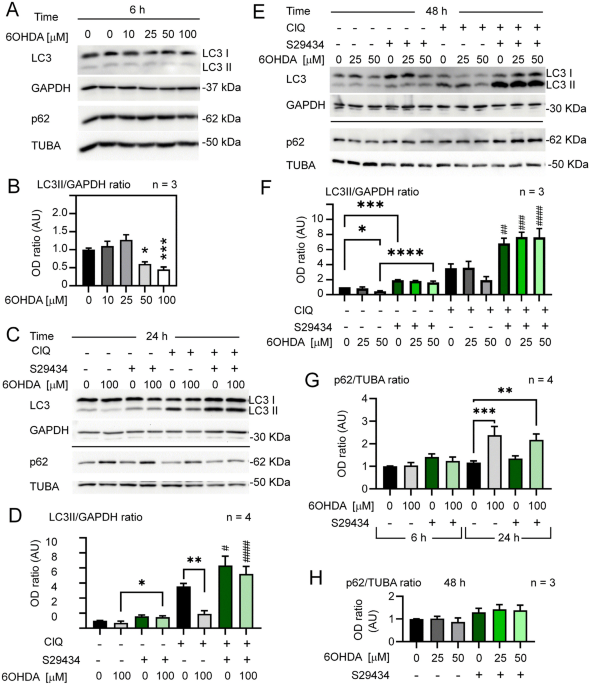
astrocytes, suggesting it as a potential pharmacological target in PD. RESULTS DOSE-DEPENDENT DOWNREGULATION OF AUTOPHAGY BY 6OHDA AND STIMULATION OF AUTOPHAGY BY NQO2 INHIBITION To examine
the effects of 6OHDA on autophagy in astrocytes, we assessed LC3II levels in U373 cells cultured under optimal growth conditions (70–80% confluency, 4.5 g/L glucose). LC3 II levels were not
significantly modulated at lower 6OHDA concentrations and were significantly reduced by 50 and 100 µM toxin 6 h after the treatment (Fig. 1A,B) as well as at 24 h (Fig. 1C,D). As expected,
we observed an increased reduction in LC3II at 24 h with respect to 6 h (Supplementary Fig. S1). To assess whether the reduction in LC3II in response to 6OHDA was a true and long-lasting
inhibition of autophagic flux, we examined the LC3II levels in the presence and absence of the lysosomal inhibitor chloroquine (ClQ) at 24 h after treatment. In the same experiment, U373
cells were cotreated with the NQO2 inhibitor S29434 to address whether LC3II modulation by 6OHDA was NQO2-dependent. Indeed, 6OHDA (100 mM) had a strong negative effect on autophagic flux at
24 h (Fig. 1C,D), as well as 50 mM at 48 h (Fig. 1E,F). Treatment with 25 mM 6OHDA had no significant effect on LC3II induction (Fig. 1E,F). The control samples in the presence of ClQ were
several times higher but reduced by more than 50% or unchanged in 6OHDA-treated samples, depending on the dose, thus confirming a suppression of LC3II by higher concentrations of this toxin.
Remarkably, the addition of S29434 potently restored LC3II levels in 6OHDA-intoxicated U373 cells compared to cells without S29434 (Fig. 1C–F). This was associated with a significant
increase in p62/SQSTM1 levels after 24 h (Fig. 1C,G) but not at 6 and 48 h posttreatment (Fig. 1A,E,G). Interestingly, a similar time-dependent pattern of induction was observed for NQO2
(Supplementary Fig. S2). THE EFFECTS OF 6OHDA ON ROS LEVELS Astrocytes are known for their capacity to counteract OS induced by various sources. Therefore, the lack of autophagy stimulation
observed here could depend on the absence of OS in response to 6OHDA. For this reason, we measured the OS induced by 6OHDA. Interestingly, the fluorescein derivative DCF-DA detected a
significant OS increase only at higher doses of 6OHDA (Fig. 2A), which effectively suppressed autophagy (Fig. 1B), suggesting that 6OHDA-induced OS inhibits autophagy, similar to what was
demonstrated for PQ-induced OS31. Next, we compared PQ- to 6OHDA-induced OS and the impact of S29434 on ROS formation in response to both compounds, as detected by the fluorescent probe
MitoSox. This dye monitors the temporal changes in hydroxyradical and superoxide anions in the mitochondria and cytoplasm. 6OHDA (100 µM) induced a time-dependent increase in MitoSox
fluorescence, with a remarkable peak of activity 15 h posttreatment (Fig. 2B), much stronger than in the case of 100 µM PQ. Cotreatment with S29434 (10 µM) attenuated ROS induction by 6OHDA
at each tested time point, but the effect was only partial, ranging from a 30% to 50% reduction, compared to the almost full attenuation of the MitoSox signal in the presence of PQ and
S29434 (Fig. 2B). Thus, OS induced by 6OHDA is more potent but less persistent and less sensitive to S29434 than PQ-induced OS. Nevertheless, the findings in Figs. 1 and 2 indicate that
6OHDA-induced toxicity suppresses basal autophagy, which can be equally well restored by S29434 in astrocytes as previously shown for PQ, despite important differences in OS dynamics between
both parkinsonian toxins. 6OHDA EFFECTS ON CELLULAR AND IN VITRO NQO2 ACTIVITY Next, we addressed whether 6OHDA can influence cellular NQO2 activity in astroglial cells when added to cell
culture. To this end, U373 cells were treated for 24 h with 6OHDA (50 and 100 mM), and cell lysates were assayed for their capacity to reduce menadione (K3) and oxidase BNAH, which are
commonly used substrates and cosubstrates, respectively, for the detection of NQO2 activity. U373 cell lysates pretreated with 6OHDA showed higher NQO2 activity than control cells in the
presence of K3 but also when no exogenous substrate was used (Fig. 3A,B). Next, we addressed whether 6OHDA or its autooxidation products, which are rapidly produced in solution41, might be
substrates of the quinone oxidoreductase activity in vitro. 6OHDA was added as the only substrate in the reaction mixture containing purified recombinant human NQO2 and BNAH as a
cosubstrate. 6OHDA dose-dependently tended to accelerate BNAH oxidation, but this effect was not statistically significant when compared to spontaneous BNAH decay in the presence of the
enzyme (without substrate) and more than 50 times lower when compared to NQO2 activity in the presence of K3 (100 mM), which was used as a positive control (Fig. 3C,D). These data indicate
that 6OHDA is not a substrate of NQO2, but 6OHDA dose-dependently induces NQO2 activity in astrocytes, which can be well explained by an increased expression of NQO2 in response to 6OHDA
(Supplementary Fig. S2). U373 ASTROCYTES ARE RESISTANT TO 6OHDA-INDUCED TOXICITY IN OPTIMAL CULTURE CONDITIONS IN DOPAMINERGIC NEUROBLASTOMA CELLS The negative effect on autophagy in U373
astroglial cells might be a consequence of general 6OHDA-induced toxicity leading to cell death. Nevertheless, we were able to detect only background cell death (≈2%) in optimal culture
conditions, which were standard conditions applied in all autophagy experiments (Fig. 4A). To detect significant cell mortality in response to 6OHDA, U373 cells had to be exposed to
additional stress factors, such as incubation in low glucose after high-glucose medium (Fig. 4B) or seeding at very low density (Fig. 4C). Interestingly, S29434 efficiently reduced cell
death, also in suboptimal growth conditions, partially dependent on the presence of the toxin (Fig. 4B,C), with a significant effect at lower concentrations of 6OHDA (25 and 50 µM) in
sparsely seeded U373 cells (Fig. 4C). Importantly, U373 cells overexpressing NQO2, were more sensitive to 6OHDA and showed higher mortality than U373 cells when assessed at a low cell
density (Supplementary Fig. S3), thus supporting a toxifying effect of NQO2 in cells exposed to 6OHDA. Next, we examined the potential involvement of NQO2 in 6OHDA–induced toxicity in
neuroblastoma cells (SH-SY5Y), a commonly used in vitro model for dopaminergic neurons. SH-SY5Y cells were much more sensitive to 6OHDA than U373 cells and were massively dying 24 h after
the addition of 10 µM 6OHDA. Treatment with S29434 did not prevent cell death in SH-SY5Y cells (Fig. 4D). Thus, 6OHDA reduces autophagic flux in U373 cells, even though these cells are
highly resistant to 6OHDA-induced cell death. This contrasts with SH-SY5Y cells that rapidly die in response to 6OHDA and are not protected by S29434. SUPPRESSION OF BASAL AUTOPHAGY BY 6OHDA
IN U373 CELLS AND PRIMARY MURINE ASTROCYTES IS DEPENDENT ON NQO2 STATUS Next, we determined whether NQO2 is a key player in 6OHDA-induced autophagy dysfunction. To answer this question, we
silenced NQO2 in 6OHDA-treated U373 cells. Our results indicated a dose-dependent decrease in LC3II levels in cells 24 h after treatment with 25–50 µM 6OHDA (Fig. 5A,B). However, _NQO2_
silencing restored or enhanced autophagy in cells exposed to higher (50 µM) and lower (25 µM) doses of toxin, respectively, although the effect was statistically significant only with 50 µM
6OHDA (Fig. 5B). In fact, a stimulatory effect on LC3II levels was statistically significant in cells expressing lower levels of NQO2 (si_NQO2_) in response to 25 µM 6OHDA doses when
analyzed by less stringent statistical tests (unpaired T test, P = 0.0418; one-way ANOVA and uncorrected Fisher’s LSD test, P = 0.0145). In conclusion, the silencing of NQO2 partially
restores autophagy in cells exposed to 6OHDA. These data support the importance of NQO2 in toxin-induced autophagy defects in human astrocytes. To confirm the glioprotective effect of S29434
in astrocytes, we established primary cultures of cortical astrocytes from C3H mice and treated them with 6OHDA (50 and 100 µM) in the absence and presence of S29434. These concentrations
of the parkinsonian toxin were not able to induce any significant cell death (data not shown), but WB analysis revealed that 24 h treatment with 6OHDA inhibited autophagic flux by decreasing
LC3II levels in a dose-dependent fashion. This was evident only in the presence of ClQ, since in these experimental conditions (90–100% confluent monolayers), it was more difficult to
detect LC3II without preincubation with ClQ (Fig. 5C). Of note, S29434 increased LC3II levels in cells exposed to 6OHDA, indicating the involvement of NQO2 in the negative regulation of
autophagy in murine astrocytes under 6OHDA stress, although it did not induce a statistically significant increase in LC3II levels in control cells (Fig. 5D). To further assess the role of
NQO2 in autophagic flux regulation upon 6OHDA intoxication, we analyzed LC3II levels in primary cortical astrocytes from mice lacking NQO2 (_NQO2-/-_) and compared them with their
_wild-type_ (_WT_) counterparts. These cells were plated and analyzed under the standard protocol, but another anti-LC3 antibody was employed, allowing LC3II detection w/o ClQ. The analysis
of LC3 expression upon 24 h of treatment with 6OHDA revealed a downregulation of LC3II only in _WT_ but not in KO astrocytes (Fig. 5E), and the difference was statistically significant in
the presence of ClQ (Fig. 5F). The absence of NQO2 also attenuated the upregulation of p62 in response to 6OHDA (Fig. 5E,G), which suggests a role of NQO2 in the regulation of autophagy in
astrocytes in response to oxidative insults. S29434 ENHANCES THE PROTECTIVE EFFECT OF U373 CELLS ON DOPAMINERGIC SH-SY5Y CELLS AGAINST 6OHDA-INDUCED CELL DEATH IN COCULTURE EXPERIMENTS The
primary role of astrocytes is to protect neurons from oxidative insults. Their neuroprotective capacity can be assessed in vitro in human astrocyte-neuron cocultures. We developed a simple
coculture assay by overlaying U373 astrocyte monolayers with SH-SY5Y cells and scoring the viability of the respective cell populations in the presence of toxic insults31. In this assay,
SH-SY5Y cells were labeled with a green-fluorescent dye (DFCA) before mixing the two cell types. Subsequently, the monolayers of naïve U373 or U373 cells overexpressing NQO2 (N-over cells,
Fig. 6E) were overlaid with a DFCA-labeled dopaminergic cells. After 24 h, cocultured cells were exposed to 6OHDA for the next 48 h in the presence or absence of 10 µM S29434 (Fig. 6B). Cell
mortality was assessed by flow cytometry with 7-AAD staining (Fig. 6A). We found that dopaminergic SH-SY5Y cells were potently protected by U373 monolayers from 6OHDA-induced cell death,
and we could observe SH-SY5Y mortality only at very high doses of 6OHDA (50 and 100 µM), while N-over U373 cells were more sensitive to the neurotoxin and less protective with respect to
SH-SY5Y cells (Fig. 6A,C). Importantly, the protective effect of U373 astrocytes was strongly enhanced by S29434, both in normal cells and in N-over cells (Fig. 6A–C). NQO2 overexpression as
well as its inhibition by S29434 in astrocytes exerted a highly significant effect, especially for SH-SY5Y cells, when analyzed by three-way ANOVA (Fig. 6D). As expected, the extent of the
protection by S29434, calculated after subtracting basal mortality of SH-SY5Y cells in coculture with normal U373 cells (around 3 to 5%), was dependent on the concentration of 6OHDA and was
around 44% at 100 µM, but reached full protection at lower doses, such as 10 µM (Fig. S5A and S5B). These experiments demonstrate that modulating NQO2 activity and protein levels has a
strong impact on dopaminergic neuron survival when in coculture with astrocytes, in contrast to monoculture, as shown in Fig. 4D. THE ANALYSIS OF NQO2 GENE EXPRESSION IN PD PATIENTS REVEALS
DIFFERENCES COMPARED TO HEALTHY INDIVIDUALS Thus far, our data suggest that high NQO2 levels or activity may increase the sensitivity of astrocytes and nearby neurons to OS and parkinsonian
toxins. However, it is not clear whether high NQO2 levels may predispose patients to PD or other OS-dependent neurodegenerative diseases because _NQO2_ gene expression and protein levels
have never been analyzed in PD patients. Public gene expression databases are unexplored resources in this respect. We analyzed the Gene Expression Omnibus (GEO) datasets of Affymetrix Human
Genome arrays. We identified a few studies that analyzed gene expression in PD patients of thousands of genes, including _NQO2_. One study analyzed genome-wide gene expression in the whole
blood of early-stage PD patients (n = 50) and compared them to healthy donors (n = 22)42. The analysis of GSE6613 datasets revealed a high variability and nonnormal distribution of _NQO2_
expression in early PD patients, but most cases [73%] showed higher expression than the central tendency in healthy patients, which was statistically significant with and without two
potential outliers (Fig. 7A). We also observed a higher median _NQO2_ expression in white blood cells of PD patients in a smaller dataset (GSE100054) comprising 10 cases and 9 controls, but
it was not significant (Fig. 7B)43. Finally, we analyzed the GSE8397 datasets from postmortem brain specimens of 24 late-stage PD patients and 16 controls44,45 available for two probes
targeting the _NQO2_ gene (GSE8397-U133A and U133B). Surprisingly, lateral and media _substantia nigra_ (L/M-SN) from PD patients presented significantly lower expression of _NQO2_ than that
from healthy donors, even after excluding three very low expression outliers (p = 0.04, n = 21 vs n = 15). In this case, 71% of PD cases showed lower NQO2 expression compared to the mean of
control cases (Fig. 7C). In contrast, the expression levels detected by the second probe, corresponding to long noncoding RNA (lncRNA) of _NQO2_ (_lncNQO2_), showed no significant increase
in PD samples compared to the mean _lncNQO2_ expression levels in healthy samples (Fig. 7D). The GSE8397 dataset also contains the data from the _superior frontal gyrus_ (SFG), but owing to
the low number of samples (n = 5), it is difficult to draw any conclusions (Fig. 7C,D). These data strongly support a possible involvement of NQO2 in PD etiology. DISCUSSION The important
finding of the present work is the identification of NQO2 as a player in autophagy and in the impairment of neuroprotective capacity in astrocytes induced by 6OHDA. This compound is a model
dopaminergic neurotoxicant, but may also represent toxic endogenous dopamine metabolites20,23. Similar to other parkinsonian toxins, 6OHDA reproduces the main cellular processes of PD , such
as OS, neurodegeneration, neuroinflammation, and neuronal death by apoptosis, although it is controversial whether 6OHDA-induced animal PD models accumulate a-synuclein aggregates similar
to Lewy bodies, typical features of human PD46. The mechanism of 6OHDA toxicity has been linked to ROS production by extracellular auto-oxidation47, leading to intracellular OS induction in
various cell types31,48. In agreement with this, we observed that mitochondrial and cytoplasmic ROS increased in a dose- and time-dependent fashion in U373 cells, but the intensity and
duration of the ROS burst were totally different than in the case of PQ. In fact, OS induced by 6OHDA was more rapidly increasing and disappearing and several times stronger than that
induced by PQ. Nevertheless, the effects of both neurotoxins on autophagy were similar and consistent with the concept that persistent and/or high levels of OS inhibit rather than stimulate
autophagy. Indeed, the exposure of U373 astrocytes to increasing 6OHDA doses had no effect or significantly downregulated autophagic machinery as evidenced by a more than 50% decrease in LC3
lipidation when 100 µM 6OHDA was applied for 24 h. This correlated with a significant increase in p62 levels at 24 h after the addition of 6OHDA (Fig. 1C,G) but not at 48 h (Fig. 1H), as
previously found for PQ31. It is likely, however, that the lack of p62 upregulation is due to the very low solubility of p62 aggregates that accumulate at later time points. In addition, the
exposure of astrocytes to different concentrations of 6OHDA led to no or marginal lethality under optimal culture conditions. In contrast, in our previous study, we were not able to
distinguish between PQ-induced general toxicity and PQ-dependent autophagy dysfunction in astrocytes, since a clear reduction in autophagic flux preceded massive cell death that took place
at 72 h31. Thus, despite evident differences in OS dynamics and cellular toxicity, both toxins efficiently lead to autophagy dysfunction in astrocytes. In contrast to the data presented
here, a net induction of autophagy was widely reported for dopaminergic neurons in vitro in response to 6OHDA, MPTP, and other parkinsonian toxins11. However, this apparent autophagy
stimulation in vitro by 6OHDA is not sufficient since many potent autophagy inducers protect dopaminergic neurons from cell death1,49,50. A net induction of autophagy does not occur in
astrocytes, likely because certain inhibitory pathways activated by toxins, comprising the NQO2 pathway reported here, counterbalance any proautophagic signals triggered by OS. This concept
is well supported not only by our observations in astrocytes1,31 but also by other authors in several alternative models of PD based on neurotoxicants51,52,53,54,55. However, robust evidence
of net autophagic flux modulation by 6OHDA or other dopamine metabolites in _vivo_ is missing. Only one paper that investigated 6OHDA effects on autophagy in rodents reported LC3II and p62
upregulation in lesioned SN samples, which would suggest inhibition of autophagic flux, but these findings were relative to 4 weeks upon treatment and were not confirmed by cotreatment with
lysosomotropic agents50. Importantly, all authors agree that pharmacological stimulation of autophagy by Torin1, curcumin or NRF2 inducers in 6OHDA-treated rodents correlates with
neuroprotective effects49,50,55, suggesting that 6OHDA causes defective rather than excessive autophagy. When autophagic flux is reduced by decreased LC3II production, autophagosome
transport blockade and/or defective lysosomal function, specialized forms of autophagy, such as mitophagy are also impaired, leading to the accumulation of dysfunctional mitochondria.
Autophagy and mitophagy impairment by genetic or epigenetic mechanisms is strictly linked to the progression of PD1,9, and recent reports suggest that a possible autophagy/mitophagy defect
may reside in astrocytes19,56. With emerging evidence demonstrating a broader role of glial mitochondria in detoxification processes57 and astrocyte dysfunction in PD pathogenesis16,19, it
is likely that parkinsonian toxins, such as 6OHDA and PQ, damage mitochondria and impair glial mitophagy at the same time, leading to the impairment of their neuroprotective capacity, which
heavily relies on mitochondrial function in astrocytes57,58. Therefore, it is possible that mitochondria are involved in the neuroprotective effect of the NQO2 inhibitor presented in this
work (Fig. 6), even though NQO2 is a cytoplasmic enzyme33, and preliminary experiments exclude its mitochondrial localization (E. Janda personal communication). Future studies should shed
more light on a possible role of NQO2 in mitophagy and mitochondrial function in the glial compartment. A potential role of NQO2 in the neurodegenerative process is also supported by its
upregulation of both protein levels and activity in cells exposed to 6OHDA (Fig. 3; Fig. S2). This observation is in line with our previous findings obtained with cells exposed to PQ and
then assayed in reactions containing BNAH and K332. In addition, we showed that cells pretreated with 6OHDA sustained faster BNAH oxidation in the absence of exogenous substrates, such as K3
(Fig. 3A), suggesting that 6OHDA metabolism leads to the production of quinones that act as substrates for NQO2 in vivo. Such quinones are not produced in vitro and 6OHDA itself is not a
substrate for NQO2 (Fig. 3C) but could be produced in cells_._ Our previous results obtained with PQ also support this hypothesis31, but futher experiments are required to validate it.
Another novel finding of the present work is that NQO2 contributes much less to 6OHDA-induced ROS induction than to autophagy impairment in astrocytes. In fact, S29434 almost doubled
autophagy levels in control cells and fully restored or even upregulated autophagic flux in U373 cells exposed to 6OHDA (Fig. 1D,F). In contrast, S29434 had a moderate effect on OS induced
by the toxin, reducing it by 25% at the peak oxidative burst (Fig. 2B), suggesting that the main beneficial activity of the compound is linked to autophagy stimulation. However, the murine
astrocytes were less responsive to S29434, since the compound produced a significant stimulation of autophagy only in 6OHDA-treated cells (Fig. 5C,D). Nevertheless, the experiments with
_NQO2_ − / − primary astrocytes provide key evidence for an important role of NQO2 in the negative regulation of LC3II induction. In contrast to _WT_ cells, astrocytes lacking NQO2 were
resistant to 6OHDA-induced LC3II downregulation, and the upregulation of p62 was significantly reduced in these cells, indicating that autophagic flux was less impaired by 6OHDA in the
absence of NQO2. In addition, we observed a statistically significant reduction in LC3II levels and lower p62 expression in knockout cells, indicating differences in autophagic machinery
between _WT_ and _NQO2_-deficient astrocytes. To our knowledge, this is the first analysis of autophagic marker expression in _NQO2_ − / − cells. Overall, these and our previous data as well
as a recent work reporting that negative regulation of autophagy by fluoride is NQO2-dependent59 strongly suggest that NQO2 is an important modulator of autophagy. The question of whether
it is mediated by its enzymatic activity or by the formation of NQO2 complexes with its protein partners hypothetically regulated by S29434 and its other inhibitors, as suggested
previously39, needs to be further investigated. Another important issue is the relevance of our in vitro model to complex pathogenic mechanisms in PD. The causative role of toxic dopamine
metabolites in PD pathogenesis is well supported by several observations60, but robust evidence of autophagic flux inhibition by 6OHDA or other dopamine metabolites in vivo is missing in the
literature and in the present paper. In addition, the coculture of U373 cells with SH-SY5Y cells provides a proof of concept that the toxicity of 6OHDA to neurons can be attenuated by
modulating NQO2 levels and activity in astrocytes, but it is far from modelling complex interactions that take place _in_ vivo. Thus, to address the NQO2 role in PD, it would be necessary to
examine first 6OHDA effects on autophagy in vivo and then to verify the protective effects of NQO2 inhibitors. On the other hand, induced pluripotent stem cell-derived organoids of
dopaminergic neurons and astrocytes would be a better model to study the role of NQO2 in neuroprotective capacity of astrocytes61. In spite of these weak points, our study provides important
clinical evidence supporting a possible role of NQO2 in PD progression is further supported by another important finding presented here, i.e., the difference in expression levels of _NQO2_
between PD patients and healthy donors. Higher expression of _NQO2_ in PD was first suggested by Harada et al.35, who described a positive association of a common form of PD with the D
(deletion) polymorphism in the _NQO2_ promoter region. This “gain of function” genetic variant of the promoter lacking the Sp3 transcriptional suppressor binding site62 was 3.46 times more
frequent in PD patients than in healthy subjects in the Japanese population35. Only two attempts to reproduce the findings of Harada’s work have been done so far, and one was discordant63,
while another minor work confirmed them64. More recent and extensive studies of _NQO2_ gene or protein expression in PD patients are missing, but higher NQO2 protein levels were reported in
postmortem brain specimens of patients with Alzheimer’s disease (AD)65, and elevated NQO2 expression was convincingly associated with learning deficits in rodent models38. The data retrieved
from GSE data sets suggest the opposite in postmortem SN specimens of late-stage PD patients, namely, a statistically significant reduction in _NQO2_ mRNA levels. This is in line with a
higher expression of lnc_NQO2_, which might repress _NQO2_ expression66. These data are unexpected, but do not exclude the possibility that higher _NQO2_ expression might be a trigger or
risk factor for neurodegenerative changes in the early stages of PD. This hypothesis is supported by the evidence coming from the analysis of _NQO2_ expression in whole blood of 48 early PD
cases, which revealed that the majority (35/48 (≈73%) of early PD patients express higher _NQO2_ levels than its median expression in healthy individuals, which is statistically significant
in the analyzed population, characterized by nonnormal distribution. Considering a high degree of heterogeneity among PD patients4 as well as the presence of polymorphisms leading to high or
low _NQO2_ expression, such a non-Gaussian distribution is plausible for both healthy and PD populations. We do not know if the levels of blood-born transcripts predict the brain expression
of a given gene, but higher NQO2 expression in the blood may indicate a more active promoter. According to our analysis, elevated _NQO2_ expression is almost 3 times more frequent among PD
patients than in the control group, which is in line with the observations of the study by Harada et al.35. In conclusion, our observations suggest that drugs targeting NQO2 may stimulate
autophagy as well as protect against dopamine quinone and hydroxyquinone toxicity. Thus, S29434 and other NQO2 inhibitors by acting as protective agents in astrocytes, might delay neuronal
damage in PD and other pathological conditions involving oxidative neurodegeneration. The vast literature suggesting a protective effect of several natural polyphenols in neurodegeneration
models strongly supports this idea, since NQO2 is a well-documented target of these compounds40,67. Further studies in PD animal models and brain organoids will be necessary to validate this
interesting hypothesis. METHODS REAGENTS AND ANTIBODIES Unless otherwise specified, all reagents were purchased from Sigma-Aldrich/MERCK (Darmstadt, Germany): 6OHDA (H4381, 100 mM stock in
bi-distilled H2O (H2Ob)), paraquat (M2254; 100 mM stock in H2O), and chloroquine (ClQ, C6628; 25 mM stock in H2Ob). Except for 6OHDA, which was freshly prepared for each experiment and
stored at − 80 °C for another use within one week, other reagents were kept in aliquots at − 80 °C for longer times and thawed shortly before each treatment. S29434, previously well
characterized39 and used under the name of NMDPEF31,32 was stored as a 10 mM stock in DMSO (D4540) at − 80 °C. The antibodies (Abs) used for Western blotting (WB) were: polyclonal rabbit
anti-LC3A/B (Abcam, ab1280025 used 1:2000), anti-p62 (MBL, PM045 used 1:1000), anti-GAPDH, clone FL335 (Santa Cruz Biotech., sc-25778 used 1:200), anti-NQO2 (ProteinTech, 15767-1-AP used
1:1000) and monoclonal mouse anti-α-Tubulin (Sigma-Aldrich, T6074 used 1:500). CELL CULTURE Astrocytoma U373 cells were a kind gift from M. Pollicita and S. Aquaro, Tor Vergata University,
Rome. After thawing, cells were expanded in high glucose (4.5% glucose) Dulbecco’s modified Eagle’s medium (DMEM) (Carlo Erba S.r.l., Milan, Italy, FA30WL0101500) and then maintained in
these conditions for routine culture or gradually switched to low-glucose (1% glucose) DMEM (FA30WL0060500) when indicated. Each type of DMEM was supplemented with 10% fetal bovine serum
(FBS) (Life Technologies, Monza MB, Italy, 10500-064), 1:100 penicillin–streptomycin (Pen Strep, Life Technologies, 15070-063), and 1:100 l-glutamine (PAA Laboratories GmbH, Austria,
M00409-2722) to constitute a standard medium, and cells were cultured under standard 5% CO2 conditions. If not otherwise specified for most experiments, U373 cells were seeded 2 days before
or 3 days before the treatments at a density of 32 × 103 or 16 × 103 × cm−2 (300 or 150 × 103 per 3.5 cm plate), respectively, and the last medium change was performed 24 h before the
treatment with a desired compound. For longer treatments (48 h and 72 h), 6OHDA was re-added every 24 h, and the medium change was performed every 48 h. ClQ was added 2 or 3 h before cell
lysis as indicated. The stressful culture conditions were generated by switching the cells cultured for one week in high glucose to low glucose or by seeding them at low density (6.4 × 103 ×
cm−2) for 2 days before treatments. Neuroblastoma SH-SY5Y cells were cultured in F-12 K medium plus high glucose DMEM supplemented with 10% FBS, Pen Strep and l-glutamine as standard
medium. ROS DETECTION IN LIVE CELLS ROS were detected by two alternative ROS fluorescent indicators: a dichlorofluorescein (DCF) derivative, namely, 5(6)-carboxy-2′,7′ dichlorofluorescein
diacetate (CA-DCF-DA; Sigma-Aldrich, 21884) or MitoSOX (3,8-phenanthridinediamine, 5-(6′-triphenylphosphoniumhexyl)-5,6 dihydro-6-phenyl). The first method detected cytoplasmic ROS in live
cell monolayers by fluorimeter, while the second method detected ROS in trypsin-detached cells by FACS analysis. The DCF-based method was performed as previously described31, with minor
modifications. U373 cells were plated at 2 × 104/cm2 in 96-well plates 2 days before treatments, but phenol-free DMEM (Gibco, Life Tech. 11054020) was used as the basal medium. 6OHDA was
added 6 h before analysis in triplicate for each concentration. Cell staining with CA-DCF-DA was performed 2 h before analysis. For this purpose, the conditioned medium +/− 6OHDA was
temporarily removed, and the cells were incubated with prewarmed basal phenol-free DMEM (Gibco, Life Tech. 11054020) containing 20 mM CA-DCF-DA or vehicle (DMSO) for 30 min. Stained cells
were rinsed in phenol-free DMEM and incubated again for 1.5 h with the conditioned medium. All medium changes and washes were performed gently to avoid any shear, osmotic or other types of
stress, drop by drop placed on the plate wall with the warm medium. Fluorescein signal detection of cell monolayers was performed on a Victor 2 multilabel fluorimeter (Perkin-Elmer,
Singapore). Background readings (cells stained with DMSO) were subtracted for the analysis of fluorescence of DCF-DA-stained cells. For detection of mitochondrial ROS, U373 cells were plated
at 1.2 × 104 cells/per well on 24 well plates in low-glucose DMEM standard medium, 2 days before treatment. MitoSox was used at a concentration of 5 µM in phenol-free DMEM. WESTERN BLOTTING
(WB) See Supplementary Methods. NQO2 ACTIVITY MEASUREMENTS NQO2 enzymatic activity assays were performed as described previously40 with the following modifications. Cellular NQO2 activity
was measured in U373 cell lysates. For this purpose, the cells were cultured for two days to reach a 70–90% density and then treated with 6OHDA for 24 h, scraped and homogenized in ice-cold
GPS buffer (50 mM Tris–HCl, 3 mM n-octyl-d-glucopyranoside pH 7.5) with 10 strokes by a tight glass douncer on ice. After clearing by centrifugation, the lysates were stored at − 80 °C.
After thawing, lysates (5 mL containing 8 to 15 mg protein) were aliquoted on 96-well plates placed on ice and then equilibrated for 10 min at RT. The reaction was initiated by the addition
of GPS buffer supplemented with 50 mM of the cosubstrate 1-benzyl-1,4-dihydro-nicotinamide (BNAH; Santa Cruz Biotech., 208609) with or without 50 mM menadione (K3) (Supelco, Merk Life
Science S.r.l., Milan, Italy) as substrate in a total volume of 200 mL. Testing of 6OHDA as an NQO2 substrate was performed with 50 or 100 ng/mL human recombinant NQO2 (Sigma-Aldrich, Q0380)
in GPS buffer plus 100 mM BNAH and different concentrations of 6OHDA or 100 mM K3 as a positive control in a total volume of 200 mL. BNAH supplemented with an appropriate amount of NQO2 and
vehicle instead of 6OHDA was used as a negative control. The reaction was initiated by the addition of the recombinant enzyme to each column of 96-well plates. The exact time of pipetting
the enzyme (time 0) was taken to calculate the delay between the fluorescence reading and the reaction start. BNAH fluorescence was monitored by a 1420 Multilabel reader Victor 2 fluorimeter
(Perkin Elmer) for 15 to 20 min. Data were analyzed by Microsoft Excel and GraphPad Prism 9.0 software (GraphPad, San Diego, CA, USA) to fit the decay curves and find unknown values by
curve interpolation. The velocity (V) of the reaction was determined from the fluorescence change as the mean reduction of mmol of BNAH (in 200 mL) per minute for the first 5 min after the
addition of the enzyme. The activity was calculated as V divided by the enzyme/protein amount added to the reaction. PREPARATION OF NQO2-OVEREXPRESSING (N-OVER) U373 CELLS See Supplementary
Methods. FLOW CYTOMETRY (FACS) ANALYSIS OF CELL VIABILITY See Supplementary Methods. COCULTURE OF ASTROGLIAL AND DOPAMINERGIC CELLS U373 cells and U373 N-over cells were seeded and cultured
on 24-well plates in high-glucose DMEM. Briefly, U373 cells were plated at a density of 40 × 103 cm−2, while U373 N-over cells were plated at a density between 30 × 103 cm−2 under standard
conditions to achieve a 90% confluent monolayer after 2 days. Before plating, SH-SY5Y cells were labeled with CellTracker Green CMFDA (chloromethyl fluorescein diacetate, Molecular Probes,
Life Tech., USA) dye. Briefly, SHSY5Y cells, grown to 80–90% confluency, were labeled with 5 µM CMFDA in PBS for 15 min. Cells were washed once in PBS, and fresh regular medium was added for
30 min. Finally, the cells were trypsinized (Trypsin–EDTA (0.05%), Gibco, Life Tech., USA), and resuspended in fresh culture medium to a desired cell concentration. Next, 5 × 104 cm−2
SH-SY5Y cells were seeded over U373 monolayers. After 24 h, the cocultures were treated for 48 h with 6OHDA and/or S29434. At the end of the treatment, the cells were washed once in PBS
(Carlo Erba S.r.l., FA30WL0615500), trypsinized for 3 min and collected in 0.5 ml of F12-K medium plus high glucose supplemented with 10% FBS in flow cytometry round tubes. The cells were
then pelleted by centrifugation for 5 min at 1000 × _g_ and resuspended in 400 µL of PBS containing 1% FBS and 0.5 mM EDTA. Cell viability and mortality were determined by 7-AAD dye (BD
Biosciences, Erenbodgem, Belgium, cat. nr 344563; used 1:10) added 2–3 min before FACS analysis on a FACSCanto II flow cytometer (BD Biosciences, Erenbodgem, Belgium). The data were acquired
in the FITC channel (for CMFDA dye) and PerCP-Cy5-5 channel (for 7-AAD dye) according to the parameters and gating criteria detailed in the Supplementary Methods and Fig. S4 (Supplementary
information link). U373 CELL TRANSFECTION AND SILENCING OF NQO2 U373 cells cultured in high glucose standard medium were seeded on 6-well plates one day before transfection at a density of 4
× 104 cells cm−2. Nonsilencing control siRNA [_siEGFP_, (EHUEGFP)] and a set of human _NQO2_-targeting _esiRNAs_ (EMU042521) were purchased from Merck, Sigma-Aldrich. Transfection of U373
cells was performed using Transfection Reagent (Merck, S1452) and Opti-MEM (Life Technologies, 51985026) according to the manufacturer's instructions. Briefly, siRNA (150 nM) and
transfection reagent (1:1 ml/pMol siRNA) were diluted in Opti-MEM and incubated for 15–20 min at RT. Cells were incubated in Opti-MEM containing siRNA-transfection reagent complexes for 4 h
and then incubated in high-glucose culture medium containing w/o PenStrep. After 20 h, the medium was exchanged for regular high-glucose medium. The treatments were initiated 48 h after
transfection and carried for the next 24 h. ClQ (25 µM) was added 3 h before the end of the experiment. After the treatments, the cells were lysed and assayed by WB. ANIMAL PROCEDURES _NQO2_
knock-out mice generated in the C3H/HeOuJ background were previously characterized38,68. Since the first description in 2004, the original knockout mice were backcrossed with _WT_ C3H/HeOuJ
mice obtained from Charles River at least 20 times. Mice used for astrocyte isolation were WT and _NQO2_ − / − pups of the C3H/HeOuJ strain born on the same day from the same litter _WT_
and _NQO2_ − / − females crossed with the same litter males. One- to two-day-old (P1/P2) pups were euthanized by head decapitation. The heads were placed into sterile PBS at RT, and the
meninges were carefully removed. The animal procedures were carried out according to EU Directive 2010/63/EU and approved by the Italian Ministry of Health (authorization Nr. 613-2017-PR).
This study complied with ARRIVE guidelines (https://arriveguidelines.org/). ISOLATION AND CULTURE OF MOUSE CORTICAL ASTROCYTES Primary astrocytes were isolated from cerebral cortices
prepared as described above and according to a previously published protocol31. Briefly, the cortices were transferred to new dishes with sterile PBS and then dissected, minced, and
mechanically dissociated. Minced cortices were centrifuged (800 g, 8 min, 4 °C), and the pellet was digested in DMEM containing 10 mg/mL type I DNase for 15 min on a benchtop orbital shaker
(100 RPM) at RT. The reaction was stopped by adding ice-cold DMEM, and the homogenate was pelleted by centrifugation (800 g, 8 min, 4 °C). The cells were plated on 10 cm dishes (1
brain/dish) in DMEM supplemented with 10% FBS and 1% P/S (Gibco, Life Tech.). The next day, the medium was changed to remove non-adherent cells. Thereafter, the medium was changed every 2
days. The cultures were maintained for approximately 1 week or until 90% confluent at 37 °C and 5% CO2. Cells were then removed from the dish with the addition of 0.05% trypsin–EDTA
(Invitrogen) and replated on 6-well plates for experiments at a density of 500,000 cells per well. The treatments were performed one day after seeding. PHASE-CONTRAST AND FLUORESCENCE
MICROSCOPY Phase-contrast and fluorescence images were acquired with LEICA DMI 4000 B fluorescent microscope, equipped with LEICA DFC 350 FX camera. MICROARRAY DATA INFORMATION AND GENE
EXPRESSION OMNIBUS (GEO) DATASET ANALYSIS See Supplementary Methods (Supplementary information link). DATA ANALYSIS AND STATISTICAL PROCEDURES Numerical data from all multigroup experiments
were evaluated by analysis of variance (ANOVA). If Brown-Forsythe’s test for equality of variances was positive, Welch’s ANOVA test was performed, otherwise ordinary one-way ANOVA or
occasionally three-way ANOVA was performed. Multiple comparisons between groups were performed by one of the suggested posttests according to GraphPad PRISM 9.0 software guidelines
http://www.graphpad.com/ or occasionally by unpaired T test as indicated in figure legends. Data are expressed as the means ± SEM for the number n of independent experiments or as the means
± SD for the number of n independent samples from a representative experiment, as indicated for each figure. For patient and healthy subject expression data, D'Agostino-Pearson and
Shapiro–Wilk normality tests were performed. If the data did not pass the normality tests or groups were lower than 10, Mann–Whitney test was applied and the data were presented as medians
+/− range; otherwise unpaired T test was used. ETHICS DECLARATION The authors declare that the current study complied with ARRIVE guidelines (https://arriveguidelines.org/). DATA
AVAILABILITY The original images of blots presented in this work are displayed in the _Supplementary Information_. All the other datasets generated and analyzed during the current study are
available from the corresponding author upon request. REFERENCES * Janda, E. _et al._ Defective autophagy in Parkinson’s disease: Role of oxidative stress. _Mol. Neurobiol._ 46(3), 639–661
(2012). Article CAS PubMed Google Scholar * Ma, L. _et al._ Parkinson’s disease: Alterations in iron and redox biology as a key to unlock therapeutic strategies. _Redox. Biol._ 41,
101896 (2021). Article CAS PubMed PubMed Central Google Scholar * Michel, P. P., Hirsch, E. C. & Hunot, S. Understanding dopaminergic cell death pathways in Parkinson disease.
_Neuron_ 90(4), 675–691 (2016). Article CAS PubMed Google Scholar * Wullner, U. _et al._ The heterogeneity of Parkinson’s disease. _J. Neural Transm._ 130(6), 827–838 (2023). Article
PubMed Google Scholar * Galluzzi, L. _et al._ Molecular definitions of autophagy and related processes. _EMBO J._ 36(13), 1811–1836 (2017). Article CAS PubMed PubMed Central Google
Scholar * Dikic, I. & Elazar, Z. Mechanism and medical implications of mammalian autophagy. _Nat. Rev. Mol. Cell. Biol._ 19(6), 349–364 (2018). Article CAS PubMed Google Scholar *
Janda, E., Boi, L. & Carta, A. R. Microglial phagocytosis and its regulation: A therapeutic target in Parkinson’s disease?. _Front. Mol. Neurosci._ 11, 144 (2018). Article PubMed
PubMed Central Google Scholar * Schondorf, D. C. _et al._ iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis.
_Nat. Commun._ 5, 4028 (2014). Article ADS PubMed Google Scholar * Karabiyik, C., Lee, M. J. & Rubinsztein, D. C. Autophagy impairment in Parkinson’s disease. _Essays Biochem._
61(6), 711–720 (2017). Article PubMed Google Scholar * Wible, D. J. & Bratton, S. B. Reciprocity in ROS and autophagic signaling. _Curr. Opin. Toxicol._ 7, 28–36 (2018). Article
PubMed Google Scholar * Dagda, R. K., Das Banerjee, T. & Janda, E. How Parkinsonian toxins dysregulate the autophagy machinery. _Int. J. Mol. Sci._ 14(11), 22163–89 (2013). Article
PubMed PubMed Central Google Scholar * McBean, G. J. Cysteine, glutathione, and thiol redox balance in astrocytes. _Antioxidants_ 6(3), 66 (2017). Article Google Scholar * Sanchez-Cano,
F., Hernandez-Kelly, L. C. & Ortega, A. The blood–brain barrier: Much more than a selective access to the brain. _Neurotox Res._ 39(6), 2154–2174 (2021). Article CAS PubMed Google
Scholar * Janda, E. _et al._ The protective effect of tianeptine on Gp120-induced apoptosis in astroglial cells: Role of GS and NOS, and NF-kappaB suppression. _Br. J. Pharmacol._ 164(6),
1590–1599 (2011). Article CAS PubMed PubMed Central Google Scholar * Linnerbauer, M., Wheeler, M. A. & Quintana, F. J. Astrocyte crosstalk in CNS inflammation. _Neuron_ 108(4),
608–622 (2020). Article CAS PubMed PubMed Central Google Scholar * Brandebura, A. N. _et al._ Astrocyte contribution to dysfunction, risk and progression in neurodegenerative disorders.
_Nat. Rev. Neurosci._ 6, 66 (2022). Google Scholar * Jenner, P. & Olanow, C. W. The pathogenesis of cell death in Parkinson’s disease. _Neurology_ 66(10 Suppl 4), S24-36 (2006). PubMed
Google Scholar * Hirsch, E. C. _et al._ Glial cell participation in the degeneration of dopaminergic neurons in Parkinson’s disease. _Adv. Neurol._ 80, 9–18 (1999). CAS PubMed Google
Scholar * Booth, H. D. E., Hirst, W. D. & Wade-Martins, R. The role of astrocyte dysfunction in Parkinson’s disease pathogenesis. _Trends Neurosci._ 40(6), 358–370 (2017). Article CAS
PubMed PubMed Central Google Scholar * Hernandez-Baltazar, D., Zavala-Flores, L. M. & Villanueva-Olivo, A. The 6-hydroxydopamine model and Parkinsonian pathophysiology: Novel
findings in an older model. _Neurologia_ 32(8), 533–539 (2017). Article CAS PubMed Google Scholar * Andrew, R. _et al._ The determination of hydroxydopamines and other trace amines in
the urine of Parkinsonian patients and normal controls. _Neurochem. Res._ 18(11), 1175–1177 (1993). Article CAS PubMed Google Scholar * Curtius, H. C. _et al._ Mass fragmentography of
dopamine and 6-hydroxydopamine. Application to the determination of dopamine in human brain biopsies from the caudate nucleus. _J Chromatographics_ 99, 529–40 (1974). Article CAS Google
Scholar * Borah, A. & Mohanakumar, K. P. Salicylic acid protects against chronic l-DOPA-induced 6-OHDA generation in experimental model of parkinsonism. _Brain Res._ 1344, 192–199
(2010). Article CAS PubMed Google Scholar * Kumar, R., Agarwal, A. K. & Seth, P. K. Free radical-generated neurotoxicity of 6-hydroxydopamine. _J. Neurochem._ 64(4), 1703–1707
(1995). Article CAS PubMed Google Scholar * Kirik, D., Rosenblad, C. & Bjorklund, A. Characterization of behavioral and neurodegenerative changes following partial lesions of the
nigrostriatal dopamine system induced by intrastriatal 6-hydroxydopamine in the rat. _Exp. Neurol._ 152(2), 259–277 (1998). Article CAS PubMed Google Scholar * Ximenes, J. C. _et al._
Valproic acid neuroprotection in the 6-OHDA model of Parkinson’s disease is possibly related to its anti-inflammatory and HDAC inhibitory properties. _J. Neurodegener. Dis._ 2015, 313702
(2015). PubMed PubMed Central Google Scholar * Mitra, S., Chakrabarti, N. & Bhattacharyya, A. Differential regional expression patterns of alpha-synuclein, TNF-alpha, and IL-1beta;
and variable status of dopaminergic neurotoxicity in mouse brain after Paraquat treatment. _J. Neuroinflamm._ 8, 163 (2011). Article CAS Google Scholar * Blum, D. _et al._ Molecular
pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: Contribution to the apoptotic theory in Parkinson’s disease. _Prog. Neurobiol._ 65(2), 135–172 (2001). Article CAS
PubMed Google Scholar * Glinka, Y., Gassen, M. & Youdim, M. B. Mechanism of 6-hydroxydopamine neurotoxicity. _J. Neural Transm. Suppl._ 50, 55–66 (1997). Article CAS PubMed Google
Scholar * Jenner, P., Schapira, A. H. & Marsden, C. D. New insights into the cause of Parkinson’s disease. _Neurology_ 42(12), 2241–2250 (1992). Article CAS PubMed Google Scholar *
Janda, E. _et al._ Parkinsonian toxin-induced oxidative stress inhibits basal autophagy in astrocytes via NQO2/quinone oxidoreductase 2: Implications for neuroprotection. _Autophagy_ 11(7),
1063–1080 (2015). Article CAS PubMed PubMed Central Google Scholar * Janda, E. _et al._ The antidote effect of quinone oxidoreductase 2 inhibitor against paraquat-induced toxicity in
vitro and in vivo. _Br. J. Pharmacol._ 168(1), 46–59 (2013). Article CAS PubMed PubMed Central Google Scholar * Janda, E. _et al._ Molecular pharmacology of NRH: Quinone oxidoreductase
2: A detoxifying enzyme acting as an undercover toxifying enzyme. _Mol. Pharmacol._ 98(5), 620–633 (2020). Article CAS PubMed Google Scholar * Cassagnes, L. E. _et al._ In cellulo
monitoring of quinone reductase activity and reactive oxygen species production during the redox cycling of 1,2 and 1,4 quinones. _Free Radic. Biol. Med._ 89, 126–134 (2015). Article CAS
PubMed Google Scholar * Harada, S. _et al._ An association between idiopathic Parkinson’s disease and polymorphisms of phase II detoxification enzymes: Glutathione S-transferase M1 and
quinone oxidoreductase 1 and 2. _Biochem. Biophys. Res. Commun._ 288(4), 887–892 (2001). Article MathSciNet CAS PubMed Google Scholar * Cassagnes, L. E. _et al._ Oxidative stress and
neurodegeneration: The possible contribution of quinone reductase 2. _Free Radic. Biol. Med._ 120, 56–61 (2018). Article CAS PubMed Google Scholar * Fu, Y., Buryanovskyy, L. & Zhang,
Z. Quinone reductase 2 is a catechol quinone reductase. _J. Biol. Chem._ 283(35), 23829–23835 (2008). Article CAS PubMed PubMed Central Google Scholar * Benoit, C. E. _et al._ Loss of
quinone reductase 2 function selectively facilitates learning behaviors. _J. Neurosci._ 30(38), 12690–12700 (2010). Article CAS PubMed PubMed Central Google Scholar * Boutin, J. A. _et
al._ S29434, a quinone reductase 2 inhibitor: Main biochemical and cellular characterization. _Mol. Pharmacol._ 95(3), 269–285 (2019). Article MathSciNet CAS PubMed Google Scholar *
Janda, E. _et al._ Apigenin and luteolin regulate autophagy by targeting NRH-quinone oxidoreductase 2 in liver cells. _Antioxidants_ 10(5), 66 (2021). Article Google Scholar * Vareslija,
D. _et al._ 6-Hydroxydopamine: A far from simple neurotoxin. _J. Neural Transm._ 127(2), 213–230 (2020). Article CAS PubMed Google Scholar * Scherzer, C. R. _et al._ Molecular markers of
early Parkinson’s disease based on gene expression in blood. _Proc. Natl. Acad. Sci. USA_ 104(3), 955–960 (2007). Article ADS CAS PubMed PubMed Central Google Scholar * Miki, Y. _et
al._ Alteration of autophagy-related proteins in peripheral blood mononuclear cells of patients with Parkinson’s disease. _Neurobiol. Aging_ 63, 33–43 (2018). Article CAS PubMed Google
Scholar * Duke, D. C. _et al._ The medial and lateral substantia nigra in Parkinson’s disease: mRNA profiles associated with higher brain tissue vulnerability. _Neurogenetics_ 8(2), 83–94
(2007). Article CAS PubMed Google Scholar * Moran, L. B. _et al._ Whole genome expression profiling of the medial and lateral substantia nigra in Parkinson’s disease. _Neurogenetics_
7(1), 1–11 (2006). Article CAS PubMed Google Scholar * Lindgren, H. S., Lelos, M. J. & Dunnett, S. B. Do alpha-synuclein vector injections provide a better model of Parkinson’s
disease than the classic 6-hydroxydopamine model?. _Exp. Neurol._ 237(1), 36–42 (2012). Article CAS PubMed Google Scholar * Hanrott, K. _et al._ 6-hydroxydopamine-induced apoptosis is
mediated via extracellular auto-oxidation and caspase 3-dependent activation of protein kinase Cdelta. _J. Biol. Chem._ 281(9), 5373–5382 (2006). Article CAS PubMed Google Scholar *
Rodriguez-Pallares, J. _et al._ Mechanism of 6-hydroxydopamine neurotoxicity: The role of NADPH oxidase and microglial activation in 6-hydroxydopamine-induced degeneration of dopaminergic
neurons. _J. Neurochem._ 103(1), 145–156 (2007). Article CAS PubMed Google Scholar * Zhuang, X. X. _et al._ Pharmacological enhancement of TFEB-mediated autophagy alleviated neuronal
death in oxidative stress-induced Parkinson’s disease models. _Cell Death Dis._ 11(2), 128 (2020). Article CAS PubMed PubMed Central Google Scholar * He, H. J. _et al._ Neuroprotective
effects of curcumin via autophagy induction in 6-hydroxydopamine Parkinson’s models. _Neurochem. Int._ 155, 105297 (2022). Article CAS PubMed Google Scholar * Chaouhan, H. S. _et al._
Calycosin alleviates paraquat-induced neurodegeneration by improving mitochondrial functions and regulating autophagy in a Drosophila model of Parkinson’s disease. _Antioxidants_ 11(2), 66
(2022). Article Google Scholar * Wills, J. _et al._ Paraquat, but not maneb, induces synucleinopathy and tauopathy in striata of mice through inhibition of proteasomal and autophagic
pathways. _PLoS ONE_ 7(1), e30745 (2012). Article ADS CAS PubMed PubMed Central Google Scholar * Wise, J. P. Jr. _et al._ Autophagy disruptions associated with altered optineurin
expression in extranigral regions in a rotenone model of Parkinson’s disease. _Front. Neurosci._ 12, 289 (2018). Article PubMed PubMed Central Google Scholar * Wu, Y. _et al._ Role of
autophagy and oxidative stress to astrocytes in fenpropathrin-induced Parkinson-like damage. _Neurochem. Int._ 145, 105000 (2021). Article CAS PubMed Google Scholar * Guo, Q., et al.,
_Activation of Nrf2 in Astrocytes Suppressed PD-Like Phenotypes via Antioxidant and Autophagy Pathways in Rat and Drosophila Models._ Cells, 2021. 10(8). * Sung, K. & Jimenez-Sanchez, M.
Autophagy in astrocytes and its implications in neurodegeneration. _J. Mol. Biol._ 432(8), 2605–2621 (2020). Article CAS PubMed Google Scholar * Ioannou, M. S. _et al._ Neuron-astrocyte
metabolic coupling protects against activity-induced fatty acid toxicity. _Cell_ 177(6), 1522.e14-1535.e14 (2019). Article Google Scholar * Miyazaki, I. & Asanuma, M. Neuron-astrocyte
interactions in Parkinson’s disease. _Cells_ 9(12), 66 (2020). Article Google Scholar * Ran, L. Y. _et al._ The influence of NQO2 on the dysfunctional autophagy and oxidative stress
induced in the hippocampus of rats and in SH-SY5Y cells by fluoride. _CNS Neurosci. Ther._ 29(4), 1129–1141 (2023). Article CAS PubMed PubMed Central Google Scholar * Zhou, Z. D. _et
al._ Role of dopamine in the pathophysiology of Parkinson’s disease. _Transl. Neurodegener._ 12(1), 44 (2023). Article CAS PubMed PubMed Central Google Scholar * Galet, B., Cheval, H.
& Ravassard, P. Patient-derived midbrain organoids to explore the molecular basis of Parkinson’s disease. _Front. Neurol._ 11, 1005 (2020). Article PubMed PubMed Central Google
Scholar * Wang, W. & Jaiswal, A. K. Sp3 repression of polymorphic human NRH:quinone oxidoreductase 2 gene promoter. _Free Radic. Biol. Med._ 37(8), 1231–1243 (2004). Article CAS
PubMed Google Scholar * Okada, S. _et al._ No associations between Parkinson’s disease and polymorphisms of the quinone oxidoreductase (NQO1, NQO2) genes. _Neurosci. Lett._ 375(3), 178–180
(2005). Article CAS PubMed Google Scholar * Wang, W. _et al._ Association of NRH:quinone oxidoreductase 2 gene promoter polymorphism with higher gene expression and increased
susceptibility to Parkinson’s disease. _J. Gerontol. A Biol. Sci. Med. Sci._ 63(2), 127–134 (2008). Article PubMed Google Scholar * Hashimoto, T. & Nakai, M. Increased hippocampal
quinone reductase 2 in Alzheimer’s disease. _Neurosci. Lett._ 502(1), 10–12 (2011). Article CAS PubMed Google Scholar * Chen, D. _et al._ Genome-wide analysis of long noncoding RNA
(lncRNA) expression in colorectal cancer tissues from patients with liver metastasis. _Cancer Med._ 5(7), 1629–1639 (2016). Article ADS CAS PubMed PubMed Central Google Scholar *
Boutin, J. A. _et al._ Quinone reductase 2 substrate specificity and inhibition pharmacology. _Chem. Biol. Interact._ 151(3), 213–228 (2005). Article CAS PubMed Google Scholar *
Mailliet, F. _et al._ Organs from mice deleted for NRH:quinone oxidoreductase 2 are deprived of the melatonin binding site MT3. _FEBS Lett._ 578(1–2), 116–120 (2004). Article CAS PubMed
Google Scholar Download references ACKNOWLEDGEMENTS Special thanks to Prof. Maria Cristina Caroleo, University Magna Graecia (UMG), Catanzaro, for critical reading of the manuscript and
useful suggestions. We also thank Prof. Walter Agosti for help with flow cytometry analysis and Dr. Concetta Riillo, UMG, Catanzaro, Italy, for technical support with cell culture and sample
preparation. Finally, we acknowledge Prof. Stefano Aquaro and D.r Michela Pollicita, University Tor Vergata, Rome, and Prof. Rossella Russo, University of Calabria, Rende (Cs), Italy, for
the kind gift of U373 cells and neuroblastoma SH-SY5Y cells, respectively. FUNDING This research was funded by the Basic Research Activity Fund (FABR) 2017 grant and Ph.D. program of Italian
Ministry of Education, University and Research (M.I.U. R), awarded to E.J.; PON03PE 00078 grant (NUTRAMED Consortium) awarded to V.M. and ADOPTION (FOE, Project ‘Nuovi Biomarker Diagnostici
e Terapeutici delle Malattie Neurodegenerative’), CNR CUP:B83C21001810005, awarded to M.A. AUTHOR INFORMATION Author notes * Maddalena Parafati Present address: Department of
Pharmacodynamics, University of Florida, Gainesville, FL 32611, USA * These authors contributed equally: Maddalena Parafati and Concetta Martino. AUTHORS AND AFFILIATIONS * Laboratory of
Cellular and Molecular Toxicology, Department of Health Science, University “Magna Græcia” of Catanzaro, 88100, Catanzaro, Italy Elzbieta Janda, Maddalena Parafati, Concetta Martino,
Francesco Crupi & Vincenzo Mollace * Center for Advanced Studies and Technology (CAST), “G. d’Annunzio” University of Chieti-Pescara, 66100, Chieti, Italy Jonahunnatha Nesson George
William * UMR 152 Pharma-Dev, Université de Toulouse III, IRD, UPS, 31400, Toulouse, France Karine Reybier * Institute for Biomedical Research and Innovation (IRIB), National Research
Council of Italy (CNR), 88100, Catanzaro, Italy Mariamena Arbitrio * Laboratory of Neuroendocrine Endocrine and Germinal Differentiation and Communication (NorDiC), Univ Rouen Normandie,
Inserm, NorDiC UMR 1239, 76000, Rouen, France Jean A. Boutin Authors * Elzbieta Janda View author publications You can also search for this author inPubMed Google Scholar * Maddalena
Parafati View author publications You can also search for this author inPubMed Google Scholar * Concetta Martino View author publications You can also search for this author inPubMed Google
Scholar * Francesco Crupi View author publications You can also search for this author inPubMed Google Scholar * Jonahunnatha Nesson George William View author publications You can also
search for this author inPubMed Google Scholar * Karine Reybier View author publications You can also search for this author inPubMed Google Scholar * Mariamena Arbitrio View author
publications You can also search for this author inPubMed Google Scholar * Vincenzo Mollace View author publications You can also search for this author inPubMed Google Scholar * Jean A.
Boutin View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS E.J. designed all experiments, supervised the work, performed experiments for Figs.
2, 3, 5 and 6, analyzed the data, wrote the main manuscript text and prepared all the figures. M.P. performed the experiments for Figs. 1 and 5 and analyzed the data, C.M. performed
experiments for Figs. 1, 4 and 6, F.C. performed experiments for Figs. 1 and 5, analyzed the data and helped with figure preparation, J.N.G.W. collected and analyzed the data for Fig. 7,
V.M., M.A. and E.J. provided the funding, J.B. and K.R. provided reagents, suggested and analyzed experiments and contributed to writing the manuscript. M. contributed to writing the revised
version of the manuscript. All authors reviewed the manuscript. CORRESPONDING AUTHORS Correspondence to Elzbieta Janda or Mariamena Arbitrio. ETHICS DECLARATIONS COMPETING INTERESTS The
authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER'S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional
affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License,
which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a
link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence,
unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory
regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Janda, E., Parafati, M., Martino, C. _et al._ Autophagy and neuroprotection in
astrocytes exposed to 6-hydroxydopamine is negatively regulated by NQO2: relevance to Parkinson’s disease. _Sci Rep_ 13, 21624 (2023). https://doi.org/10.1038/s41598-023-44666-7 Download
citation * Received: 24 January 2023 * Accepted: 11 October 2023 * Published: 07 December 2023 * DOI: https://doi.org/10.1038/s41598-023-44666-7 SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative