Play all audios:
ABSTRACT Deconvolution is an efficient approach for detecting cell-type-specific (cs) transcriptomic signals without cellular segmentation. However, this type of methods may require a
reference profile from the same molecular source and tissue type. Here, we present a method to dissect bulk proteome by leveraging tissue-matched transcriptome and proteome without using a
proteomics reference panel. Our method also selects the proteins contributing to the cellular heterogeneity shared between bulk transcriptome and proteome. The deconvoluted result enables
downstream analyses such as cs-protein Quantitative Trait Loci (cspQTL) mapping. We benchmarked the performance of this multimodal deconvolution approach through CITE-seq pseudo bulk data, a
simulation study, and the bulk multi-omics data from human brain normal tissues and breast cancer tumors, individually, showing robust and accurate cell abundance quantification across
different datasets. This algorithm is implemented in a tool MICSQTL that also provides cspQTL and multi-omics integrative visualization, available at
https://bioconductor.org/packages/MICSQTL. SIMILAR CONTENT BEING VIEWED BY OTHERS CELL-ATTRIBUTE AWARE COMMUNITY DETECTION IMPROVES DIFFERENTIAL ABUNDANCE TESTING FROM SINGLE-CELL RNA-SEQ
DATA Article Open access 05 June 2023 BENCHMARKING OF CELL TYPE DECONVOLUTION PIPELINES FOR TRANSCRIPTOMICS DATA Article Open access 06 November 2020 ENHANCED SENSITIVITY AND SCALABILITY
WITH A CHIP-TIP WORKFLOW ENABLES DEEP SINGLE-CELL PROTEOMICS Article Open access 16 January 2025 INTRODUCTION Proteomics profiling and analysis at cell type level is critical in the study of
complex biological systems with numerous applications in immunology, cancer research, and developmental biology1,2,3. Several technologies have been developed to identify and quantify
proteins at cellular resolution4. For example, the detection of proteins by CyTOF coupled with fluorescence activated cell sorting (FACS)5 and the single cell multimodal technology
CITE-seq6, which is a multimodal sequencing technique that enables simultaneous profiling of gene expression and up to 300 cell surface protein markers in individual cells, allows the
identification of rare cell types and cells that express low levels of certain genes. However, these technologies only measure limited number of proteins. The abundance of proteins not
detectable in CyTOF or CITE-seq may be strikingly different from the transcript expression of coding genes and cannot be approximated by scRNA-seq measurement because of the RNA/protein
degradation and post-translation modifications. Recent advances in liquid chromatography mass spectrometry (LC-MS)-based proteomics methods have addressed the limitations in the sensitivity
and throughput7,8, which accelerates the evolvement of single cell mass spectrometry (scMS) proteomics. One major challenge in scMS proteomics is that the number of unique samples and cells
analyzed in a single day is very limited9. For label-free scMS, samples are analyzed sequentially with analysis time ranging from 35 to 90 min. At this speed, a maximum of 40 single cells
could be analyzed in a day, which is not ideal for population-scale clinical studies due to the burden of time and costs. The scMS technology limitations and costs of cell sorting are the
hurdles for cell-type-specific inference such as differential expression (csDE) or protein quantitative trait mapping (cspQTL) that requires median or large sample size. Deconvolution
algorithms are rapidly developed to measure molecule proportions (e.g., RNA transcripts) mapped to each cell type, which is different from the cell counts composition and varies across
molecular sources. To estimate the cellular composition in human proteome, the pure cell or single cell reference proteomes (i.e., signature matrix) is needed but lacking in certain tissues
or cell types due to the challenges in cellular dissociation (e.g., astrocytes and excitatory neurons2) and the aforementioned limitations in scMS and CITE-seq. Meanwhile, the multi-omics
profiling matched by samples becomes popular in recent decade, enabling the integration across data sources and the discovery of multimodal signatures for disease1. Hence, we design a
algorithm to estimate the proteomics cell fractions by integrating bulk transcriptome-proteome without single cell reference proteome, implemented in R package MICSQTL. Our method enables
the downstream cell-type-specific protein quantitative trait loci mapping (cspQTL) based on the mixed-cell proteomes and pre-estimated proteomics cellular composition, without the need for
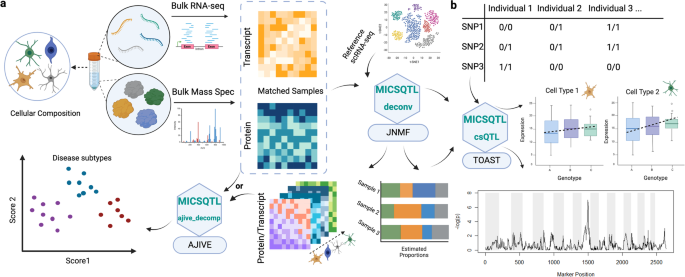
large-scale single cell sequencing10 or cell sorting. RESULTS MICSQTL ALGORITHMS AND IMPLEMENTATION Our method quantifies the cell abundances in proteins by jointly deconvoluting matched
bulk transcriptome and proteome, which can be used in downstream cspQTL mapping (Fig. 1). For each tissue sample with bulk transcriptome-proteome, we model the cellular compositions _Θ_(_i_)
in the _i_th modality as a product of tissue-specific cell counts fractions _P_ and molecule source-specific cell size factors _S_(_i_). A Joint Non-negative Matrix Factorization (JNMF)
framework was employed to link the modalities through shared cell counts _P_, allowing individualized multimodal reference panels. We employ a loss function that integrates the observed bulk
RNA and protein expressions to optimize the cell abundances in each molecular source, as described in Methods. The parameters in JNMF are initialized by an RNA signature matrix of similar
tissue type and the RNA proportions pre-estimated by CIBERSORT (CBS)11 with this signature matrix, the first of which can be obtained from scRNA-seq or sorted cell RNA-seq data accessible in
many public repositories. Hence, this joint deconvolution algorithm is semi reference-free without using a single cell or pure cell proteomics reference profile, implemented in the function
_deconv_. In proteomics deconvolution, researchers may not have a priori knowledge about the cell marker proteins to be used for certain cellular subpopulations, but the cell marker genes
in transcriptome have been broadly identified and curated in public databases12. Here, we use the AJIVE framework13 to construct a common space shared across two molecular sources: bulk RNA
expression of cell marker genes and the sample-matched whole proteome, which captures the between-sample heterogeneity caused by cellular abundance variation. Next, the observed whole
proteome is projected onto this shared space by employing the reduced-rank loadings from AJIVE, where protein dimensions and annotations are unchanged. The rank of loadings is determined by
an inherent algorithm. The potential cell marker proteins are selected based on the feature-wise Euclidean distance between the projected and observed proteomes. This cross-source feature
selection is similar to ReFACTor14, but ReFACTor is only applicable to single modality and sets the rank of loadings by the assumed number of cell types. Hence, we name our signature
selection procedure as ‘AJ-RF’ and use the selected proteins in joint deconvolution, implemented in the function _ajive_decomp_. VALIDATION USING MULTIMODAL EXPRESSION FROM CITE-SEQ We first
validate our algorithm by using the pseudo bulk multimodal expression profiles built from a public CITE-seq dataset with 161,764 human peripheral blood mononuclear cells (PBMCs). These
samples were collected from eight donors1 and processed by \(10\times {3}^{{\prime} }\) technology and Seurat v4. For each single cell, the expression of 228 surface proteins and more than
30,000 RNA genes were measured. Hence, we generated pseudo bulk expression data for a list of RNA cell marker genes and 228 surface proteins by aggregating feature-wise abundance or Unique
Molecular Identifier (UMI) counts across the cells per donor. The ground truth of cellular fractions can be achieved by the annotated cell labels, which are identical for RNA and protein in
CITE-seq. We selected four donors (samples P1 and P7 in Fig. 2a) with disparities in B cell, natural killer (NK) cell, dendritic cell (DC), and other T cell abundances to generate the pseudo
signature matrix. The cell count fractions quantified by our algorithm were similar to the true cell count fractions captured and annotated in CITE-seq (i.e., Pearson correlation (r) =
0.91, Lin’s concordance correlation coefficient (CCC) = 0.91), as demonstrated in Fig. 2b, and showing improvement over the CBS method (r = 0.88, CCC = 0.85). The individualized
cell-type-specific expression is exemplified for three cell types: B cell, CD4 T cell, and CD8 cell in Fig. 2c–h. Although the individualized cell-type-specific protein expression displayed
larger variance, it remained significantly correlated with the observed pure cell bulk expression (Fig. 2c–e). This discrepancy may arise from the distinct nature of cell surface protein
measurement through a binding strategy, as opposed to RNA UMI counts. The RNA expression levels resolved by our algorithm were well-aligned near the diagonal line with _r_ > 0.9 across
all three cellular populations (Fig. 2f–h). Further, we examined the impact of varying step sizes on the results obtained from the same input data and different rescaling approaches. The
results in Supplementary Note 1 Figs. S1–S5 suggest that despite of ambiguity in the optimal step size, moderate adjustments in step size and rescaling by log or MinMax do not lead to
substantial change in the deconvoluted cellular composition. Nevertheless, the above results demonstrate the validity and power of our algorithm without reliance on a single cell (type)
proteomics reference. ASSESSMENT USING SIMULATION DATA The above pseudo bulk data from CITE-seq only provides the ground truth for cell counts fractions instead of the cell proportions in
each molecule source. To mimic the possible differences in RNA vs. protein cellular compositions and compare the performance of distinct methods, we rigorously designed a simulation study to
generate synthetic bulk transcriptome-proteome with ground truth of modality-specific cellular compositions. The statistical models used in data generation are described in Supplementary
Note 2, in which the statistical parameters are extracted from a public scRNA-seq data of human brain15, a public single-cell-type mouse brain proteomics data16, and a bulk proteomics data
of human brain described in the next section. We designed two scenarios to allow relatively low (scenario A) and high (scenario B) correlations between the protein and RNA proportions, as
visualized in Fig. 3a, b. We compared our algorithm to three existing methods: the RNA proportions resolved by CBS (i.e., the initial cell counts fractions used in JNMF) as surrogate protein
proportions, the cellular fractions estimated by TCA with CBS RNA proportions as initial value and the bulk proteomes as target data, and a deep learning-based method scpDeconv17 using
single-cell reference data for training. To the best of our knowledge, the high-quality single cell proteomics data in human prefrontal cortex is not publicly available. Therefore, we chose
the scRNA-seq data used in the above data generation as a surrogate single cell reference for scpDeconv. The output from each method was compared to the ground truth protein proportion via
mean absolute error (MAE) in Fig. 3c–d, g–h and CCC in Fig. 3e–f, i–j. To deconvolve the synthetic bulk RNA-seq data with CBS, we constructed _n_ = 50 replicates of pseudo signature matrix
per scenario by introducing small vs. large random noises to the ground truth of subject-specific reference transcriptome. The RNA cell marker genes were chosen based on gene-wise
coefficients of variation from the true reference panel, while 700 out of 1000 proteins were selected with AJ-RF. For each pseudo signature matrix and the corresponding initial CBS estimate,
JNMF significantly improved the accuracy of cellular compositions in proteomes compared to CBS, as validated by a paired _t_ test (Fig. 3c–j). Our method also outperforms TCA and scpDeconv
across the scenarios and pseudo signature matrices, whereas a single cell (type) proteomics reference profile is lacking. Notably, the proteins selected by AJ-RF yielded similar
outperformance over the competing methods, implying the efficacy in capturing the latent cellular heterogeneity. We also benchmarked the robustness of our algorithm via initialization with
Non-Negative Least Squares (NNLS) RNA proportions and assessed the computation power of scpDeconv in bulk RNA deconvolution. Supplementary Fig. S6 presents statistically significant
improvement by JNMF in MAE and CCC, although NNLS estimate was less accurate compared to CBS initial and restricted the absolute performance of our method. The result of proteomics
deconvolution tool scpDeconv in an application to bulk RNA-seq data was even worse than NNLS because of the inherent differences between RNA and protein molecules and the disitnct profiling
technologies. Again, our algorithm demonstrated superior accuracy and robustness in predicting proteomics cell proportions compared to alternative approaches. APPLICATION TO BULK
TRANSCRIPTOME-PROTEOME IN BIPOLAR DISORDER, SCHIZOPHRENIA, AND HEALTHY CONTROLS We used tissue-matched transcriptome-proteome from postmortem human brain prefrontal cortex in a study of (_n_
= 25) bipolar disorder (BP) and (_n_ = 45) schizophrenia (SCZ) cases and (_n_ = 194) healthy controls to demonstrate the performance of MICSQTL. The details of human brain tissues, MS
proteomics and RNA-seq transcriptomics profiling are available in Supplementary Note 3. The signature genes used in competing methods were selected from previous findings16, while the
(initial) signature matrix was generated by scRNA-seq data of healthy human prefrontal cortex15. The protein proportions quantified by JNMF-AJ-RF algorithm (Fig. 4a) showed possible
disparities in astrocyte, microglia, and oligodendrocyte abundances between the (BP and/or SCZ) cases and controls whose brain tissues (death) were under 70 years of age. This result aligns
with the expected cell abundance in human prefrontal cortex15,18 and is similar to the previous findings about glia cell abundance in SCZ19,20. But there are confounding factors not
regressed out in this dataset such as sub-cohorts and aging. The validity of cellular compositions in BP and SCZ compared to controls requires more experiments or an external cohort with
adequate sample size of single cell proteomes or transcriptomes. On the other hand, the initial CBS RNA deconvolution failed to recover the expected mean abundances in inhibitory neuron and
microglia (Fig. 4b). We also applied CBS to the bulk proteomes with a proteomics signature matrix of four cell types profiled from major regions of mouse brain16, which neither decomposed
the neuron cells into excitatory and inhibitory subpopulations nor recovered the expected cell abundances (Fig. 4c). A downstream cspQTL screening was performed by cell-type-specific
differential analysis, implemented in the function _csQTL_. The input data include bulk proteomes, whole genome sequencing genetic variants (SNP), and protein proportions in Fig. 4a and
Supplementary Data 4. We selected several mutation markers reported in previous literature to illustrate the csQTL function in MICSQTL. For each protein of interest, we select the nearby
SNPs within a genomic distance of 1 million bases (Mb). Figure 4d, e shows the SNPs with false discovery rate (FDR) adjusted _p_ value < 0.2 per cell type for the proteins encoded by
genes associated with neurodevelopmental or neuropsychiatric illness: _MAST4_ and _ADCYAP1R1_. These genes or the related gene family were reported as mutations associated with increased
risks for Alzheimer’s disease21, mega-corpus-callosum syndrome22, and post-traumatic stress disorder23. The adjusted p-values were listed in Supplementary Data 4, while cspQTL results for
additional genes were shown in Figure S7. Note that the validity of cspQTL result depends on the study populations for the bulk proteomes and the accuracy of proteomics deconvolution, which
should be investigated through a rigorous single cell study measuring the proteins of interest with adequate sample size. Last but not least, our tool MICSQTL outputs the multivariate Common
Normalized Scores (CNS) from AJIVE that represents the sample-specific variation shared across transcriptome and proteome, which may uncover the heterogeneity across disease phenotypes and
the hidden drivers. The multi-omics human brain tissue samples were jointly visualized in Fig. 4f by using the concatenated CNS, which outperforms the single modality visualization (Fig. 4g,
h). APPLICATION TO MULTI-OMICS DATA IN BREAST CANCER To demonstrate our method in different tissue types, we utilized the patient-matched bulk RNA-seq and MS proteomics data from _n_ = 122
breast cancer (BC) tumor tissues24 along with the single cell multi-omics data in an external BC cohort25. We first applied CBS to the bulk RNA-seq data24 and a signature matrix built from
the external scRNA-seq profiles25, which served as the initial values in JNMF algorithm. The aforementioned deep learning deconvolution tool scpDeconv was applied to the bulk MS proteomics
data, using the single cell CyTOF proteomics profiles from external BC tissues25 to train the autoencoder. The input single cell and bulk data were transformed by log scale for CBS,
JNMF-AJ-RF and z-score for scpDeconv. The deconvolution result by each method was benchmarked by the annotated cell fractions in the scRNA-seq data of external BC tissues25 (Fig. 5a).
Obviously, the tumor microenvironment deconvolved by CBS with bulk transcriptomes (Fig. 5b) was substantially improved by applying JNMF-AJ-RF to the bulk multi-omics profiles (Fig. 5c and
Supplementary Data 5). The proteomics tumor microenvironment composition resolved by a scpDeconv submodule that only uses the (seven) proteins detected in both single cell CyTOF panel and
bulk MS proteomes was the best estimate (Fig. 5d). However, this outperformance of scpDeconv may rely on the accurate measurement or low dimension of marker proteins in the CyTOF reference
panel and not necessarily hold for the untargeted single cell reference proteomics data (e.g., scMS). To illustrate this caveat, we ran another submodule of scpDeconv that imputed the single
cell reference for additional (20 or 50) highly variable proteins (HVP) only detected in the bulk proteome. For either set of HVP, this module was rerun ten times to assess the variation of
performance. The replicates of deconvoluted proportions in Supplementary Note 1 Figs. S8–S9 demonstrate that predicting the abundance of proteins not available in CyTOF reference panel may
randomly reduce the accuracy of deconvolution. In other words, the performance of scpDeconv depends on the availability and quality of protein markers measured at single cell level.
DISCUSSION Our pipeline offers three primary functions to perform multi-omics cell abundance quantification with or without marker protein selection, integrative visualization, and cspQTL
mapping. The semi reference-free joint quantification of cellular compositions in RNA and proteins were benchmarked by multiple datasets. That is pseudo bulk RNA and protein expression
constructed from CITE-seq single cell data, synthetic bulk multimodal expression generated by statistical models, and real human brain bulk transcriptome-proteome with external snRNA-seq
data. Overall, JNMF coupled with cross-modality signature protein selection significantly improves the cell abundance quantification of MS proteomes compared to CBS, TCA, and scpDeconv with
imputation. Our algorithm also identifies the proteins contributing to the latent cellular heterogeneity in bulk multi-omics profiles, which may elucidate the cell marker proteins for
certain tissue types. This multimodal deconvolution framework is more favorable in population-scale studies compared to the single modal deconvolution or single cell profiling, since it
neither relies on a single cell proteomics reference profile nor requires cell clustering and labeling. The hyperparameter (PGD step size) in our algorithm has marginal impact on the
estimated proportions and can be adjusted according to the scale of input data. In the current version of MICSQTL, we do not suggest the optimal scaling approach since the accuracy of
purified data varies and may affect the downstream analysis such as integrating purified multi-omics profiles. According to the extensive experiments in CITE-seq pseudo bulk data, the impact
of step size on cellular fractions was reduced by the MinMax rescaling across all the features, but the purified RNA expression was distorted. Another potential utility of our tool is the
high-resolution purification of individualized pure cell expression, which is an essential component in the output of our algorithm and paves the way for deep proteome profiling at single
cell type resolution. However, the current implementation of joint deconvolution algorithm emphasizes the accuracy of cell abundance quantification, which may sacrifice the power of
high-resolution purification. A possible solution to improve individualized multimodal pure cell expression is to train a deep learning model (such as autoencoder26) on the observed bulk
proteomes and the reference panels pre-estimated by JNMF algorithm. Meanwhile, it’s worth employing Stochastic Gradient Descent in the future to reduce the errors in pre-estimated reference
panels. Altogether, the JNMF deconvolution algorithm substantially improves the cell abundance estimation in bulk proteome by integrating modalities and using PGD for high-dimensional
parameter optimization. The impact of PGD optimization on the quantified cell abundances and individualized purification will be assessed extensively in future. Our tool MICSQTL not only
fills the methodological gap in bulk proteomics deconvolution without using single cell proteomics data, but also sheds light on the design of a comprehensive experiment that profiles single
cell MS proteomes matched to bulk samples to benchmark the performance of different deconvolution tools. METHODS MULTIMODAL JOINT DECONVOLUTION For the tissue biospecimen of individual _i_,
we measure the expressions of protein _g_ (_g_ = 1, …, _G_) and mRNA transcript (or gene) _m_ (_m_ = 1, …, _M_), respectively, denoted by \({y}_{gi}^{(1)}\), \({y}_{mi}^{(2)}\). The
unobserved and individualized pure cell expressions are denoted by \({x}_{gik}^{(1)},{x}_{mik}^{(2)}\). The molecular source-specific cellular fractions for cell type _k_ are \({\theta
}_{ik}^{(1)}\), \({\theta }_{ik}^{(2)}\) (_k_ = 1, …, _K_), determined by the common tissue-specific cell counts (fractions) _p__i__k_ and source-specific cell size factors
\({s}_{ik}^{(1)}\), \({s}_{ik}^{(2)}\). That is \({\theta }_{ik}^{(1)}={p}_{ik}{s}_{ik}^{(1)}\), \({\theta }_{ik}^{(2)}={p}_{ik}{s}_{ik}^{(2)}\). Thus, the bulk multi-modal expression data
are modeled as $$E({y}_{gi}^{(1)})=\mathop{\sum }\limits_{k=1}^{K}{x}_{gik}^{(1)}{\theta }_{ik}^{(1)}=\mathop{\sum }\limits_{k=1}^{K}{x}_{gik}^{(1)}{p}_{ik}{s}_{ik}^{(1)},$$
$$E({y}_{mi}^{(2)})=\mathop{\sum }\limits_{k=1}^{K}{x}_{mik}^{(2)}{\theta }_{ik}^{(2)}=\mathop{\sum }\limits_{k=1}^{K}{x}_{mik}^{(2)}{p}_{ik}{s}_{ik}^{(2)}.$$ We denote
\({{{{{{{{\boldsymbol{y}}}}}}}}}_{i}^{(1)}={\left[{y}_{gi}^{(1)}\right]}_{G\times 1}\), \({{{{{{{{\boldsymbol{X}}}}}}}}}_{i}^{(1)}={\left[{x}_{gik}^{(1)}\right]}_{G\times K}\),
\({{{{{{{{\boldsymbol{y}}}}}}}}}_{i}^{(2)}={\left[{y}_{mi}^{(2)}\right]}_{M\times 1}\), \({{{{{{{{\boldsymbol{X}}}}}}}}}_{i}^{(2)}={\left[{x}_{mik}^{(2)}\right]}_{M\times K}\), _P__i_ =
diag(_p__i_1, …, _p__i__K_), \({{{{{{{{\boldsymbol{s}}}}}}}}}_{i}^{(1)}={\left[{s}_{ik}^{(1)}\right]}_{K\times 1}\) and
\({{{{{{{{\boldsymbol{s}}}}}}}}}_{i}^{(2)}={\left[{s}_{ik}^{(2)}\right]}_{K\times 1}\). The above source-specific models are linked by the common tissue-specific cell counts fractions
_P__i_. Hence, we propose to jointly estimate the high-dimensional non-negative parameters \({{{{{{{{\boldsymbol{\eta
}}}}}}}}}_{i}=\{{{{{{{{{\boldsymbol{X}}}}}}}}}_{i}^{(1)},{{{{{{{{\boldsymbol{X}}}}}}}}}_{i}^{(2)},{{{{{{{{\boldsymbol{p}}}}}}}}}_{i},{{{{{{{{\boldsymbol{s}}}}}}}}}_{i}^{(1)},{{{{{{{{\boldsymbol{s}}}}}}}}}_{i}^{(2)}\}\)
by minimizing a loss function that integrates \({{{{{{{{\boldsymbol{y}}}}}}}}}_{i}^{(1)}\), \({{{{{{{{\boldsymbol{y}}}}}}}}}_{i}^{(2)}\). This is achieved by solving
$${{{{\hat{{{{{\boldsymbol{\eta }}}}}}}}}}_{i}=\arg \mathop{\min }\limits_{{{{{{{{{\boldsymbol{\eta }}}}}}}}}_{i}}\left\{{\left\Vert
{{{{{{{{\boldsymbol{y}}}}}}}}}_{i}^{(1)}-{{{{{{{{\boldsymbol{X}}}}}}}}}_{i}^{(1)}{{{{{{{{\boldsymbol{p}}}}}}}}}_{i}{{{{{{{{\boldsymbol{s}}}}}}}}}_{i}^{(1)}\right\Vert }^{2}+{\left\Vert
{{{{{{{{\boldsymbol{y}}}}}}}}}_{i}^{(2)}-{{{{{{{{\boldsymbol{X}}}}}}}}}_{i}^{(2)}{{{{{{{{\boldsymbol{p}}}}}}}}}_{i}{{{{{{{{\boldsymbol{s}}}}}}}}}_{i}^{(2)}\right\Vert }^{2}\right\}$$ subject
to \(\min \{{{{{{{{{\boldsymbol{\eta }}}}}}}}}_{i}\}\ge 0\). This loss function integrates the observed multimodal bulk data by the shared cell counts _P__i_ as an extension to the
Non-negative Matrix Factorization. This algorithm initializes the multi-omics reference panels \({{{{{{{{\boldsymbol{X}}}}}}}}}_{i}^{(1)},{{{{{{{{\boldsymbol{X}}}}}}}}}_{i}^{(2)}\) and the
cell counts fractions _P__i_ with an external RNA-seq signature matrix and the RNA proportions pre-estimated by CBS. These sample-wise parameters of multimodal reference panels and cellular
compositions are then optimized by the Projected Gradient Descent algorithm27, being simultaneously updated and adapted to tissue-specific bulk multi-omics profiles. ALGORITHM * 1. The
initial values of \({{{{{{{{\boldsymbol{X}}}}}}}}}_{i}^{(1)},{{{{{{{{\boldsymbol{X}}}}}}}}}_{i}^{(2)}\) are the cell-type-specific expression from an external RNA signature matrix, while the
initial values of \({s}_{ik}^{(1)}\), \({s}_{ik}^{(2)}\) are ones. The genes in initial \({{{{{{{{\boldsymbol{X}}}}}}}}}_{i}^{(2)}\) should be mapped to the features in bulk proteome
\({{{{{{{{\boldsymbol{y}}}}}}}}}_{i}^{(2)}\). Initialize _P__i_ by applying CIBERSORT to the target bulk RNA-seq data with the above (RNA) signature matrix. * 2. Evaluate the loss function’s
first gradient at current estimates: \({{{{{{{\boldsymbol{l}}}}}}}}({{{{{{{{\boldsymbol{\eta }}}}}}}}}_{i}^{(s)})\). * 3. Update parameters with non-negative bounds:
\({{{{{{{{\boldsymbol{\eta }}}}}}}}}_{i}^{(s+1)}={[{{{{{{{{\boldsymbol{\eta }}}}}}}}}_{i}^{(s)}-\Delta {{{{{{{\boldsymbol{l}}}}}}}}({{{{{{{{\boldsymbol{\eta }}}}}}}}}_{i}^{(s)})]}_{+}\),
where Δ is step size. * 4. Repeat steps 2-3 until convergence: \(\max \left\{| {{{{{{{{\boldsymbol{\eta }}}}}}}}}^{(s+1)}-{{{{{{{{\boldsymbol{\eta }}}}}}}}}^{(s)}| \right\}\, < \,
\epsilon\) or a limit of iterations (e.g., 1000). * 5. Normalize the cellular fractions as \({\theta }_{ik}^{(1)}=\frac{{p}_{ik}{s}_{ik}^{(1)}}{\mathop{\sum }\nolimits_{k =
1}^{K}{p}_{ik}{s}_{ik}^{(1)}}\) and \({\theta }_{ik}^{(2)}=\frac{{p}_{ik}{s}_{ik}^{(2)}}{\mathop{\sum }\nolimits_{k = 1}^{K}{p}_{ik}{s}_{ik}^{(2)}}\). INTEGRATIVE SIGNATURE SELECTION The
decomposition provided by AJIVE enables the identification of underlying biological patterns that are common to all molecular modalities. Suppose there are _J_ molecule sources for the same
sets of _N_ samples but different features from each source. For the bulk expression matrices _Y_(_j_) (_j_ = 1, …, _J_) in distinct modalities, AJIVE integrates _Y_(_j_)’s to reconstruct
each by three components: _Y_(_j_) = _C_(_j_) + _I_(_j_) + _E_(_j_), where _C_(_j_) represents the common variation originating from the _j_th modality, _I_(_j_) and _E_(_j_) are the
source-specific structured variation and the residual noise, respectively. The top proteins contributing to the common variation shared between proteome (_Y_(1)) and signature genes (_Y_(2))
are selected by using the loadings _V_(1) obtained from the singular value decomposition (SVD) of matrix _C_(1) = _V_(1)_D_(1)_U_(1), where _V_(1) is _G_ × _r_ with _r_ as reduced rank
estimated by the Wedin bound procedure13. Next, we compute
\({{{{\tilde{{{{{\boldsymbol{Y}}}}}}}}}}^{(1)}={{{{{{{{\boldsymbol{V}}}}}}}}}^{(1)}{{{{{{{{\boldsymbol{V}}}}}}}}}^{{(1)}^{T}}{{{{{{{{\boldsymbol{Y}}}}}}}}}^{(1)}\) as a projected
approximation to the observed bulk proteome with rank _r_. For each protein _g_, we calculate the distance between \({{{{{{{{\boldsymbol{y}}}}}}}}}_{g.}^{(1)}\) and
\({{{{\tilde{{{{{\boldsymbol{y}}}}}}}}}}_{g.}^{(1)}\) by the Euclidean distance \({d}_{g}=\Vert
{{{{{{{{\boldsymbol{y}}}}}}}}}_{g.}^{(1)}-{{{{\tilde{{{{{\boldsymbol{y}}}}}}}}}}_{g.}^{(1)}\Vert\). To select the proteins that contribute most significantly to the shared variation, we
choose the proteins with the smallest distances _d__g_. CELL-TYPE-SPECIFIC PROTEIN QTL MAPPING The potential errors produced in sample-wise deconvoluted proteomes may lead to bias in
cell-type-specific QTL mapping. Hence, we apply a published method implemented in TOAST28 to perform cell-type-specific protein differential analysis for genotypes based on the bulk
proteomes, estimated protein cell proportions, and whole genome sequencing variants. This method uses pre-estimated cell proportions and a linear model to describe the cell-type-specific
differential expression pattern in bulk data, and then performs F-test on the hypothesized cell-type-specific changes across three genotype groups29, with false discovery rate being well
controlled according to the previous simulation studies30,31. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this
article. DATA AVAILABILITY The processed signature matrices derived from both real datasets are available for access on the GitHub repository:
https://github.com/YuePan027/MICSQTL/tree/main/processed_signature. The CITE-seq data is available on GEO under accession number GSE164378. The human brain prefrontal cortex mass
spectrometry proteomics data and RNA-seq data are available in the Synapse database https://www.synapse.org under accession code syn32136022. The breast cancer bulk multi-omics data are
available as supplementary files in25, while the scRNA-seq and single cell CyTOF data of breast cancer are available at GEO: GSE180878 and https://data.mendeley.com/datasets/vs8m5gkyfn/1.
The source data necessary for creating the main figures can be found in Supplementary Data 1-5. CODE AVAILABILITY MICSQTL is a Bioconductor package with license GPL-3, available at
https://bioconductor.org/packages/MICSQTL. CIBERSORT (https://cibersortx.stanford.edu/). TOAST (https://bioconductor.org/packages/TOAST). TCA (https://cran.r-project.org/web/packages/TCA).
REFERENCES * Hao, Y. et al. Integrated analysis of multimodal single-cell data. _Cell_ 184, 3573–3587 (2021). Article CAS PubMed PubMed Central Google Scholar * Kumar, P. et al.
Single-cell transcriptomics and surface epitope detection in human brain epileptic lesions identifies pro-inflammatory signaling. _Nat. Neurosci._ 25, 956–966 (2022). Article CAS PubMed
PubMed Central Google Scholar * Sun, X. et al. Deep single-cell-type proteome profiling of mouse brain by nonsurgical aav-mediated proximity labeling. _Anal. Chem._ 94, 5325–5334 (2022).
Article CAS PubMed PubMed Central Google Scholar * Perkel, J. M. Single-cell proteomics takes centre stage. _Nature_ 597, 580–582 (2021). Article CAS PubMed Google Scholar * Cheung,
R. K. & Utz, P. J. Cytof—the next generation of cell detection. _Nat. Rev. Rheumatol._ 7, 502–503 (2011). Article PubMed PubMed Central Google Scholar * Frangieh, C. J. et al.
Multimodal pooled perturb-cite-seq screens in patient models define mechanisms of cancer immune evasion. _Nat. Genet._ 53, 332–341 (2021). Article CAS PubMed PubMed Central Google
Scholar * Zhu, Y. et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. _Nat. Commun._ 9, 882 (2018). Article PubMed PubMed
Central Google Scholar * Petelski, A. A. et al. Multiplexed single-cell proteomics using scope2. _Nat. Protocols_ 16, 5398–5425 (2021). Article CAS PubMed Google Scholar * Bennett, H.
M., Stephenson, W., Rose, C. M. & Darmanis, S. Single-cell proteomics enabled by next-generation sequencing or mass spectrometry. _Nat. Methods_ 20, 363–374 (2023). Article CAS PubMed
Google Scholar * Ben-David, E. et al. Whole-organism eqtl mapping at cellular resolution with single-cell sequencing. _Elife_ 10, e65857 (2021). Article CAS PubMed PubMed Central
Google Scholar * Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. _Nat. Methods_ 12, 453–457 (2015). Article CAS PubMed PubMed Central Google
Scholar * Hu, C. et al. Cellmarker 2.0: an updated database of manually curated cell markers in human/mouse and web tools based on scrna-seq data. _Nucleic Acids Res._ 51, D870–D876 (2023).
Article CAS PubMed Google Scholar * Feng, Q., Jiang, M., Hannig, J. & Marron, J. Angle-based joint and individual variation explained. _J. Multivariate Anal._ 166, 241–265 (2018).
Article Google Scholar * Rahmani, E. et al. Sparse pca corrects for cell type heterogeneity in epigenome-wide association studies. _Nat. Methods_ 13, 443–445 (2016). Article CAS PubMed
PubMed Central Google Scholar * Trobisch, T. et al. Cross-regional homeostatic and reactive glial signatures in multiple sclerosis. _Acta Neuropathol._ 144, 987–1003 (2022). Article CAS
PubMed PubMed Central Google Scholar * Sharma, K. et al. Cell type–and brain region–resolved mouse brain proteome. _Nat. Neurosci._ 18, 1819–1831 (2015). Article CAS PubMed PubMed
Central Google Scholar * Wang, F. et al. Deep domain adversarial neural network for the deconvolution of cell type mixtures in tissue proteome profiling. _Nat. Mach. Intell._ 5, 1236–1249
(2023). Article Google Scholar * Ruzicka, W. B. et al. Single-cell multi-cohort dissection of the schizophrenia transcriptome. _medRxiv_ 2022–08 https://doi.org/10.1101/2022.08.31.22279406
(2022). * Notaras, M. et al. Schizophrenia is defined by cell-specific neuropathology and multiple neurodevelopmental mechanisms in patient-derived cerebral organoids. _Mol. Psychiatry_ 27,
1416–1434 (2022). Article CAS PubMed Google Scholar * Puvogel, S. et al. Single-nucleus rna sequencing of midbrain blood-brain barrier cells in schizophrenia reveals subtle
transcriptional changes with overall preservation of cellular proportions and phenotypes. _Mol. Psychiatry_ 27, 4731–4740 (2022). Article CAS PubMed PubMed Central Google Scholar *
Hibar, D. P. et al. Novel genetic loci associated with hippocampal volume. _Nat. Commun._ 8, 13624 (2017). Article CAS PubMed PubMed Central Google Scholar * Tripathy, R. et al.
Mutations in mast1 cause mega-corpus-callosum syndrome with cerebellar hypoplasia and cortical malformations. _Neuron_ 100, 1354–1368 (2018). Article CAS PubMed PubMed Central Google
Scholar * Ressler, K. J. et al. Post-traumatic stress disorder is associated with pacap and the pac1 receptor. _Nature_ 470, 492–497 (2011). Article CAS PubMed PubMed Central Google
Scholar * Krug, K. et al. Proteogenomic landscape of breast cancer tumorigenesis and targeted therapy. _Cell_ 183, 1436–1456 (2020). Article CAS PubMed PubMed Central Google Scholar *
Gray, G. K. et al. A human breast atlas integrating single-cell proteomics and transcriptomics. _Dev. Cell_ 57, 1400–1420 (2022). Article CAS PubMed PubMed Central Google Scholar *
Chen, Y. et al. Deep autoencoder for interpretable tissue-adaptive deconvolution and cell-type-specific gene analysis. _Nat. Commun._ 13, 6735 (2022). Article CAS PubMed PubMed Central
Google Scholar * Polyak, R. A. Projected gradient method for non-negative least square. _Contemp Math_ 636, 167–179 (2015). Article Google Scholar * Li, Z. & Wu, H. Toast: improving
reference-free cell composition estimation by cross-cell type differential analysis. _Genome Biol._ 20, 1–17 (2019). Article Google Scholar * Li, Z., Wu, Z., Jin, P. & Wu, H.
Dissecting differential signals in high-throughput data from complex tissues. _Bioinformatics_ 35, 3898–3905 (2019). Article CAS PubMed PubMed Central Google Scholar * Meng, G., Tang,
W., Huang, E., Li, Z. & Feng, H. A comprehensive assessment of cell type-specific differential expression methods in bulk data. _Briefings Bioinf._ 24, bbac516 (2023). Article Google
Scholar * Feng, H. et al. Islet: individual-specific reference panel recovery improves cell-type-specific inference. _Genome Biol._ 24, 174 (2023). Article CAS PubMed PubMed Central
Google Scholar Download references ACKNOWLEDGEMENTS This work was partially supported by Cancer Center Support Grant P30CA21765 (Y.P., Q.L.), the American Lebanese Syrian Associated
Charities (Y.P., X.W., J.S., J.P., Q.L.), and NIH R01MH110920 grant (C.L.). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Biostatistics, St. Jude Children’s Research Hospital,
Memphis, TN, 38105, USA Yue Pan, Jiao Sun & Qian Li * Center for Proteomics and Metabolomics, St. Jude Children’s Research Hospital, Memphis, TN, 38105, USA Xusheng Wang * Department of
Genetics, Genomics & Informatics, University of Tennessee Health Science Center, Memphis, TN, 38105, USA Xusheng Wang * Department of Psychiatry, SUNY Upstate Medical University,
Syracuse, NY, 13210, USA Chunyu Liu * Department of Structural Biology, St. Jude Children’s Research Hospital, Memphis, TN, 38105, USA Junmin Peng * Department of Developmental Neurobiology,
St. Jude Children’s Research Hospital, Memphis, TN, 38105, USA Junmin Peng Authors * Yue Pan View author publications You can also search for this author inPubMed Google Scholar * Xusheng
Wang View author publications You can also search for this author inPubMed Google Scholar * Jiao Sun View author publications You can also search for this author inPubMed Google Scholar *
Chunyu Liu View author publications You can also search for this author inPubMed Google Scholar * Junmin Peng View author publications You can also search for this author inPubMed Google
Scholar * Qian Li View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Y.P., X.W., J.P., and Q.L. conceive this study. Y.P. develops the
algorithms and R package as maintainer, performs simulation and real data analyses, and visualizes analysis results. Q.L. proposes the algorithm and designs R package, simulation study, and
real data analysis. J.S. runs experiments on scpDeconv in simulation study and breast cancer data, and visualizes the results. Q.L. and Y.P. write the manuscript. X.W., C.L., and J.P.
generate and share the real data, contribute to methodology discussion, and interpret real data analysis results. All authors reviewed the manuscript. CORRESPONDING AUTHOR Correspondence to
Qian Li. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION Communications Biology thanks Oliver Crook and the other,
anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Yuedong Yang and Luke R. Grinham. A peer review file is available. ADDITIONAL
INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION PEER REVIEW FILE
SUPPLEMENTARY FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 SUPPLEMENTARY DATA 3 SUPPLEMENTARY DATA 4 SUPPLEMENTARY DATA 5 REPORTING SUMMARY
RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and
reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes
were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If
material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain
permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS
ARTICLE Pan, Y., Wang, X., Sun, J. _et al._ Multimodal joint deconvolution and integrative signature selection in proteomics. _Commun Biol_ 7, 493 (2024).
https://doi.org/10.1038/s42003-024-06155-z Download citation * Received: 13 October 2023 * Accepted: 08 April 2024 * Published: 24 April 2024 * DOI:
https://doi.org/10.1038/s42003-024-06155-z SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative