Play all audios:
ABSTRACT BIVV003 is a gene-edited autologous cell therapy in clinical development for the potential treatment of sickle cell disease (SCD). Hematopoietic stem cells (HSC) are genetically
modified with mRNA encoding zinc finger nucleases (ZFN) that target and disrupt a specific regulatory GATAA motif in the _BCL11A_ erythroid enhancer to reactivate fetal hemoglobin (HbF). We
characterized ZFN-edited HSC from healthy donors and donors with SCD. Results of preclinical studies show that ZFN-mediated editing is highly efficient, with enriched biallelic editing and
high frequency of on-target indels, producing HSC capable of long-term multilineage engraftment in vivo, and express HbF in erythroid progeny. Interim results from the Phase 1/2 PRECIZN-1
study demonstrated that BIVV003 was well-tolerated in seven participants with SCD, of whom five of the six with more than 3 months of follow-up displayed increased total hemoglobin and HbF,
and no severe vaso-occlusive crises. Our data suggest BIVV003 represents a compelling and novel cell therapy for the potential treatment of SCD. SIMILAR CONTENT BEING VIEWED BY OTHERS SEVERE
INFLAMMATION AND LINEAGE SKEWING ARE ASSOCIATED WITH POOR ENGRAFTMENT OF ENGINEERED HEMATOPOIETIC STEM CELLS IN PATIENTS WITH SICKLE CELL DISEASE Article Open access 01 April 2025 BASE
EDITING OF HAEMATOPOIETIC STEM CELLS RESCUES SICKLE CELL DISEASE IN MICE Article 02 June 2021 CRISPR–CAS9-MEDIATED GENE EDITING OF THE _BCL11A_ ENHANCER FOR PEDIATRIC Β0/Β0
TRANSFUSION-DEPENDENT Β-THALASSEMIA Article 04 August 2022 INTRODUCTION Sickle cell disease (SCD) is a hereditary hemoglobinopathy characterized by inflammation, hemolytic anemia,
vaso-occlusive crises, organ damage, and early mortality1,2,3,4. Elevated expression of fetal hemoglobin (HbF) has a well-established beneficial effect on the clinical course of SCD, with
decreased disease severity in patients with naturally occurring genetic variants or mutations leading to a persistence of HbF into adulthood5,6,7,8. The ameliorative effect of HbF in
patients with SCD has prompted assessment of genetic and pharmacological therapeutic approaches aimed at inducing HbF9,10,11,12. Allogeneic hematopoietic stem cell (HSC) transplant remains
the only curative option for SCD but is limited to the minority of patients with a matched donor and carries a risk of graft failure or rejection and graft-versus-host disease13,14,15,16.
Transplantation of autologous, genetically modified HSC has therefore been pursued as an alternative. Several gene-addition and gene-editing strategies using autologous HSC are currently
approved or in clinical development in patients with SCD, including approaches to reactivate HbF9,11,12,17,18. Several of these share a common approach of targeting the _BCL11A_
erythroid-specific enhancer (ESE), a key regulatory element in the fetal-to-adult hemoglobin (Hb) switch, to selectively generate erythroid cells with reduced _BCL11A _expression (and
induced HbF production) without affecting non-erythroid cells9,11,12,19,20,21,22. Other gene editing strategies to reactivate HbF include mimicking mutations in the promoter of the
gamma-globin gene that lead to high persistence of fetal hemoglobin (HPFH)23,24,25,26. BIVV003 is a novel gene-edited autologous cell therapy in clinical development for the potential
treatment of SCD, in which HSC are genetically modified with mRNA encoding zinc finger nucleases (ZFN) that target and disrupt a specific regulatory GATAA motif in the _BCL11A _ESE to
reactivate HbF27,28. Zinc finger proteins combined with the nuclease domain of the restriction endonuclease FokI create double-strand breaks (DSBs) at precisely defined genomic locations.
Repair of the DSBs via non-homologous end joining (NHEJ) or microhomology-mediated end joining (MMEJ) results in target sequence disruption29. ZFN-mediated gene editing of the _BCL11A _ESE
in HSC from healthy donors and donors with β-thalassemia reactivates HbF in erythroid cells without compromising in vivo engraftment30,31. In these studies, we show that ZFN-mediated editing
is highly efficient and produces HSC capable of long-term multilineage engraftment in vivo_,_independent of mobilization strategy or disease state, and robustly express HbF in the erythroid
progeny. Through clonal analysis of edited HSC from healthy donors and donors with SCD, we have also shown enriched biallelic editing with a high frequency of small indels, generating a
high efficiency of ZFN gene editing. Together with preliminary clinical studies32,33, our data suggest BIVV003 represents a novel cell therapy for the potential treatment of SCD and support
ongoing clinical development of this approach (NCT03653247). RESULTS ZFN-MEDIATED GENE EDITING IS HIGHLY EFFICIENT AND LEADS TO HBF INDUCTION EX VIVO INDEPENDENTLY OF MOBILIZATION STRATEGY
OR DISEASE STATE In individuals with β-thalassemia, the combination of granulocyte colony–stimulating factor (G-CSF) with plerixafor is used for stem cell mobilization34,35. However G-CSF is
not recommended for hematopoietic stem-progenitor cell (HSPC) mobilization in patients with SCD due to the risk of vaso-occlusive complications associated with leukocytosis, therefore
single-agent plerixafor mobilization is utilized36. We characterized ZFN-edited HSPC from healthy donors from single (plerixafor)- or dual-agent (plerixafor plus G-CSF) mobilization.
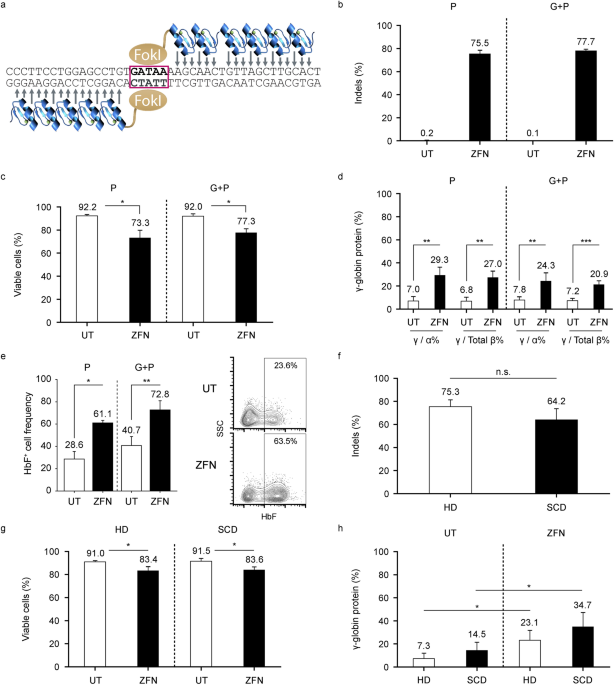
Modification of the _BCL11A_ ESE target site (Fig. 1a) was efficient using both mobilization strategies, with > 75% of alleles modified as measured by frequency of indels (i.e.,
insertions and deletions) and high post-editing viability (Figs. 1b and 1c). For both mobilization strategies, we observed approximately 3–fold increases in γ-globin protein and the
frequency of HbF+ cells after erythroid differentiation of ZFN-edited HSPC, compared with unedited control cells (Figs. 1d and 1e). ZFN transfection in HSPC from four healthy donors and
three participants with SCD (all from single-agent plerixafor mobilization) resulted in comparable _BCL11A_ ESE target site editing in a majority of both healthy and SCD donor HSPC (75.3%
and 64.2%, respectively; p > 0.05; Fig. 1f), with high post-editing viability (Fig. 1g). Editing led to approximate 2–threefold increases in γ-globin levels in erythroid progenies from
healthy and SCD donor HSPC at day 18 of differentiation (Fig. 1h). Owing to unexpected differential growth kinetics between SCD donors and premature cell death of unknown cause in two SCD
donor HSPC preparations, we were only able to evaluate HbF+ cell frequency in HSC derived erythroid cells from one SCD donor. We found comparable increases in HbF+ cell frequency after
editing in healthy (n = 1) and SCD (n = 1) donor HSPC derived erythroid cells (Figure S1a). Red blood cell (RBC) sickling from the same SCD donor under hypoxic conditions was lower after
editing (Figure S1b-d). ZFN-MEDIATED GENE EDITING DOES NOT COMPROMISE THE FUNCTION OR PROGENIES OF HSC AND RESULTS IN STABLE EDITING LEVELS AFTER ENGRAFTMENT IN IN VIVO MOUSE MODELS We next
measured colony-forming unit (CFU) production in single-agent mobilized HSPC from healthy donors in vitro_._ CFU composition was similar between edited and unedited cells 2 days post-ZFN
editing (Fig. 2a; n = 3). Furthermore, there were no significant differences in frequency of enucleated cells after 20 days of in vitro erythroid differentiation (n = 4, p > 0.05; Fig.
2b). To determine the effect of ZFN editing on the function of HSC and engraftment potential, we transplanted ZFN-edited and unedited dual-agent mobilized control HSPC from two healthy
donors into immune-deficient NOD scid gamma (NSG) mice. Mice treated with edited or unedited HSPC had similar chimerism (Figs. 2c and 2d). We observed a small increase in HSPC and committed
progenitors in the bone marrow of mice engrafted with the first donor between edited and unedited cells after engraftment in NSG mice (p < 0.05; Fig. 2e). No significant difference was
observed in the bone marrow of mice engrafted with cells from the second donor (Fig. 2f). Comparable indel levels were observed in both healthy donor HSPC in the cell input and after
transplantation at Week 19 in peripheral blood (Fig. 3a). No significant difference in indel levels were measured in _BCL11A_-dependent (CD19+ B cells; CD34+CD38+ primitive progenitors) and
_BCL11A_-independent (CD33+ myeloid cells) lineages at Week 19 after engraftment of HSC from both donors (p > 0.05; Fig. 3b). Human in vitro differentiated erythroid cells derived from
engrafted HSPC showed similar indel levels to the other lineages (p > 0.05; Fig. 3b). In addition, no significant differences in terminal erythroid differentiation in vitro were observed
based on the proportion of enucleated cells in recipients treated with edited or unedited HSPC at Week 19 post-engraftment (p > 0.05; Fig. 3c). A significant increase in γ-globin in the
erythroid progeny was observed in edited versus unedited HSPC (Fig. 3d). We determined that the most common indel sequences at input resulting from editing in both donors were comparable,
with 15-, 13-, and 2-bp deletions the most frequently observed (Fig. 3e). The frequencies of indel patterns and sizes of insertions or deletions after 19 weeks of engraftment were similar
between the two donors (Fig. 3g), with no difference between the fractions of total indels likely derived from microhomology-mediated end-joining (MMEJ; Fig. 3f). The frequency of -13 and
-15 MMEJ indels dropped significantly after engraftment in both donors, likely reflecting loss of short-term HSC and committed progenitors (Fig. 3f and 3g, Figure S2). Insufficient cell
preparations were available to conduct these experiments for HSPC from sickle cell donors. ZFN-MEDIATED GENE EDITING LEADS TO ENRICHED BIALLELIC-EDITED CELLS AND ALLELE-ADDITIVE INCREASES IN
HBF We sorted single-agent mobilized, ZFN-edited HSPC from healthy (n = 5) and SCD (n = 3) donors into single cells and performed erythroid differentiation on each resulting colony (Fig.
4)37. We genotyped 1,175 single-cell–derived HSPC colonies by next-generation sequencing (Table S1a). Similar levels of editing, up to 95%, were observed after erythroid differentiation in
single cells from healthy and SCD donor HSPC (Fig. 4a). Indel levels in single-cell derived colonies were generally higher than in bulk HSPC or in vitro differentiated erythroid cells
(Figure S3). The most observed indel patterns in healthy donor and SCD donor samples were comparable, with the 15-bp, 2-bp, and 13-bp deletions the most frequent (Fig. 4b). The distribution
of wild-type (WT), monoallelic-, and biallelic-edited genotypes was highly skewed towards biallelic-edited cells in both healthy donor and SCD donor HSPC, representing > 90% and 88% of
edited clones, respectively, and indicating that ZFN-edited cells contained edits at both alleles (Fig. 4c and 4d; Table S1b and S1c). Background HbF protein levels varied across donors,
which were processed in batches, ranging from 7 to 22% in WT clones from healthy donors and from 22 to 51% for WT clones from SCD donors. Nevertheless, we observed a similar increase in HbF
across donors: HbF protein measurements indicated that each edited allele contributed additively to an increase of γ/(γ + β) ratio of ~ 15% (Figs. 4e and 4f). Biallelic-edited clones from
healthy donors and SCD donors had a 29% and 27% increase, respectively, of γ/(γ + β) ratio, compared with basal levels in unedited clones. To understand the contribution of different indel
patterns to HbF induction, we separated indels according to whether they disrupted the internal GATAA motif. We observed a similar additive increase in HbF in clones with GATAA-disrupting
indels in both healthy (n = 5) and SCD (n = 3) donor cells compared with WT, with biallelic clones expressing ~ 34% more HbF than WT (Fig. 4g, Figure S4). In contrast, biallelic-edited
clones with both GATAA motifs intact expressed ~ 13% more HbF compared with WT, suggesting that non–GATAA-disrupting indels contribute to HbF up-regulation, albeit to a lower extent (Fig.
4g). To determine the effect of ZFN editing on erythroid function, we examined the frequency of cell enucleation based on genotype and whether the indel pattern disrupted the internal GATAA
motif. In healthy donor HSC (n = 3), biallelic-edited cells showed a slight decrease in enucleation frequency compared with WT (Figure S5a). However, biallelic-edited cells with intact GATAA
sites (“WT/WT”) did not show differences in enucleation compared with biallelic-edited cells with disrupted GATAA sites (Figure S5b), suggesting that the observed differences in enucleation
may be due to the process of transfection and editing rather than specific effects of _BCL11A_ knockdown. No differences in enucleation frequency were observed among various genotypes or
indel patterns within GATAA sites in SCD donor HSPC (n = 3; Figures S5c and S5d). ZFN-MEDIATED GENE EDITING IS CHARACTERIZED BY HIGH FREQUENCY OF SMALL INDELS It was possible that
biallelic-edited homozygous clones (N = 162) and clones that failed to genotype (N = 16 with visible colonies and good HbF detection) harbored larger deletions at the target sites in one of
the alleles, encompassing the primer sites used for the amplicon, and thus were not captured by sequencing. Therefore, we further genotyped these clones from healthy donors (n = 4) using an
amplicon design that included both the target site and an informative heterozygous single nucleotide polymorphism that allowed the discrimination of both alleles. We subsequently sequenced
clones that remained indeterminate using a larger 12 kb Nextera XT amplicon assay centered on the target site (Fig. 5a). Overall, the majority (89.7%) of edited alleles were small indels
(1–50 bp; N = 963; Fig. 5b). Large deletions and insertions > 1 kb accounted for less than 1% and 0.3%, respectively. Similarly, we observed a very small number of complex events
involving both insertions and deletions (0.5%). The largest deletion was 5.9 kb within the _BCL11A_ ESE (Fig. 5c). However, this was an outlier considering that the second largest deletion
was ~ 2.5 kb and the median size of large deletions (> 1 kb) was 1.4 kb. The largest insertion was estimated to be 2.9 kb. All large insertions mapped to other genomic loci. Overall,
between 79.9% and 86.6% of total indels disrupted the GATAA motif (Fig. 5d). To understand the effect of indel size on HbF, we correlated the mean indel length per clone with HbF using a
linear model accounting for sample differences. There was a significant effect of mean size on HbF, although this was attenuated when accounting for GATAA-disrupting indels (Figure S6a and
S6b). There was no significant correlation between edit length and enucleation rate, suggesting that larger indels do not negatively impact erythroid function (Figure S6c). Finally, we
hypothesized that deletions > 50 bp may be more frequent in either committed progenitors or HSC. We observed an enrichment of MMEJ indel patterns in the second allele of clones harboring
deletions > 50 bp compared with clones with NHEJ small indels (Fig. 5e) and observed an increased frequency of microhomologies around deletions > 50 bp compared to smaller deletions
(< 50 bp; p < 0.00005). Since MMEJ mediated repair is less frequent in long-term engrafting HSC, these results suggest that large deletions are less likely to arise in this population.
INTERIM RESULTS FROM THE PHASE 1/2 PRECIZN-1 STUDY To show how the preclinical research data shown above translate to the clinical setting, we are summarizing the current results from the
PRECIZN-1 study, which was conducted following extensive safety studies. PARTICIPANT DISPOSITION At the cutoff date (August 15, 2023), seven participants with SCD had received infusions of
BIVV003 (Table S2)32,33. Group 1 comprised four participants administered BIVV003 using the original manufacturing process, each with up to 30 months of follow-up as of the cutoff.
Participants in Group 2 received BIVV003 manufactured according to an improved process designed to increase the number of long-term progenitors present at the end of the process without
affecting their editing frequency. One participant in Group 2 had 1 month of follow-up at the cutoff. Six of 13 enrolled participants who underwent mobilization and apheresis at least once
did not collect sufficient CD34 + HSPC on the first attempt to manufacture the minimal dose of BIVV003 and discontinued from the study without being dosed (TABLE S3). Four of 13 (31%)
participants underwent two cycles of mobilization and apheresis; two collected sufficiently through the repeat cycle to manufacture a dose of BIVV003 and subsequently received the infusion.
HEMATOPOIETIC RECONSTITUTION AND ENGRAFTMENT OF EDITED HSPC Seven participants underwent busulfan conditioning and infusion of autologous, ZFN-edited HSPC. The mean hemoglobin on the day of
BIVV003 infusion was 9.9 g/dL. Median times for neutrophil and platelet recovery were 21 days and 29 days, respectively. Participants received RBC transfusion support up to Day 21
post-transplant. After hematopoietic recovery, all participants were transfusion-free except for Participant 3, who required sporadic transfusions. The indel frequency in the BIVV003 drug
product infused ranged from 56–78% (Table S4). At Week 26, indel frequency ranged from 17–34% in unsorted bone marrow in all four Group 1 participants, and 37% and 33% in two Group 2
participants. The corresponding values for Week 52 were 17–32%. While both input indel percentages and indel percentages at equilibrium tended to be slightly lower in the clinical study,
with no strict correlation between the two values for individual samples, the indel patterns in the patient samples mirror those observed after engraftment of ZFN treated human HSC in mice
(Fig. 3) with a steep reduction of the percentage of the MMEJ derived 15 and 13 bp deletions compared to the drug product and a high degree of complexity with no specific indel dominant but
the 2 bp deletion as the most common indel observed post-infusion in the bone marrow or PBMC derived samples from all patients (data not shown). HB FRACTIONATION FOLLOWING BIVV003 INFUSION
Total hemoglobin (Fig. 6a) and clinical markers of hemolysis (Figure S7) stabilized by Week 26 post- BIVV003 infusion in all six participants with ≥ 26 weeks of follow-up. In Group 1,
percent HbF level (1–11% at screening) increased to 14–39% by Week 26 in all four participants. Notably, one participant had an HbF level which dropped below 15% from Week 26 onwards (Fig.
6a). In Group 2, percent HbF level increased to 45–54% at Week 26. Percent F cells increased up to 99% and reached a stable level by 26 weeks of follow-up in six infused participants (one
subject having reached only Day 29 post-infusion by the cutoff date). F cell levels persisted at 77% or greater in five participants for up to 104 weeks of follow-up (Fig. 6b). At Week 26,
five of six participants had stably achieved the average level of ≥ 10 pg of HbF/F cells that inhibits sickle hemoglobin (HbS) polymerization25 and sustained this level for up to 104 weeks
of follow-up (Fig. 6c). Group 2 participants generally attained higher HbF/F cell levels than Group 1 participants. (Fig. 6c). SAFETY AND TOLERABILITY Overall, the AEs reported in the study
were generally consistent with mobilization, apheresis, and conditioning treatment (TABLE S5). Two SAEs of sickle cell anemia with crisis in two patients were reported by the investigator as
related to plerixafor. One SAE of nausea was reported by the investigator as related to busulfan. There were no related SAEs and only one Grade 2 AE of anxiety assessed by the investigator
as related to BIVV003. As of the cutoff, three vaso-occlusive crises had been reported ~ 9 ,17 and 22 months after BIVV003 infusion in one participant in Group 1 (Fig. 6D). No other
SCD-related events were reported. DISCUSSION BIVV003 is a novel gene-edited autologous cell therapy in development for SCD (NCT03653247), in which HSPC are genetically modified with mRNA
encoding ZFNs that disrupt a specific regulatory GATAA motif in the _BCL11A_ ESE to reactivate HbF. ZFN-mediated gene editing does not require viral transduction or the use of DNA-annealing
oligonucleotides, such as guide RNAs. In our preclinical studies, ZFN editing was highly efficient, as evidenced by high indel frequency, and led to HbF induction ex vivo, in
plerixafor-mobilized HSC from both healthy donors and donors with SCD. ZFN-mediated gene editing did not impact terminal erythroid differentiation, consistent with previously published
approaches targeting the ESE of _BCL11A_30,31,38,39,40,41. Erythroid progenies from ZFN gene-edited HSC from an SCD donor showed attenuated in vitro sickling, supporting the therapeutic
impact of ZFN editing and HbF induction, and expanding on our previous studies in β-thalassemia31. ZFN-mediated gene editing did not compromise the function and potency of hematopoietic
progenitors, consistent with our previous studies30,31. Following engraftment of ZFN-edited HSPC into immunodeficient mice, similar human chimerism in circulation and bone marrow was
observed compared to non-edited HSPC. Furthermore, the frequency of engrafted, gene-edited cells in total and sorted bone marrow populations persisted_._ Erythroid cells differentiated ex
vivo from the chimeric bone marrow of primary recipients showed that ZFN-mediated gene editing resulted in effective HbF reactivation with no impact on enucleation, consistent with our in
vitro findings. Our studies show a long-term and sustainable effect in an in vivo mouse model. Long-term HSC were shown to preferentially undergo NHEJ-mediated repair following ZFN-mediated
gene editing. Sequencing of input cells showed the most common edits were 15- and 13-bp deletions, due to MMEJ repair. After in vivo engraftment, we observed a reduced frequency of MMEJ
repair alleles in engrafted versus input cells, suggesting that long-term HSC favor repair by NHEJ. Consistently, preclinical data from CRISPR-mediated gene editing in HSC suggest that
long-term HSC favor NHEJ-repair and that MMEJ may be less common in these cell types40,41. A major consideration for the clinical development of gene therapies is high efficiency of
therapeutic editing and the reduction of off-target editing. We demonstrate that ZFN-mediated gene editing in HSC from healthy donors and donors with SCD results in highly efficient and
precise disruption of the _BCL11A_ ESE. Genotype distributions of edited HSC were highly skewed towards biallelic editing, suggesting that ZFN-expressing cells are characterized by complete
allelic disruption. Colonies with biallelic modifications also exhibited increased HbF levels in an edited allele- and GATAA site disruptive-dependent fashion. The high precision of
ZFN-mediated gene editing was confirmed by sequencing a 12 kb region around the target site. High rates of unintended on-target large deletion of genes, including _BCL11A_, in CRISPR/Cas9
gene-edited SCD HSC were recently reported42,43. We observed that indels resulting from ZFN editing of healthy donor and SCD donor HSC were generally small and located well within the
intronic region of _BCL11__A_. The largest deletion was contained within the _BCL11A _ESE and all large insertions appeared to be random integrations. Our data also suggest that large
deletions are less likely to arise in the long-term engrafting HSC as large deletion rates positively correlate with MMEJ repair usage44. However, a limitation of our study is that
long-range sequencing was not conducted, which would have provided a more accurate characterization of potential large deletions, insertions, and complex events. Interim safety and efficacy
results confirm the potential therapeutic value of ZFN-mediated modification of the _BCL11A_ ESE region and BIVV003 infusion to address current unmet needs of individuals with SCD. Five of
six infused participants with more than 3 months of follow-up showed sustained increases in total Hb, HbF, and percent F cells. Participant 3, the one who displayed a non-sustained level of
HbF expression, likely did not have adequate myeloablation and/or engraftment of ex vivo LT-HSC despite receiving a cell product with a high percentage of edited cells. This is supported by
the large difference between the indel percentage in the drug product and after infusion (Table S4). BIVV003 was well tolerated in all seven participants infused to date, with all three
post-infusion severe VOC events reported in one participant (participant 3) after treatment, which were associated with HbF levels below what is inhibitory of HbS polymerization. The
manufacturing process used in group 1 has been shown to result in a reduction in the LT-HSPC population in the drug product (data not shown). This manufacturing process was also used in our
study of ZFN-edited autologous HSPC transplant in patients with transfusion-dependent beta-thalassemia (NCT03432364), and, in addition to the different mobilization procedure and patient
population, may have factored into the short-lived increases in HbF in RBC observed in that study. It may also have factored in the non-sustained HbF expression in participant 3, although
unique aspects of the treatment of this patient or their biology may have contributed to this outcome. In group 2, a refined manufacturing process with improved culture conditions during
CD34 + HSPC isolation and culture to leading to an increased number of LT-HSC in the drug product has been used. Remaining participants to be dosed in the Phase 1/2 study will receive drug
product manufactured using the improved process. Despite this manufacturing challenge, there was natural enrichment of modified erythroid lineage cells in the blood with elevated HbF,
sufficient for a clinical benefit in all participants, in terms of prevention of severe VOC and not requiring transfusion therapy with the exception of participant 3. Our results show that
ZFN-mediated disruption of the _BCL11A_ ESE results in highly efficient editing and produces HSC that are capable of long-term multilineage engraftment in vivo_,_ independent of mobilization
strategy or disease state, and express increased levels of HbF as erythroid progeny. The HbF and Hb levels achieved in group 2 to date indicate an improvement compared to group 1 and appear
comparable to those observed with exa-cel, a CRISPR-based, _BCL11A_ ESE editing approach that targets the same site and that recently received marketing approval. Through clonal analysis of
edited HSPC from healthy donors and donors with SCD, we have also shown enriched biallelic editing and a high frequency of small indels, supporting the high efficiency of ZFN gene editing.
While highlighting the challenges of consistently manufacturing clinical grade, autologous, gene-edited HSPC therapy for patients with SCD, our preliminary data support further clinical
development of this approach and suggest that BIVV003 represents a novel cell therapy for the potential treatment of SCD. ONLINE METHODS Experimental approaches used in this study have been
described previously30. All experiments were performed in accordance with relevant guidelines and regulations. All animal procedures were approved by the Sanofi Institutional Animal Care and
Use Committee (IACUC) and performed in accordance. The study is reported in accordance with the ARRIVE guidelines (https://arriveguidelines.org). The PRECIZN-1 study was performed in
accordance with the Declaration of Helsinki and local regulations and was approved by independent ethics committees or institutional review boards as described in the PRECIZN-1 section. CELL
CULTURE Human CD34 + cells were mobilized from de-identified healthy or sickle cell disease volunteers using granulocyte colony stimulating factor (G-CSF) and plerixafor or plerixafor as
previously described35. Mobilized CD34 + HSPC and/or chimeric bone marrow cells were cultured for 3 days in a maintenance media consisting of X-VIVO 10 (VWR), 100 U/mL
penicillin–streptomycin (ThermoFisher), 2 mM L-glutamine (Fisher Scientific), 100 ng/mL Recombinant Human Stem Cell Factor (SCF; ThermoFisher), 100 ng/mL Recombinant Human Thrombopoietin
(TPO; ThermoFisher), and 100 ng/mL Recombinant Human Flt-3 Ligand (Flt-3L; ThermoFisher). Cells were then differentiated into erythroid cells using a three-step differentiation protocol
developed by Giarratana et al. (2005)45. In brief, CD34 + cells were cultured for 7 days in Step1 media consisting of Iscove’s modified Dulbecco’s medium (IMDM) (ThermoFisher) supplemented
with 1X GlutaMAX, 100 U/mL penicillin–streptomycin (ThermoFisher), 5% human AB + plasma, 330 ug/mL human holo-transferrin, 10 ug/mL human insulin, 2 U/mL heparin, 1 uM hydrocortisone
(Sigma-Aldrich), 3 U/mL recombinant human erythropoietin (EPO) (ThermoFisher), 100 ng/mL SCF (ThermoFisher), and 5 ng/mL interleukin 3 (IL3; Sigma-Aldrich). On Day 7, cells were transferred
to Step2 media i.e., Step1 media without hydrocortisone and IL3, and cultured for 3–4 days. Lastly, cells were cultured for 8–9 days in Step3 media i.e., Step2 media without SCF. _BCL11A
EDITING OF HUMAN CD34_ + _CELLS USING ZFNS_ ZINC-FINGER NUCLEASES (ZFN) ZFN were designed and assembled as previously described28. The ZFN are composed of a triple flag domain, a nuclear
localization signal, an engineered DNA-binding domain and a mutated obligate heterodimer FokI nuclease domain. The pGEM-based expression vectors contain a T7 promoter 5′ and 3′ UTRs and a
synthetic 64-bp poly(A) stretch for optimized mRNA production. Compared to a ZFN pair described earlier (Psatha, Ref. 31) ZFN activity and specificity was further optimized. On the one hand,
this was achieved by fine tuning DNA binding affinity through extensive screening of zinc finger domain variants containing mutations which remove nonspecific phosphate contacts to the DNA
backbone. The ZFNs described in this publication both contain three variant amino acids in the zinc finger domain compared to the original left and right BCL11A targeting ZFNs. On the other
hand, ZFN performance was further improved via optimization of the DNA cleavage kinetics by mutating one residue in the FokI domain of the right ZFN. Both approaches are described in
Reference 29 (Miller et al.). The amino acid sequence in the zinc finger DNA binding domains is as follows: F1 F2 F3 F4 F% F6 Left ZFN DQSNLRA RNFSTM STGNTN TSGSLTR DQSNLRA AQCCLFH Right ZFN
DQSNLRA QKAHLIR QKGTLGE RGRDLSR RRDNLHS ELECTROPORATION To edit CD34 + cells, cells were thawed and cultured in the CD34 maintenance media described above for 1 or 2 days at 37C, 5% CO2
in a humidified incubator. CD34 + cells were electroporated using a MaxCyte GT transfection instrument (MacCyte) or Lonza Nucleofector 4D in the presence of the ZFN mRNAs. After
transfection, cells were incubated in the CD34 maintenance media at 30C for 18–24 h and then at 37C for 24 h. Two days after electroporation, cells were harvested, resuspended in CryoStor
CS10 (10% dimethyl sulfoxide [DMSO]), and transferred to cryovials. The cells were then cryopreserved and stored in liquid nitrogen. _TRANSPLANTATION OF HUMAN CD34_ + _HSPC IN NSG MICE_
Female NOD.Cg-_Prkdc_scid IL2rgtm1Wjl/SzJ (NSG) mice (Jackson Laboratories) at the age of 6–7 weeks, pre-treated with 10 mg/kg/day Baytril water for two days were sub-lethally irradiated at
300 rad 1 day before transplant. For primary recipients, human CD34 + cells were injected into the mice via the tail vein at 1 million viable cells per mouse. FLOW CYTOMETRY ENUCLEATION To
determine the enucleation rate of the erythroid differentiation culture at Day 20, cells were stained with NucRed (used for living cells; ThermoFisher) per the manufacturer’s instructions (2
drops/mL) and fluorescein isothocyanate (FITC)-conjugated anti-CD235 (Glycophorin A) antibody (Clone JC159-Dako-Agilent), washed with PBS (ThermoFisher) supplemented with 0.5% BSA
(Sigma-Aldrich). HBF STAINING To determine the percentage of HbF + cells, cells were fixed and permeabilized according to ThermoFisher protocol. Cells were also stained with Phycoerythrin
(PE)-conjugated anti CD235 antibody (ThermoFisher) and NucBlue (Hoechst 33,342, used for fixed cells; ThermoFisher). HbF levels were detected using Allophycocyanin (APC)-conjugated anti-HbF
antibody (ThermoFisher) after gating on the enucleated (Hoechst low) CD235 positive cell fraction. For enucleation and HBF staining methods, the acquisition of stained cells was performed on
FACSCanto and the analysis was done using FlowJo software. EX VIVO STAINING AND SORTING To assess the degree of human chimerism, fractions of cells in peripheral blood (at 8-, 12-, 16-, and
19-weeks post-engraftment) and bone marrow (at 12 and 19 weeks post-engraftment) were stained with mCD45 (Biolegend) and hCD45 (BD Biosciences) antibodies respectively and FACS analysis was
performed. In addition, hematopoietic lineage analysis was performed by staining bone marrow cells with anti-hCD3 (clone UCHT1), anti-hCD19 (clone SJ25C1), anti-hCD45, anti-hCD33,
anti-hCD38, anti-hCD34, anti-hCD71, and anti-hCD56 antibodies (BD Biosciences) and lineage cocktail (Biolegend) (Table S6). To purify and sort HSC populations, an enrichment/depletion
strategy using magnetic cell separation (MACS) was used (Table S7). Bone marrow cells were first stained with hCD19-biotin (clone HIB19), hCD3-biotin (clone HIT3a), mB220-biotin,
mTER119biotin and -m-ckitbiotin (BD Biosciences) and then incubated with -antibiotin- beads (Miltenyi Biotec). The positive fraction and depleted fraction were separated using LS columns
(Miltenyi Biotec) placed in the magnetic field of a MACS. After separation, the positive fraction was stained with StreptavidinAPC-, hCD3-FITC, hCD19-PE, hCD45-BV510 (BD Biosciences) and the
depleted fraction was stained with hCD34-FITC (BD Biosciences), hCD19APC (BD, clone SJ25C1), -LinAPC- (Biolegend), StreptavidinAPC-, hCD45-BV510, hCD33-PE-CF594 (BD Biosciences), and
hCD38-PECy-7 (Biolegend). Cell populations were sorted and processed for MiSeq analysis. IN VITRO_ HSPC FLOW STAINING_ Purified CD34 + cells were stained with anti-CD34, anti-CD38,
anti-CD45rA, anti CD49f. (BD), anti CD123, anti CD90 (ebioscience) and CD34hCD38-CD90 + CD45RA-CD49f. + (LT-HSC), CD34hCD38-CD90-CD45RA-CD49f.- (MPP), CD34hCD38-CD90-CD45RA-CD49f. +
(ST-HSC), CD34hCD38-CD90 + CD45RA- (HSC),CD34hCD38-CD90-CD45RA + (LMPP), CD34hCD38 + CD123 + CD45RA- (CMP), CD34hCD38 + CD123-CD45RA- (MEP), CD34hCD38 + CD123 + CD45RA + (GMP), CD34 low and
CD34- were sorted and processed for MiSeq. ASSESSMENT OF RBC SICKLING Sickling of CD34 + HSPC-derived RBCs was assessed under hypoxic and normoxic conditions. Approximately 2–10 × 106 RBCs
were pelleted by centrifugation and resuspended in 800 µL phosphate buffered saline (PBS) supplemented with 10 mmol/L-1 glucose and 0.2% w/v (bovine serum albumin (BSA). Two duplicate
24-well plates were prepared using 400 µL of each sample. The hypoxia plate was incubated at 1% oxygen for 2 h in a hypoxia chamber and the control plate incubated in normoxic conditions.
Following incubation, all cell samples were fixed with 40 µL of fresh 25% glutaraldehyde (EM grade), before pelleting by centrifugation and resuspension in 50 µL. Sickling was measured by
imaging flow cytometry (ImageStream). The number of sickled or normal cells is reported as a percentage of all cells. REVERSE PHASE UPLC Reverse phase high-performance liquid chromatography
(RP UPLC or RPLC) of human globin chains was used to determine any changes in the ratios of γ-globin, β-globin, and α-globin protein. Under conditions of varying pH (such as used in the UPLC
gradient), the globin chains (β, δ, α, G γ and A γ) were denatured and separated from the heme groups. Hemolysates from 1.0 × 106 RBCs obtained from Day 18 or 21 cultures were washed twice
with PBS and resuspended in 50 µL of UPLC grade water for lysis. The sample was then vortexed and the hemolysate clarified by centrifugation at 10,000 × g for 5 min at 4 °C. The hemolysate
supernatant was then transferred to a new tube or plate and stored at -80 °C until analysis. For the UPLC runs, 20 µL of hemolysate was injected directly into the column for each assay. UPLC
analyses were performed on a Waters Acquity BEH300 C4 column (C4 Reversed Phase, 300 A Pore Size 2.1 × 100 mm, 1.7 μm particle size; Waters Acquity; Product No. 186004496) on an Agilent
1290 Infinity II Ultra High-Performance Liquid Chromatographer (Agilent). Globin chains were eluted with a two solvent system (100% UPLC H2O + 0.1% trifluoroacetic acid and solvent B, 100%
acetonitrile + 0.1% trifluoroacetic acid) and a multi-step RPLC elution program starting at 35% Solvent B and gradually increasing to 38% Solvent B over a time period of 4 min, increasing
again to 42.5% Solvent B over a time period of 18 min, an increase to 70% Solvent B in 4 min followed by a 4 min flush of the column at 70% Solvent B and re-equilibration down to 35% solvent
B within 5 min. The flow rate was 0.2 mL/min. Quantitation of the amounts of the various globins was obtained by integration of the peak areas monitored by absorbance at 220 nm with
reference wavelength of 360 nm using software provided with the UPLC unit (Agilent OpenLab Chemstation Rev. C.01.07). COLONY FORMING UNIT CFU) ASSAY Human CD34 + enriched were washed with
PBS (1X) / 0.5% BSA and added to 300 mL media (IMDM with GlutaMAX; Invitrogen; Cat No. 31980030). The cells were then mixed in semi solid methycellulose media supplemented with cytokines
(MethoCult H4435 Classic; StemCell Technologies; Cat No. 04445) per the manufacturer’s instructions. In brief, 500 cells per well were plated in duplicates (1.1 mL cell culture per well) in
six well meniscus free cell culture plates (SmartDish; StemCell Technologies; Cat No. 27301). The plates were incubated for 14 days in a humidified incubator at 37 °C with 5% CO2 and then
scored for total progenitors and type of colony using an automated colony counter (STEMvision; StemCell Technologies; Cat No. 22000). The automated colony counter recorded the total number
of erythroid colony forming units (CFU E), erythroid burst forming units (BFU E), granulocyte colony forming unit / macrophage colony forming unit / granulocyte, macrophage colony forming
units (CFU G/M/GM) and granulocyte, erythrocyte, macrophage, megakaryocyte forming units (CFU GEMM) in each well. MISEQ INDEL ANALYSIS (OR GENOTYPING THE TARGETED LOCI BY DEEP SEQUENCING)
Disruption of targeted _BCL11A _ESE loci was quantitated by MiSeq deep sequencing of a 140-bp amplicon using procedures described previously30. The primer sequences bearing sample-specific
barcodes and the Illumina flow cell-specific sequences (P5 and P7) are provided in Methods Table 1. We used a MiSeq Reagent Kit v2 (300 cycles) for deep sequencing of the amplicon. N: mixed
base containing 25% of each nucleotide of ACGT (to increase sequence diversity). We merged paired-end reads using SeqPrep (https://github.com/jstjohn/SeqPrep), then trimmed reads using a
custom script to remove any remaining adapter sequence as well as the 4 random nucleotides from the left primer. The script also filtered out sequences where the first and last 20 bp do not
match the reference amplicon sequence. We then removed reads with lower quality bases using prinseq46(-min_qual_score 15) and aligned remaining sequences to the human genome (hg38) using
bowtie247. We filtered out sequences that aligned outside the target site but kept reads that failed to align. As a last filtering step, we removed reads ≤ 70 bp long to filter out potential
PCR artifacts. Finally, we aligned sequences to the expected amplicon using the Needleman-Wunsch algorithm as implemented in seq-align (https://github.com/noporpoise/seq-align). To count
indels, a window of 21 bp was defined around the central GATAA motif (GATAA ± 8 bp each side), and contiguous gaps in either the query or reference sequence that overlapped this window were
considered indels. SINGLE-CELL CLONAL EXPANSION ANALYSIS Single-cell clonal expansion analysis was performed as described previously30. Briefly, ZFN-edited CD34 + cells were seeded at 2
cells/well into 96-well non-TC treated plates (Corning) using FACSAria III. The cells were subjected to 3-step expansion in erythroid differentiation media for 20 days; genomic DNA and
protein lysate were collected at Day 14 and 20, for deep sequencing and RP-UPLC analysis, respectively. Additionally, at Day 20, cells were stained with GlyA-FITC antibody and Nuc Red for
enucleation rate. We called genotypes of single clones as above, with the following differences: For a subset of clones from sample 1 and all clones from samples 3 and 4, we used a larger
amplicon (471 bp) (MethodsTable 2) that covered common SNPs to potentially distinguish homozygous clones from clones where one allele failed to amplify due to a deletion covering the primer
sites. For the individuals with informative heterozygous SNP, this method allows confirmation of whether the originally assigned homozygous clones (biallelic edits with same editing pattern)
are true homozygotes or false homozygous where one allele failed to amplify due to a deletion covering the primer sites. We used a MiSeq Reagent Kit v2 (500 cycles) for deep sequencing of
the larger amplicon. We also included reads that failed merge with SeqPrep due to the overlap being too small. We called SNPs using bcftools mpileup48 on all clones. We only considered
clones with read counts > 50 reads and allele frequencies consistent with a clonal population (i.e. 2 major alleles with read counts of 50% each or one major allele at ~ 100% frequency).
Specifically, we used the following criteria: For heterozygotes, the top 2 indels needed to each comprise 40% to 60% of reads and together more than 85%, and the third most common indel
constitute less than 5% of read sequences. For homozygotes, the most common sequence needed to comprise more than 85% of reads, with the second indel less than 5%. For clones with
uncharacterized alleles (including homozygotes and clones that failed to amplify), we amplified a ~ 12 kb region centered on the target site (Methods Table 3) with PrimeSTAR® GXL DNA
Polymerase (R050A, Takara). After purification of the ~ 12 kb PCR products, we fragmented and tagged the DNA with adapter sequences using Nextera XT DNA library prep kit (FC-131–1096,
Illumina), following the instruction from the manufacturer. We amplified the library using Nextera XT Index Kit v2 Set A (FC-131–2001, Illumina) and pooled the library for deep sequencing
with MiSeq Reagent Kit v2 (300 cycles). We trimmed adapters from reads using Trimmomatic49, then aligned reads to the genome (hg38) using SpeedSeq align50. We used LUMPY51to call large
indels and bcftools mpileup48 to call small indels and SNPs. Moreover, each alignment was visually inspected in Integrated Genomics Viewer (IGV) to assure that editing events were not missed
by the variant callers, and to filter out events likely reflecting PCR artifacts (e.g. structural variants at the ends of the amplicon). ASSOCIATION OF HBF WITH EDITING IN SINGLE CLONES We
used linear regression to test the association of indels with HbF, adjusting for the differences between donors by incorporating a donor-specific covariate, specifically: $$\text{Y}={\beta
}_{0}+ {\beta }_{indels}{X}_{editing}+{\beta }_{donor}{X}_{donor}$$ where Y represents the HbF levels, \({\beta }_{0}\) is the intercept, \({\beta }_{indels}\) is the slope (beta)
coefficient for the editing level variable and \({\beta }_{donor}\) is the slope coefficient for the donor effect variable\({X}_{donor}\) . We represented HbF levels as the ratio of
gamma-globin over all beta-like globins. BIALLELIC ENRICHMENT ANALYSIS To calculate the enrichment of biallelic clones, we first calculated the expected number of clones using an equation
analogous to the Hardy–Weinberg equilibrium52: $${f}_{unedited}^{2}+ {2f}_{unedited}{f}_{edited}+{f}_{edited}^{2}=1$$ where \({f}_{unedited}\) and \({f}_{edited}\) are the frequencies of
unedited and edited alleles respectively. \({f}_{unedited}^{2}\) represents the expected frequency of unedited clones,\({2f}_{unedited}{f}_{edited}\) the expected frequency of monoallelic
edited clones and \({f}_{edited}^{2}\) the expected frequency of biallelic edited clones. We multiplied the frequencies by the total number of clones to obtain the expected number of
wild-type, monoallelic edited, and biallelic edited clones. Finally, we constructed a contingency table with the observed and expected number of clones and calculated significant deviations
using a chi-squared test. This analysis was done separately for each donor. HOMOZYGOTE ENRICHMENT ANALYSIS To calculate the enrichment of homozygote clones, we first permuted each allele
100,000 times. Then, we derived a _P_ value by counting the number of permutations that lead to a count of homozygotes higher than observed and dividing that number by 100,000. MICROHOMOLOGY
INDELS IDENTIFICATION We used the CRISPR RGEN Microhomology online tool53 to predict MMEJ indels, using the following sequence as input: CGCCCCCA CCCTAATCA GAGGCCAA ACCCTTC CTGGAGCC
TGTGATAA AAGCAAC TGTTAG CTTGCAC TAGACTA GCTTCA AAGTT GTATT. We used Pearson correlation to relate pattern scores with MMEJ-based indel frequencies. In addition, we inferred microhomology
sequences around small (< 50 bp) and large (> 50 bp) deletions using mhscanR44. PRECIZN-1: BIVV003 PHASE 1/2 STUDY DESIGN AND METHODOLOGY PRECIZN-1 (NCT03653247) is an ongoing,
first-in-human, open-label, single arm, multi-site, US-based study to evaluate safety, tolerability, and efficacy of BIVV003 in adult participants age 18–40 years with severe SCD .
Eligibility was based on having HbSS or HbSβ0 genotype, and at least one of the following: (1) history of clinically significant neurologic event (stroke) or any neurological deficit lasting
> 24 h; (2) history of 2 or more episodes of ACS in the 2-year period preceding informed consent; (3) six or more pain crises (requiring intravenous [IV] pain management) in the prior 2
years; (4) 2 or more episodes of priapism with subject seeking medical care or following a pre-approved management plan in the prior 2 years; (5) regular (≥ 8) RBC transfusions in the
preceding year to prevent vaso-occlusive clinical complications; (6) tricuspid valve regurgitant jet velocity ≥ 2.5 m/s by echocardiography. Hydroxyurea (HU) use was permitted but one had to
be willing to discontinue HU at least 30 days prior to stem cell mobilization through Day 100 post-transplantation. There was an initial 12-week screening period to assess participants’
suitability for the study. At the screening visit, participant 2 reported cerebral infarction, participant 4 reported stroke, and participant 7 reported cerebral microembolism. No stroke was
reported after the screening visit in the study. Transfusion therapy was used within 2 months of mobilization and prior to myeloablative conditioning to target levels of HbS < 30% and
total Hb at least 9–10 g/dL. Enrolled participants underwent plerixafor mobilization (240 μg/kg/day for up to 3 days) and apheresis to collect autologous CD34 + HSC with a target of 10 × 106
CD34 + HSC/kg for manufacturing BIVV003 using a LOVO device. Additional apheresis cycles were allowed to collect the minimum cell dose and unmodified rescue aliquots. Selected CD34 + cells
were cultured in the presence of cytokines (SCF, FLT-3L, TPO). Two days after CD34 + cell isolation autologous HSC were transfected ex vivo with ZFN messenger ribonucleic acids (mRNAs)
targeting the ESE region of the _BCL11A_ locus using a MaxCyte device. Transfected cells were cultured in the presence of cytokines for another two days and then harvested, formulated in
cryopreservation medium, aliquoted, frozen using a controlled-rate freezer and stored in the vapor phase of liquid nitrogen at ≤ −150 °C. For CD34 + cell culture in Group 1 Lonza X-Vivo 10
Medium and ThermoFisher cytokines were used, in Group 2 Cell Genix media and cytokines. In addition, for Group 2 manufacturing the LOVO instrument was upgraded with software version 3.0
which improved CD34 + cell recovery. Myeloablation of the participant’s bone marrow was performed using busulfan 3.2 mg/kg/day IV × 4 days, with pharmacokinetically adjusted dosing. At least
72 h after the final busulfan dose, a single IV infusion of 3–20 × 106 CD34 + HSC/kg was administered. Participants were monitored for hematopoietic engraftment and recovery, adverse events
(AE), clinical and laboratory hemolysis markers, total Hb and HbF, percentage of F cells, and sickle-cell-related events post-BIVV003 infusion. KEY STUDY ENDPOINTS Primary endpoints (to
evaluate safety and tolerability of BIVV003) were survival at post transplantation Day 100, Week 52, and Week 104 (last study visit), successful engraftment, and the occurrence of adverse
events and serious adverse events. Secondary endpoints (to assess the success and kinetics of stem-cell collection, manufacturing, and engraftment) were CD34 + HSC yield from stem-cell
mobilization, yield of ZFN-edited CD34 + HSC, time to initial neutrophil recovery following infusion (first of three consecutive days with absolute neutrophil count ≥ 500/μL), time to
platelet recovery post infusion (first of three consecutive measurements with a platelet count ≥ 50,000/μL at least 7 days after last platelet transfusion) and clinical assessments after
BIVV003 infusion. Data cutoff for study endpoints was September 2022. STATISTICAL ANALYSES No formal statistics were performed. All statistics are presented as descriptive. DATA AVAILABILITY
Next-generation sequencing data generated during the current study are available in the Sequence Read Archive (BioProject PRJNA1053294,
https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1053294). Qualified researchers may request access to patient level data and related study documents. Patient level data will be anonymized, and
study documents will be redacted to protect the privacy of our trial participants. Further details on Sanofi’s data sharing criteria, eligible studies, and process for requesting access can
be found at: https://www.vivli.org/.Other datasets generated during the current study are available from the corresponding author on reasonable request. REFERENCES * Frenette, P. S. &
Atweh, G. F. Sickle cell disease: old discoveries, new concepts, and future promise. _J Clin Invest_ 117, 850–858 (2007). Article CAS PubMed Google Scholar * Hebbel, R. P., Osarogiagbon,
R. & Kaul, D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. _Microcirculation_ 11, 129–151 (2004). Article CAS PubMed Google Scholar *
Kato, G. J. _et al._ Sickle cell disease. _Nat Rev Dis Primers_ 4, 18010 (2018). Article PubMed Google Scholar * Khemani, K., Katoch, D. & Krishnamurti, L. Curative Therapies for
Sickle Cell Disease. _Ochsner J_ 19, 131–137 (2019). Article PubMed Google Scholar * Lettre, G. & Bauer, D. E. Fetal haemoglobin in sickle-cell disease: from genetic epidemiology to
new therapeutic strategies. _Lancet_ 387, 2554–2564 (2016). Article CAS PubMed Google Scholar * Perrine, R. P., Brown, M. J., Clegg, J. B., Weatherall, D. J. & May, A. Benign
sickle-cell anaemia. _Lancet_ 2, 1163–1167 (1972). Article CAS PubMed Google Scholar * Kar, B. C. _et al._ Sickle cell disease in Orissa State. _India. Lancet_ 2, 1198–1201 (1986).
Article CAS PubMed Google Scholar * Pincez, T. _et al._ Variation and impact of polygenic hematologic traits in monogenic sickle cell disease. _Haematologica_ 108, 870–881 (2023).
Article CAS PubMed Google Scholar * Drysdale, C. M. _et al._ Hematopoietic-Stem-Cell-Targeted Gene-Addition and Gene-Editing Strategies for beta-hemoglobinopathies. _Cell Stem Cell_ 28,
191–208 (2021). Article CAS PubMed Google Scholar * Steinberg, M.H. et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. _Am J
Hematol_ 85, 403–408 (2010). * Venkatesan, V., Srinivasan, S., Babu, P. & Thangavel, S. Manipulation of Developmental Gamma-Globin Gene Expression: an Approach for Healing
Hemoglobinopathies. _Mol Cell Biol_ 41 (2020). * Wienert, B., Martyn, G. E., Funnell, A. P. W., Quinlan, K. G. R. & Crossley, M. Wake-up Sleepy Gene: Reactivating Fetal Globin for
beta-Hemoglobinopathies. _Trends Genet_ 34, 927–940 (2018). Article CAS PubMed Google Scholar * Hsieh, M. M. _et al._ Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem
cell transplantation for severe sickle cell phenotype. _JAMA_ 312, 48–56 (2014). Article PubMed Google Scholar * Hsieh, M. M. _et al._ Allogeneic hematopoietic stem-cell transplantation
for sickle cell disease. _N Engl J Med_ 361, 2309–2317 (2009). Article CAS PubMed Google Scholar * Mentzer, W. C., Heller, S., Pearle, P. R., Hackney, E. & Vichinsky, E. Availability
of related donors for bone marrow transplantation in sickle cell anemia. _Am J Pediatr Hematol Oncol_ 16, 27–29 (1994). CAS PubMed Google Scholar * Robinson, T. M. & Fuchs, E. J.
Allogeneic stem cell transplantation for sickle cell disease. _Curr Opin Hematol_ 23, 524–529 (2016). Article CAS PubMed Google Scholar * Esrick, E. B. _et al._ Post-Transcriptional
Genetic Silencing of BCL11A to Treat Sickle Cell Disease. _N Engl J Med_ 384, 205–215 (2021). Article CAS PubMed Google Scholar * Frangoul, H. _et al._ CRISPR-Cas9 Gene Editing for
Sickle Cell Disease and beta-Thalassemia. _N Engl J Med_ 384, 252–260 (2021). Article CAS PubMed Google Scholar * Basak, A. _et al._ BCL11A deletions result in fetal hemoglobin
persistence and neurodevelopmental alterations. _J Clin Invest_ 125, 2363–2368 (2015). Article PubMed Google Scholar * Sankaran, V. G. _et al._ Human fetal hemoglobin expression is
regulated by the developmental stage-specific repressor BCL11A. _Science_ 322, 1839–1842 (2008). Article ADS CAS PubMed Google Scholar * Sankaran, V. G. _et al._ Developmental and
species-divergent globin switching are driven by BCL11A. _Nature_ 460, 1093–1097 (2009). Article ADS CAS PubMed Google Scholar * Bauer, D. E. _et al._ An erythroid enhancer of BCL11A
subject to genetic variation determines fetal hemoglobin level. _Science_ 342, 253–257 (2013). Article ADS CAS PubMed Google Scholar * Weber, L. et al. Editing a γ-globin repressor
binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. _Sci Adv_ 6 (2020). * Metais, J. Y. _et al._ Genome editing of HBG1 and HBG2 to induce fetal
hemoglobin. _Blood Adv_ 3, 3379–3392 (2019). Article PubMed Google Scholar * Li, C. _et al._ In vivo HSPC gene therapy with base editors allows for efficient reactivation of fetal
gamma-globin in beta-YAC mice. _Blood Adv_ 5, 1122–1135 (2021). Article CAS PubMed Google Scholar * Humbert, O. et al. Therapeutically relevant engraftment of a CRISPR-Cas9-edited
HSC-enriched population with HbF reactivation in nonhuman primates. _Sci Transl Med_ 11 (2019). * Canver, M. C. _et al._ BCL11A enhancer dissection by Cas9-mediated in situ saturating
mutagenesis. _Nature_ 527, 192–197 (2015). Article ADS CAS PubMed Google Scholar * Vierstra, J. _et al._ Functional footprinting of regulatory DNA. _Nat Methods_ 12, 927–930 (2015).
Article CAS PubMed Google Scholar * Miller, J. C. _et al._ Enhancing gene editing specificity by attenuating DNA cleavage kinetics. _Nat Biotechnol_ 37, 945–952 (2019). Article CAS
PubMed Google Scholar * Chang, K. H. _et al._ Long-Term Engraftment and Fetal Globin Induction upon BCL11A Gene Editing in Bone-Marrow-Derived CD34(+) Hematopoietic Stem and Progenitor
Cells. _Mol Ther Methods Clin Dev_ 4, 137–148 (2017). Article CAS PubMed Google Scholar * Psatha, N. _et al._ Disruption of the BCL11A Erythroid Enhancer Reactivates Fetal Hemoglobin in
Erythroid Cells of Patients with beta-Thalassemia Major. _Mol Ther Methods Clin Dev_ 10, 313–326 (2018). Article CAS PubMed Google Scholar * Alavi, A. _et al._ Interim Safety and
Efficacy Results from a Phase 1/2 Study of Zinc Finger Nuclease-Modified Autologous Hematopoietic Stem Cells for Sickle Cell Disease (PRECIZN-1). _Blood_ 140, 4907–4909 (2022). Article
Google Scholar * Alavi, A. _et al._ Preliminary Safety and Efficacy Results from Precizn-1: An Ongoing Phase 1/2 Study on Zinc Finger Nuclease-Modified Autologous CD34+ HSPCs for Sickle
Cell Disease (SCD). _Blood_ 138, 2930–2930 (2021). Article Google Scholar * Yannaki, E. _et al._ Hematopoietic stem cell mobilization for gene therapy: superior mobilization by the
combination of granulocyte-colony stimulating factor plus plerixafor in patients with beta-thalassemia major. _Hum Gene Ther_ 24, 852–860 (2013). Article CAS PubMed Google Scholar *
Yannaki, E. _et al._ Hematopoietic stem cell mobilization for gene therapy of adult patients with severe beta-thalassemia: results of clinical trials using G-CSF or plerixafor in
splenectomized and nonsplenectomized subjects. _Mol Ther_ 20, 230–238 (2012). Article CAS PubMed Google Scholar * Hsieh, M. M. & Tisdale, J. F. Hematopoietic stem cell mobilization
with plerixafor in sickle cell disease. _Haematologica_ 103, 749–750 (2018). Article CAS PubMed Google Scholar * Lessard, S. _et al._ Zinc Finger Nuclease-Mediated Disruption of the
BCL11A Erythroid Enhancer Results in Enriched Biallelic Editing, Increased Fetal Hemoglobin, and Reduced Sickling in Erythroid Cells Derived from Sickle Cell Disease Patients. _Blood_ 134,
974–974 (2019). Article Google Scholar * Brendel, C. _et al._ Lineage-specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype. _J Clin Invest_ 126, 3868–3878 (2016).
Article PubMed Google Scholar * Brendel, C. _et al._ Preclinical Evaluation of a Novel Lentiviral Vector Driving Lineage-Specific BCL11A Knockdown for Sickle Cell Gene Therapy. _Mol Ther
Methods Clin Dev_ 17, 589–600 (2020). Article CAS PubMed Google Scholar * Demirci, S. _et al._ BCL11A enhancer-edited hematopoietic stem cells persist in rhesus monkeys without
toxicity. _J Clin Invest_ 130, 6677–6687 (2020). Article CAS PubMed Google Scholar * Wu, Y. _et al._ Highly efficient therapeutic gene editing of human hematopoietic stem cells. _Nat
Med_ 25, 776–783 (2019). Article CAS PubMed Google Scholar * Leibowitz, M. L. _et al._ Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. _Nat Genet_ 53, 895–905
(2021). Article CAS PubMed Google Scholar * Park, S.H. et al. Comprehensive analysis and accurate quantification of unintended large gene modifications induced by CRISPR-Cas9 gene
editing. _Sci Adv_ 8, eabo7676 (2022). * Owens, D. D. G. _et al._ Microhomologies are prevalent at Cas9-induced larger deletions. _Nucleic Acids Res_ 47, 7402–7417 (2019). Article CAS
PubMed Google Scholar * Giarratana, M. C. _et al._ Ex vivo generation of fully mature human red blood cells from hematopoietic stem cells. _Nat Biotechnol_ 23, 69–74 (2005). Article CAS
PubMed Google Scholar * Schmieder, R. & Edwards, R. Quality control and preprocessing of metagenomic datasets. _Bioinformatics_ 27, 863–864 (2011). Article CAS PubMed Google Scholar
* Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. _Nat Methods_ 9, 357–359 (2012). Article CAS PubMed Google Scholar * Danecek, P. et al. Twelve years of
SAMtools and BCFtools. _Gigascience_ 10 (2021). * Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. _Bioinformatics_ 30, 2114–2120 (2014).
Article CAS PubMed Google Scholar * Chiang, C. _et al._ SpeedSeq: ultra-fast personal genome analysis and interpretation. _Nat Methods_ 12, 966–968 (2015). Article CAS PubMed Google
Scholar * Layer, R. M., Chiang, C., Quinlan, A. R. & Hall, I. M. LUMPY: a probabilistic framework for structural variant discovery. _Genome Biol_ 15, R84 (2014). Article PubMed Google
Scholar * Canver, M. C. _et al._ Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in
mammalian cells. _J Biol Chem_ 292, 2556 (2017). Article CAS PubMed Google Scholar * Bae, S., Kweon, J., Kim, H. S. & Kim, J. S. Microhomology-based choice of Cas9 nuclease target
sites. _Nat Methods_ 11, 705–706 (2014). Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS This study was funded by Biogen, Sangamo, and Sanofi. We would like to
thank the investigators, research staff, donors, and trial volunteers who participated in these studies. Medical writing and editing support were provided by Neil M. Thomas and Rebecca S.
Jones of Fishawack Communications Ltd., part of Fishawack Health. This support was funded by Sanofi. FUNDING Sanofi, Biogen, Sangamo Therapeutics. AUTHOR INFORMATION Author notes * Samuel
Lessard and Pauline Rimmelé contributed equally to this work. AUTHORS AND AFFILIATIONS * Rare Blood Disorders, Sanofi, Waltham, MA, 02451, USA Samuel Lessard, Pauline Rimmelé, Hui Ling,
Kevin Moran, Benjamin Vieira, Yi-Dong Lin, Gaurav Manohar Rajani, Vu Hong, Isobelle Galeon, David Reiner, Lin Wang, Anne Ramezi, Pablo Rendo, Dana Levasseur, Robert Peters, Timothy Harris
& Alexandra Hicks * Sangamo Therapeutics, Richmond, CA, 94804, USA Andreas Reik, Richard Boismenu, Ben Hsu, Michael Chen & Bettina M. Cockroft * Cellular and Molecular Therapeutics
Branch, National Heart, Lung, and Blood Institutes/National Institute of Diabetes and Digestive and Kidney Diseases, National Heart, National Institutes of Health (NIH), Bethesda, MD, USA
Naoya Uchida & John Tisdale * Henry Ford Cancer Institute, Detroit, MI, USA Asif Alavi * Emory University, Aflac Cancer and Blood Disorders Center, Children’s Healthcare of Atlanta,
Atlanta, GA, USA Lakshmanan Krishnamurti * University of California-Davis Medical Center, Sacramento, CA, USA Mehrdad Abedi * University of California San Francisco Benioff Children’s
Hospital, Oakland, CA, USA Mark C. Walters * Precision Medicine and Computational Biology, Sanofi, Cambridge, MA, 02141, USA Samuel Lessard * Immunology and Inflammation, Sanofi, Cambridge,
MA, 02141, USA Alexandra Hicks Authors * Samuel Lessard View author publications You can also search for this author inPubMed Google Scholar * Pauline Rimmelé View author publications You
can also search for this author inPubMed Google Scholar * Hui Ling View author publications You can also search for this author inPubMed Google Scholar * Kevin Moran View author publications
You can also search for this author inPubMed Google Scholar * Benjamin Vieira View author publications You can also search for this author inPubMed Google Scholar * Yi-Dong Lin View author
publications You can also search for this author inPubMed Google Scholar * Gaurav Manohar Rajani View author publications You can also search for this author inPubMed Google Scholar * Vu
Hong View author publications You can also search for this author inPubMed Google Scholar * Andreas Reik View author publications You can also search for this author inPubMed Google Scholar
* Richard Boismenu View author publications You can also search for this author inPubMed Google Scholar * Ben Hsu View author publications You can also search for this author inPubMed Google
Scholar * Michael Chen View author publications You can also search for this author inPubMed Google Scholar * Bettina M. Cockroft View author publications You can also search for this
author inPubMed Google Scholar * Naoya Uchida View author publications You can also search for this author inPubMed Google Scholar * John Tisdale View author publications You can also search
for this author inPubMed Google Scholar * Asif Alavi View author publications You can also search for this author inPubMed Google Scholar * Lakshmanan Krishnamurti View author publications
You can also search for this author inPubMed Google Scholar * Mehrdad Abedi View author publications You can also search for this author inPubMed Google Scholar * Isobelle Galeon View author
publications You can also search for this author inPubMed Google Scholar * David Reiner View author publications You can also search for this author inPubMed Google Scholar * Lin Wang View
author publications You can also search for this author inPubMed Google Scholar * Anne Ramezi View author publications You can also search for this author inPubMed Google Scholar * Pablo
Rendo View author publications You can also search for this author inPubMed Google Scholar * Mark C. Walters View author publications You can also search for this author inPubMed Google
Scholar * Dana Levasseur View author publications You can also search for this author inPubMed Google Scholar * Robert Peters View author publications You can also search for this author
inPubMed Google Scholar * Timothy Harris View author publications You can also search for this author inPubMed Google Scholar * Alexandra Hicks View author publications You can also search
for this author inPubMed Google Scholar CONTRIBUTIONS All authors had access to study data, had full editorial control of the manuscript, and provided their final approval of all content.
CORRESPONDING AUTHOR Correspondence to Samuel Lessard. ETHICS DECLARATIONS COMPETING INTERESTS AH and SL are employees of Sanofi and may hold shares and/or stock options in the company. HL,
KM, BV, DL, DR, PR, RP, TH, YL, GMR, IG and VH were employees of Sanofi at the time of the study. AR, MC, and BH are employees of Sangamo and may hold shares and/or stock options in the
company. RB and BC were employees of Sangamo at the time of study. NU and JT have no conflict of interest. MA receives honorarium from Bristol Myers Squibb, hold stock from Cytodine Inc, and
participates in speaker bureaus for Bristol Myers Squibb, Gilead, and Abbvie. MCW has served as consultant for Ensoma, Inc., Vertex pharmaceuticals, AllCells, Inc, and BioLabs, Inc.
INFORMED CONSENT The study (NCT03653247) was performed in accordance with the Declaration of Helsinki and local regulations. The study was approved by an independent ethics committee or
institutional review board at each trial site (UCSF Benioff Children’s Hospital Oakland 2018–041; National Institutes of Health 526,145; Western Institutional Review Board
1–1,134,061-1/20,181,878; UC Davis Clinical Committee 1,363,599–4). All participants provided written informed consent. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains
neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION Below is the link to the electronic supplementary material.
SUPPLEMENTARY MATERIAL 1 (DOCX 1279.1 KB) SUPPLEMENTARY MATERIAL 2 (XLSX 152.6 KB) RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission
under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons
licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Lessard, S., Rimmelé, P., Ling, H. _et al._ Zinc finger nuclease-mediated
gene editing in hematopoietic stem cells results in reactivation of fetal hemoglobin in sickle cell disease. _Sci Rep_ 14, 24298 (2024). https://doi.org/10.1038/s41598-024-74716-7 Download
citation * Received: 13 November 2023 * Accepted: 27 September 2024 * Published: 16 October 2024 * DOI: https://doi.org/10.1038/s41598-024-74716-7 SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative