Play all audios:
ABSTRACT Osteosarcoma (OS) is a heterogeneous, aggressive malignancy of the bone that disproportionally affects children and adolescents. Therapeutic interventions for OS are limited, which
is in part due to the complex tumor microenvironment (TME). As such, we used single-cell RNA sequencing (scRNA-seq) to describe the cellular and molecular composition of the TME in 6
treatment-naïve dogs with spontaneously occurring primary OS. Through analysis of 35,310 cells, we identified 41 transcriptomically distinct cell types including the characterization of
follicular helper T cells, mature regulatory dendritic cells (mregDCs), and 8 tumor-associated macrophage (TAM) populations. Cell-cell interaction analysis predicted that mregDCs and TAMs
play key roles in modulating T cell mediated immunity. Furthermore, we completed cross-species cell type gene signature homology analysis and found a high degree of similarity between human
and canine OS. The data presented here act as a roadmap of canine OS which can be applied to advance translational immuno-oncology research. SIMILAR CONTENT BEING VIEWED BY OTHERS
CHARACTERIZING THE TUMOR MICROENVIRONMENT AT THE SINGLE-CELL LEVEL REVEALS A NOVEL IMMUNE EVASION MECHANISM IN OSTEOSARCOMA Article Open access 03 January 2023 SINGLE-CELL RNA LANDSCAPE OF
INTRATUMORAL HETEROGENEITY AND IMMUNOSUPPRESSIVE MICROENVIRONMENT IN ADVANCED OSTEOSARCOMA Article Open access 10 December 2020 IMMUNE PATHWAYS AND _TP53_ MISSENSE MUTATIONS ARE ASSOCIATED
WITH LONGER SURVIVAL IN CANINE OSTEOSARCOMA Article Open access 11 October 2021 INTRODUCTION Osteosarcoma (OS) is an aggressive malignancy of the bone that disproportionally impacts children
and adolescences. Despite a profound impact on the lives of affected individuals, effective therapeutics are lacking, with minimal advancements since the introduction of combined surgical
excision and adjuvant chemotherapy in 19861. Slow advancements in the development of OS therapeutics are, in part, due to the relatively rare incidence which limits patient accrual into
clinical trials. In recent years, there has been increased interest in using large animal models to evaluate and validate the potential of immunotherapeutics for various cancers2,3.
Spontaneously occurring canine OS is regarded as an ideal model of human OS due to higher disease prevalence in dogs, similar genetics and pathology, and the immune competent status of
dogs4. Although dogs have been identified as a valuable pre-clinical model, species-specific reagent limitations have restricted researcher’s ability to fully characterize the canine OS
tumor microenvironment (TME). Osteosarcoma has a complex TME that consists of malignant osteoblasts, osteoclasts, fibroblasts, macrophages, and lymphocytes as well as numerous other stromal
and immune components. Together, the OS TME creates an immune suppressive milieu that hinders antitumor immune responses5. Researchers have turned to the TME with the objective of
understanding and targeting the cellular constituents that promote immune suppression6,7. Unlike many other cancer types, there have been reports in both humans and dogs that suggest
increased macrophage abundance in OS reduces metastatic rate and enhances survival8,9,10. This unexpected finding and the ill-defined mechanisms of immune suppression in the OS TME
highlights the need for a deeper understanding of OS pathobiology. In recent years, single-cell RNA sequencing (scRNA-seq) has emerged as a valuable tool to investigate the transcriptomes of
individual cells within heterogenous tissues. The approach overcomes species-specific regent limitations by relying on a universal transcript capture method that is only limited by the
completeness of genome annotations11. Importantly, the human scRNA-seq landscapes of primary, recurrent, and metastatic OS have recently been described and act as a point of reference for
cell type homology analysis between canine and human OS12,13. The aim of the current study was to use scRNA-seq to complete a molecular dissection of the canine OS TME and evaluate cell type
transcriptomic homologies between humans and dogs. In the present study, we generated a single-cell RNA sequencing reference dataset of six treatment-naïve dogs with primary osteosarcoma.
Our analysis revealed the presence of 41 transcriptomically distinct cell types in canine OS and provided evidence of conserved cell type gene signatures between human and canine OS.
Overall, the data generated here can be used to inform the identification of conserved OS TME features and facilitate further study of the canine osteosarcoma tumor microenvironment. RESULTS
ESTABLISHMENT OF A TREATMENT-NAÏVE CANINE OSTEOSARCOMA REFERENCE DATABASE To establish a treatment-naïve canine osteosarcoma reference, we completed single-cell RNA sequencing (scRNA-seq)
on 6 dogs and collected data on a total of 35,310 cells. The average number of cells collected per tumor was 5885 and on average each cell was sequenced to a depth of 72,649 reads per cell
(Supplementary Table 3). All tumors were confirmed to be osteosarcoma by histology and histological subtyping was completed on each tumor (Table 1). In total, the curated dataset consisted
of 1 fibroblastic, 1 chondroblast, and 4 osteoblastic tumors, with one dog exhibiting radiographical evidence of lung metastasis. Initial low-resolution cell type annotation revealed the
presence of 7 major cell types consisting of T cells, B cells, tumor infiltrating monocytes (TIMs)/tumor associated macrophage (TAMs), dendritic cells (DCs), osteoclasts (OCs), tumor cells,
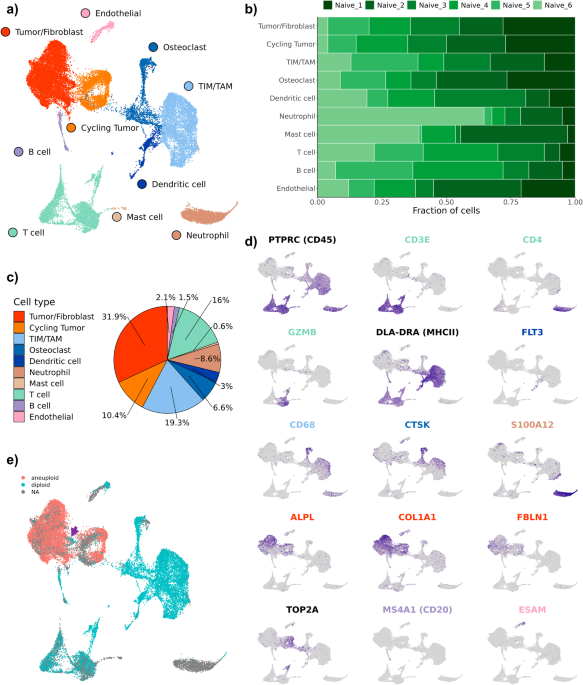
cycling tumor cells, and an additional 3 minor cell populations consisting of neutrophils, mast cells, and endothelial cells (Fig. 1a). Evaluation of the dataset for evidence of batch
effects indicated uniform distribution of cell types between biological replicates (Fig. 1b). The one exception was that naïve dog 6 had a higher proportion of neutrophils compared to the
other study dogs. This skew might be a result of sampling bias in which necrotic tumor, blood, or bone marrow contamination was introduced during sampling. Subsequent analysis of cell type
proportions revealed 42.3% of the dataset consisted of tumor cells or fibroblasts, 2.1% of the dataset was endothelial cells, and the remaining 55.6% was composed of immune cells (Fig. 1c).
Cell types were annotated based on expression of canonical markers, reference mapping using a human OS dataset, and gene set enrichment analysis (Fig. 1d). Cell type gene lists used by Liu
et. al. to define cell populations in human OS were applied using module scoring to provide further support for cell classifications (Supplementary Fig. 1a)12. These approaches consistently
enabled the identification of T cells, B cells, osteoclasts, and endothelial cells. However, our high-level unsupervised clustering failed to distinguish between stromal fibroblasts and
malignant osteoblasts. This unexpected observation may be in part due to the presence of a fibroblastic osteosarcoma tumor in our dataset and the broad expression of fibroblast markers
(_FAP_, _FBLN1_) across all tumor cell clusters (Supplementary Fig. 1b). Due to the inability to identify a distinct fibroblast population using feature expression, we applied CopyKAT and
inferCNV to complete copy number variation (CNV) analysis to infer which cells exhibited aneuploidy based on their global transcriptional properties (Fig. 1e)14,15. The analysis revealed
that the majority of cells in the tumor/fibroblast cluster exhibited evidence of CNV aberrations with only a small subset of cells predicted to be diploid (Fig. 1e; purple arrow,
Supplementary Figs. 2–9). The diploid cells were determined to represent a small cluster of fibroblasts which were investigated further through subclustering analysis. DISSECTION OF THE
TUMOR AND STROMAL POPULATIONS REVEALS A DISTINCT FIBROBLAST CLUSTER Subclustering analysis on cycling tumor cells and tumor/fibroblasts identified 10 distinct cell clusters which we defined
as 4 cycling malignant osteoblasts clusters, 5 non-cycling malignant osteoblast clusters, and 1 fibroblast cluster (Fig. 2a, Supplementary Fig. 10a). The defining features for each cluster
were identified using a Wilcoxon Rank Sum test and the top 3–5 unique features were visualized using a heatmap and feature plots (Fig. 2b, c). Overall, the malignant osteoblasts exhibited a
unique gene expression profile with collagen genes and alkaline phosphatase (_ALPL_) contributing to the gene signatures. We observed a small cluster of tumor cells (c9) that exhibited a
gene expression pattern (_OAS1_, _ISG15_, _OAS2_) consistent with an interferon (IFN) response gene signature (Fig. 2b). This observation was further supported through completion of GSEA
using Hallmarks gene set terms (Fig. 2d). Similar IFN signature enriched cells have been reported among immune cells, but the observation of such a cluster in a tumor population has not been
previously reported in human OS studies12,13,16,17. Interpretation of GSEA further revealed that fibroblasts (c6) exhibited the most pronounced “epithelial-mesenchymal transition” (EMT) and
“angiogenesis” signatures, which suggests the fibroblasts might play a role in promoting tumor growth. Additionally, GSEA supported the annotation of hypoxic osteoblasts (c4), as the
cluster exhibited the strongest hypoxic transcriptomic signature. To confirm the identification of fibroblasts, we used module scoring with a human fibroblast gene list18. This analysis
confirmed Cluster 6 exhibited the strongest fibroblast gene signature (Supplementary Fig. 10b). We then completed differential gene expression (DGE) analysis contrasting fibroblasts (c6) and
non-hypoxic osteoblasts (c0, c1, and c2) to better define the canine fibroblast gene signature (Fig. 2e; Supplementary Data 3). While key fibroblast markers such as _FAP_ and _ACTA2_ were
identified, the top features consisted of _SFRP2_ and _PRSS23_ which were recently reported to be associated with a fibroblast population involved in wound healing19. To conclude our
analysis of tumor cells, we sought to further investigate the transcriptomic signature of hypoxic osteoblasts (c4) by contrasting with non-hypoxic osteoblasts (c0, c1, and c2). Few
differentially expressed genes were identified, suggesting the cell types are similar, but subsequent pathway analysis identified enrichment of “response to oxygen levels” to be a top
enriched pathway, suggesting that the tumor cells were indeed hypoxic (Fig. 2f, Supplementary Data 3, Supplementary Fig. 10c). In summary, we were able to resolve a population of fibroblasts
through completion of subclustering analysis, as well as define the transcriptional heterogeneity within malignant osteoblasts. SUBCLUSTERING ANALYSIS REVEALS A POPULATION OF _CXCL13_ +
FOLLICULAR HELPER CD4 T CELLS To ensure we captured all biologically relevant T cell populations, we completed subclustering analysis, which led to the identification of 10
transcriptomically distinct clusters: 3 CD8 T cell, 4 CD4 T cell, 1 NK cell, and 2 mixed CD4/CD8 T cell clusters (Fig. 3a, b). Next, we interrogated T cell subtypes using an approach that
has been applied in human breast cancer and OS scRNA-seq datasets to describe T cell populations13,20. We modified the gene lists used in previous applications to include signatures for
cycling T cells, NK cells, and IFN-signature T cells that we recently established in circulating canine leukocytes21. Overall, this approach proved to be consistent with annotations assigned
using canonical markers (Fig. 3c). Although the gene signatures were definitive for naïve CD4 T cells and cytotoxic CD8 T cells, other gene signature scores provided weaker support for
their corresponding cell types. For instance, regulatory T cells (Tregs) and follicular helper CD4 T cells (CD4fh) both exhibited moderate enrichment for exhausted and costimulatory terms,
with minimal distinction between the two T cell types. The analysis also revealed the presence of a T cell cluster with an IFN gene signature, a population that has been reported to be
hypersensitive to stimulation17. After identifying each T cell subset, we completed pseudobulk conversion and DGE analysis to further establish the transcriptomic signatures of Tregs and
CD4fh. Comparisons between Tregs (c3) and activated CD4 T cells (c2) revealed overexpression of _IL21R_, _TNFRSF4_, and _TNFRSF18_, with _CTLA4_ being the most definitive marker of Tregs
(Fig. 3d, Supplementary Data 3). When repeating this analysis on CD4fh (c5) cells, we identified _CXCL13_, _IL4I1_, and _TMEM176A_ to be defining features (Fig. 3e, Supplementary Data 3).
Although intratumoral CD4fh cells exhibited a distinct exhaustion profile (_PDCD1_, _TOX_, _TOX2_, _IL4I1_), they also displayed a gene signature consistent with follicular helper T cells
(_CXCL13_, _IL21_, _CD70_)22,23. A similar population of _CXCL13_+ CD4fh T cells has been identified in multiple human tumors and the cell type has been implicated in tertiary lymphoid
follicle formation and modification of intratumoral adaptive immune responses24,25,26. Our analysis confirms expression of _CTLA4_/_FOXP3_ on Tregs and _CXCL13_/_IL21_ on CD4fh is conserved
across species, while also providing complete gene signatures for the canine T cell subtypes18. Following initial cell classification, we determined the cellular composition of each cell
type as a percentage of immune cells and of all cells within each sample (Fig. 3f, Supplementary Table 4). This analysis revealed exhausted CD8 T cells (CD8ex) and effector CD8 T cells
(CD8eff) to be among the most abundant populations, along with activated CD4 T cells and Tregs. We then curated a heatmap of defining features predicted to be expressed on the cell surface
with the objective of identifying potential cell markers to be used in alternative cell identification approaches, such as flow cytometry (Fig. 3g, Supplementary Fig. 11, Supplementary Data
4)27. With the caveat that transcript presence does not always correlate with protein expression, the analysis suggested that _TNFRSF4_ (OX-40), _TNFSF8_ (CD153), and _TMEM140_ may represent
valuable surface markers for further investigation of canine Tregs, CD4fh, and TIFN-sig, respectively. Together, the relative cellular percentages and potential surface markers provide a
foundation for further functional study of the cell types identified in our transcriptomic analysis. MATURE REGULATORY DENDRITIC CELLS ARE PRESENT IN CANINE OS AND ARE PREDICTED TO MODULATE
T CELL MEDIATED IMMUNITY Five dendritic cell (DC) subtypes were identified when completing subclustering analysis on FLT3+ cells. The subtypes identified included conventional DC2s (cDC2;
c0), mature regulatory DCs (c1; mregDC), cDC1s (c2), plasmacytoid DCs (c3; pDC), and precursor DCs (c4; preDC) (Fig. 4a). Key features used to assign cell type identities included _DNASE1L3_
(cDC1), _CCR7_/_IL4I1_ (mregDCs), _CD1C_ (cDC2), _IL3RA_ (preDC), and _IGKC_ (pDC) (Fig. 4b)28,29. The population of canine preDCs closely resembled a recently redefined human preDC cell
type that exhibits a tendency to cluster with pDCs when investigated using scRNA-seq28. Of note, we previously identified cDC2, cDC1, preDC, and pDC cell types in canine peripheral blood,
however mregDCs (c1) were not observed, suggesting a potential tissue specificity21. The identification of mregDCs, also reported as migratory (mig) DCs, is of note as this cell type is
predicted to modulate T cell responses30,31. Thus, we provide evidence that a key cell type reported to have immune regulatory properties is present in canine OS. We next used hierarchical
clustering and Toll-like receptor expression to investigate differences between preDCs and pDCs. Hierarchical clustering indicated preDCs are closely related to myeloid cDC2s and cDC1s,
while pDCs were located on a unique clade (Fig. 4c). In humans, pDCs are reported to exhibit high expression of _TLR9_ and _TLR7_, which we identified to be highly expressed on canine pDCs
(Fig. 4d)32. To ensure that none of the DC populations were of B cell origin, we evaluated _MS4A1_ (CD20) expression and found it to be minimally expressed (Supplementary Fig. 12a). We then
used pySCENIC to predict active regulons in each DC subtype (Supplementary Fig. 12b). This analysis revealed _TCF4_ and _RUNX2_, master regulators of pDC development, to be enriched in both
pDCs and preDCs33. Overall, these findings suggest canine preDCs are closely related to the recently defined plasmacytoid-like human preDCs28. To confirm mregDCs exhibited a mature, immune
regulatory transcriptomic signature, we used module scoring with gene lists previously applied to investigate human DC subtypes30,34. This analysis revealed that mregDCs had a marked
enrichment for migration, regulatory, and maturation associated gene signatures (Fig. 4e). Subsequent, DGE analysis of canine mregDCs (c1) relative to cDC2s (c0) revealed a distinct mregDC
signature of _CCR7_, _IL4I1_, _CCL19_, and _FSCN1_ with substantial overlap to the human mregDC transcriptional program (Fig. 4f, Supplementary Data 3)30. With the precedent that mregDCs
interact with intratumoral T cells to shape adaptive immune responses in humans, we wanted to determine whether a similar interaction might occur between mregDC and T cells in dogs24,25. We
used CellChat to evaluate interactions between mregDCs and T/NK cells35. This analysis revealed enriched PD-1/PD-L1 and CTLA4/CD80 interactions between mregDCs and CD4 Tregs, Tfh cells, and
naïve T cells (Fig. 4g). In summary, we present the transcriptomic signature of canine mregDCs and provide evidence of intratumoral interactions between canine mregDCs and T cells.
MACROPHAGE TRANSCRIPTOMIC STATES SUPPORT A SPECTRUM OF CELL TYPES Due to the transcriptional overlap between tumor associated macrophages (TAMs) and osteoclasts (OCs), we analyzed these two
cell types in the same UMAP space. In doing so, our analysis highlighted the relatedness of OCs and TAMs which would have been overlooked if analyzed independently. Through subclustering
analysis we identified 8 transcriptomically distinct macrophage/monocyte populations which were annotated using modified nomenclature derived from Ma et al. (Fig. 5a–c)36. Activated TAMs
(c0, TAM_ACT) and intermediate TAMs (c1, TAM_INT) did not fit into any of the macrophage subtypes presented in Ma et al. so they were instead annotated based on an activated signature
(_CD5L_, _CD40_, _CD80_) and an intermediate polarization signature, respectively. Tumor infiltrating monocytes (TIMs) were divided into two populations based on CD4 expression; a division
of monocytes unique to dogs21,37. Unsupervised clustering divided lipid-associated (LA-) TAMs into two subclusters defined by either _C1QC_hi expression (c3) or _SPP2_hi expression (c2). To
better define the distinctions between the two LA-TAM populations we completed pseudobulk-based DGE analysis (Fig. 5d, Supplementary Data 3). The analysis revealed _IL2RA_, _CXCL10_, and
_SERPING1_ as key markers of C1QChi LA-TAMs, while _ENO1_, _LGALS3_, and _RBP4_ defined SPP2hi LA-TAMs. Based on the analysis, _C1QC_hi LA-TAMs appear to most closely resemble the
definitions of human LA-TAMs provided by Ma et al. In addition to the recently proposed TAM nomenclature, we used module scoring with pro- and anti-inflammatory gene lists to investigate the
macrophage populations in a more traditional dichotomy (Supplementary Table 5)38. We identified the C1QChi LA-TAM (c3) cluster to have the strongest anti-inflammatory transcriptomic
signature while CD4+ monocytes (c11) exhibited the most prominent pro-inflammatory transcriptomic signature (Fig. 5e). To further investigate the gene signatures of Clusters 11 and 3 we
completed pseudobulk-based DGE analysis (Fig. 5f, Supplementary Data 3). The genes upregulated in Cluster 11 exhibited overlap with the predefined gene set that was used to identify the
cluster as pro-inflammatory, while also revealing _IL1B_, _S100A12_, _LTF_, and _VCAN_ as defining features. DGE analysis of the anti-inflammatory cluster (c3) exhibited less overlap with
the gene list originally used to identify the cluster as anti-inflammatory (with _MRC1_ the only overlapping feature), but the analysis further revealed _APOE_, _IGF1_, and complement
receptors _C1QA/B/C_ as enriched markers. The top features identified when contrasting Clusters 11 and 3 were then used to generate a heatmap to evaluate how the expression of these features
varied across all macrophage clusters (Fig. 5g). Findings from the analysis suggested that there is a spectrum of macrophage phenotypes, which is consistent with human macrophage
literature39. As such, we next sought to better define the heterogeneity of the macrophage populations without relying on predefined cell type gene signatures. Gene set enrichment analysis
was used to provide further insights into the inferred functional capacity of each macrophage subtype (Fig. 5h). Cell clusters 4, 7, and 11 clustered together based on pathway enrichment
scores suggesting the three transcriptionally distinct clusters have similar underlying gene signatures. LA-TAMs (c2, c3) and intermediate TAMs (c1) exhibited the strongest scavenger
receptor associated activation, suggesting a mature macrophage population with immune suppressive properties40. Several terms were identified suggesting that both SPP2hi LA-TAMS and
intermediate TAMs preferentially utilize oxidative phosphorylation and mitochondrial metabolic pathways. C1QChi LA-TAMs had a distinct profile suggestive of lipid and polysaccharide
metabolism. Lastly, GSEA confirmed c10 to be consistent with IFN-TAMs based on strong enrichment of IFN signaling associated terms. In summary, we described the transcriptional profiles of
macrophages in the canine OS TME which provides a foundation for further investigation of the functional relevance of each cell type. ANALYSIS OF CANINE OSTEOCLASTS REVEALS FOUR
TRANSCRIPTOMICALLY DISTINCT POPULATIONS Within the same UMAP space, we next shifted our focus to further characterize osteoclast heterogeneity. Consistent with human and murine reports using
scRNA-seq to characterize OCs, we identified 4 transcriptiomically distinct OC populations12,13,41. The cycling OCs (c5/c8) in our canine OS dataset likely correspond to previously reported
pre/progenitor OCs, while the mature OCs (c6) are consistent with previous reports (Fig. 6a, b). CD320+ OCs (transcobalamin receptor expressing OCs, c9) had not been described in macrophage
or osteoclast clusters from human and mouse tissues and may represent a canine specific cell type, or possibly a previously unresolved OC subtype (Fig. 6a, b). Due to the similarity of OCs
and macrophages we completed hierarchical clustering to confirm the unsupervised clustering results (Fig. 6c). The secondary analysis was consistent with unsupervised clustering and further
suggested Clusters 5, 6, 8, and 9 are distinct from the macrophage clusters. To confirm the mature OC classification and provide a canine specific transcriptomic signature, we completed DGE
analysis. When comparing mature OCs (c6) to macrophages (c0, c1, c2, c3) we identified canine mature OCs to be defined by _ATP6V1C1_, _CD84_, _HYAL1_, and _CAMTA2_ expression, which
subsequent GSEA analysis revealed an association with bone resorption and remodeling (Fig. 6d, Supplementary Data 3, Supplementary Fig. 13a). We next completed DGE analysis contrasting
CD320+ OCs (c9) with macrophages and mature OCs. (Supplementary Data 3, Supplementary Fig. 13b, c). By evaluating the intersection of the differentially expressed genes we determined CD320+
OCs are defined by _HMGA1_, _TNIP3_, and _CD320_ expression (Fig. 6e). The analysis also provided further evidence that c9 is an OC cluster based on _TNFRSF11A_ (RANK) enrichment when
contrasted with macrophage, but not when contrasted to mature OCs42. Lastly, we used pySCENIC’s regulon specificity scoring to better define the transcription factors active in mature OCs
and CD320+ OCs. We identified _ZEB1_ and _NFATC1_, known regulators of OC development, to be enriched in mature OCs, while _SNAI1_ and _ETV3/6/7_ were enriched in CD320+ OCs suggesting a
differentiating cell type (Fig. 6f, g)43,44,45. Together our analysis indicates, CD320+ OCs are a distinct population from mature OCs that may represent an OC precursor. TRANSCRIPT ABUNDANCE
OF WIDELY USED IMMUNOHISTOCHEMISTRY MACROPHAGE MARKERS EXHIBIT DISTINCT SPECIFICITY TO MYELOID CELLS In contrast to other tumor types, there have been multiple reports in humans and dogs
suggesting that increased TAM infiltrates in OS are correlated with reduced metastasis rates and increased patient survival8,9. Despite these reports, other groups completing similar
analysis have concluded that increased macrophage infiltrates have a negative impact on OS clinical outcomes46. Given the conflicting nature of previous reports we sought to employ our
dataset to investigate which cell types express the transcript of the prototypical macrophage markers used for IHC analysis. To complete this analysis, we profiled TIMs, TAMs, DCs, and OCs
for the expression of widely used canine (_MSR1_ aka CD204 and _AIF1_ aka Iba1) and human (_CD163_ and _CD68_) macrophage markers (Fig. 7a). With the caveat that this analysis is limited to
transcript abundance and does not evaluate protein expression, we found that _CD163_ transcript expression was the most specific for macrophages. _CD68_ expression was detected in TIMs,
TAMs, and OCs, with remarkably high expression levels in mature OCs. The expression of _CD68_ on mature OCs is consistent with human literature47. _AIF1_ (Iba1) was the most non-specific
marker with diffuse expression across all cell types, except for mature OCs. Lastly, _MSR1_ (CD204) was determined to be largely specific to TAMs, but the expression also extended to CD320+
OCs and CD4+ monocytes. To investigate the translational relevance of this finding, we evaluated expression of the markers in human OS (Supplementary Fig. 14). We observed similar expression
patterns, with marked variability in specificity of each marker, suggesting the variability is conserved across species. Given the degree of heterogeneity within the myeloid compartment in
the OS TME, we used a Wilcoxon Rank Sum test to identify features that define each cell type, then selected for features predicted to be expressed on the cell surface (Fig. 7b, Supplementary
Fig. 15, Supplementary Data 5). Overall, the analysis suggested there is substantial overlap in expression of most features. Despite the overlap, we were able to identify candidate markers
which included _ADAM28_ for LA-TAM_C1QChi, _TNFSF13B_ for IFN-TAMs, and _CD84_ for mature OCs. Lastly, we calculated the relative percentages of each cell type to further facilitate cell
identification (Fig. 7c, Supplementary Table 6). Together, the data presented here act as a foundation to further investigate the role of myeloid cells in OS biology. CELL-CELL INTERACTION
ANALYSIS INDICATES TAMS ARE INVOLVED IN IMMUNE REGULATORY PATHWAYS Following cell identification through subclustering analysis of major cell types, we evaluated the cell-cell interaction
networks using CellChat. Between the 41 cell types included in the analysis, we identified a total of 13,235 inferred interactions across 59 signaling networks. The number of interactions
and the predicted interaction strength of incoming (express receptor) versus outgoing (express ligand) signals were used to infer the activity of cells within the TME (Fig. 8a, Supplementary
Fig. 16a). The top three cell types predicted to have the strongest interactions were fibroblasts, mature OCs, and endothelial cells. We next categorized the significantly enriched networks
as “immune specific”, “immune related”, and “non-immune” to investigate if certain cell types were more active in a subset of networks (Fig. 8b). We found that malignant osteoblasts and
stromal cells were largely predicted to be involved in “non-immune” interactions, while “immune specific” interactions were largely confined to TAMs and DCs with strong outgoing
interactions. By subsetting on immune cells and evaluating interactions of known immune regulatory pathways we identified mregDCs and IFN-TAMs to have the most interactions, while activated
(_CD5L_+) macrophages and C1QChi LA-TAMs were predicted to have the strongest outgoing signals (Fig. 8c). It was further predicted that follicular helper and regulatory CD4 T cells make up
the populations receiving most of the signals originating from myeloid cells. When evaluating the PD-L1 network, we identified mregDCs, TIMs, and IFN-TAMs to have the highest expression of
PD-L1 and were predicted to interact with Tfh, Tregs, and exhausted CD8 T cells (Fig. 8d, Supplementary Fig. 16b). The CD80 and CD86 networks involved a larger portion of myeloid cells, with
all CD4 T cells predicted to be influenced by the interactions (Fig. 8e, Supplementary Fig. 16c, d). Overall, activated TAMs, IFN-TAMs, and C1QChi LA-TAMs are predicted to be key
contributors to shaping T cell mediated immunity. COMPARISON OF HUMAN AND CANINE SCRNA-SEQ OS DATASETS REVEAL A HIGH DEGREE OF SIMILARITY IN CELL TYPE GENE SIGNATURES BETWEEN SPECIES Lastly,
we obtained 6 publicly available treatment-naïve human OS scRNA-seq samples to complete a cross-species analysis (GSE162454)12. The two datasets were integrated using a Seurat alignment
workflow which is reported to overcome genomic annotation differences between species48. Hierarchical clustering of the integrated SCTransform normalized data revealed a high degree of
similarity between species, with major clades containing similar cell types based on pre-integration annotations (Fig. 9a). Evaluation of similarities in cell type gene signatures using
Jaccard similarity index produced similar results (Supplementary Fig. 17). All canine lymphocyte subtypes paired 1:1 with their human counterpart, as did endothelial cells and fibroblasts.
Discrepancies between species included the placement of plasmacytoid dendritic cells (pDCs), which clustered into separate clades, and weak Jaccard similarity index values for pDCs and mast
cells across species. Overall, macrophages clustered in the same clade, but due to differences in annotation levels, many cell types did not pair off into terminal clades. To further compare
transcriptional programs across species we used an analysis approach adopted from Scheyltjens et al.49. Briefly, the approach used DGE analysis between two cell populations in each species,
then signing of the adjusted _P_ value to determine if transcriptomic signatures were conserved. When contrasting fibroblasts and endothelial cells, we found substantial overlap in gene
expression patterns with key endothelial cell markers (_PLVAP_, _CD34_, and _PECAM1_) enriched in both species (Fig. 9b, Supplementary Data 6). Top features conserved in fibroblasts included
_VCAN_, _COL6A1_, and _LUM_, while key features such as _FAP_ and _ACTA2_ were also conserved. Interesting discrepancies included the expression of _HYAL2_ and _NOTCH_ as defining features
in human endothelial cells, but nonsignificant in canine endothelial cells. Completion of the same analysis on plasmacytoid DCs and cDC2s revealed _TCF4_ to be enriched in pDCs and _BATF_
expression enriched in cDC2s, which is consistent with human literature (Fig. 9c, Supplementary Data 7)29. An intriguing distinction between species included the high expression of _GZMB_
and _PTGDS_ (prostaglandin D2 synthase) in human pDCs, but not in canine pDCs. Lastly, we applied the same approach to compare mature OCs with TIMs (Fig. 9d, Supplementary Data 8). As
expected, mature osteoclasts were defined by _CSTK_, _ACP5_, and _ATP6V0D2_ expression, while monocytes in both species were defined by _CXCL8_, _OSM_, and _LYZ_ expression. Notable
differences included canine monocytes exhibiting high expression of _SLAMF9_ and _PLBD1_, while human monocytes had high _S100A8_ and _HCST_ expression. In summary, we present a
comprehensive comparison of human and canine OS cell types, which suggests a high degree of consistency in cell type gene signatures across the two species, however we also present evidence
of distinct transcriptional programs in pDCs, mast cells, and monocytes. DISCUSSION In the present study, we completed a comprehensive analysis of canine osteosarcoma (OS) using single-cell
RNA sequencing which revealed the complex network of cells within the tumor microenvironment (TME). Through analysis of 6 treatment-naïve canine OS samples we were able to identify 30
distinct immune cell types, 9 unique malignant osteoblast populations, 1 cluster of fibroblasts, and 1 population of endothelial cells (Supplementary Data 1). We described the transcriptomic
heterogeneity within malignant osteoblasts, identified cell types that have not been previously reported in dogs, and applied our data set to investigate the transcript abundance of widely
used macrophage surface markers. Ultimately, the data presented here act as a molecular roadmap of the canine OS tumor microenvironment which provides canine specific cell type gene
signatures that can be applied to guide immunological reagent development and further investigations of the canine OS TME. Prior to this study, evidence of a conserved OS TME between humans
and dogs was limited, with the most recent human-canine comparisons being presented in Mannheimer et al.50. To compare the human and canine OS TME more directly we obtained a publicly
available scRNA-seq human OS dataset which enabled evaluation of the relative relatedness of cell types between species. The analysis revealed that lymphocytes exhibited the highest degree
of conservation between species, while the gene signatures of major cell types also exhibited substantial overlap. The cell types with the most distinct transcriptional signatures included
plasmacytoid dendritic cells and mast cells. These species differences could be the result of either distinct transcriptional profiles or, less interestingly, discordant annotations between
species. In both datasets plasmacytoid dendritic cells (pDCs) were defined by _FLT3_, _IGHM_, and _TCF4_ which suggests consistent annotation in both species51. Therefore, the difference in
gene signatures within pDCs may represent distinct transcriptional profiles. The mast cells identified in each species were defined by _GATA2_ and _MS4A2_ expression, but the population had
the weakest Jaccard similarity index suggesting poor conservation of gene signatures. _GATA2_ (a transcription factor with implications in basophil and mast cell differentiation) and _MS4A2_
(the IgE receptor found on basophils and mast cells) are not specific to mast cells, so it is possible that the cluster could instead represent basophils or possibly eosinophils52,53,54.
Further functional and transcriptomic investigation of these cell populations is warranted. Our analysis revealed the presence of many rare cell populations, including mregDCs, CD4fh T
cells, and IFN-TAMs, opening avenues for further investigation of these cell populations. With the caveat that transcript expression may not correlate with protein expression, we used the
surfaceome reference database to identify possible surface markers for further study of these cell types27. In addition to antibody-based assays, the transcriptomic signatures presented here
provide a reference for the application of deconvolution algorithms (such as CIBERSORTx and TIMER) when evaluating bulk RNA sequencing data obtained from canine OS samples55,56. Mature
regulatory dendritic cells represent a recently defined cell type which has been identified across several human tumor types, including OS31,57. The biological role of mregDCs is still being
identified, but recent reports suggest a potential role in shaping T cell antitumor immune responses24,25. In our analysis, we were able to identify a _CCR7_+/_IL4I1_+/_FSCN1_+ dendritic
cell population which closely resembles the descriptions of human mregDCs. We found that canine mregDCs express high levels of immune suppressive and costimulatory molecules, which may play
a role in modulation of adaptive immune responses through communication with follicular helper and regulatory T cells. This study provides evidence that mregDCs are present in canine OS,
demonstrating a conserved role within osteosarcoma across species. The heterogeneity within the myeloid compartment of OS is an area of intense interest, in particular the role of TAMs in
regulating OS biological behavior and clinical outcomes is debated58. To provide further context for the discrepant findings reported in the literature regarding the role of TAMs, we
evaluated the transcript abundances of key macrophage markers used in human and canine analysis. Although the analysis was completed at the transcript level, we observed notable differences
in the specificity of each cell type marker within given myeloid populations. Inconsistencies between TAM cell types using immunohistochemistry (IHC) could explain why some groups identify
negative prognostic correlates with TAM density, while other groups report that TAM density is correlated with positive outcomes8,9,46. Further validation of the variability in cell type
markers could be completed using refined IHC panels coupled with spatial transcriptomics. Ultimately, a better understanding of the prognostic and functional roles of myeloid cells within
the TME will aid in the development of effective myeloid cell targeted therapeutics. While the single-cell RNA reference presented here provides key insights into canine OS, the dataset is
not without limitations. First, although we sampled male and female dogs across a range of ages, our dataset still only consisted of 6 dogs and may not fully represent all cell populations
found in canine OS. Secondly, the tumor sample obtained from one dog (dog 6) exhibited markedly more neutrophils relative to other samples which may suggest sample contamination with blood,
bone marrow, or necrotic tissue. Lastly, cellular annotations largely relied on human gene signatures due to the lack of canine specific data available. This may have exaggerated
similarities between species and may have resulted in the forcing of distinct canine-specific cell types into subtypes derived from human nomenclature. Thus, the discordant findings
regarding mast cell gene signatures represents an important distinction that should be investigated further and considered when using the dog as a model for human disease. The data presented
here represent a valuable resource for comparative oncology research using the canine cancer model. A major goal of this project was to make the data accessible to the greater research
community and multiple avenues are provided for researchers to explore and use the dataset (see data availability statement). Our comparisons between human and canine OS revealed the
conserved nature of cell type gene signatures in OS while also identifying potential differences. Overall, our analysis further supports the value of the dog as a model for human OS research
and provides an important reference dataset to advance canine immuno-oncology research. METHODS STUDY ANIMALS Dogs included in the study were selected based on the presence of an
appendicular primary tumor and the absence of previous therapeutic intervention. All dogs presented with radiographic evidence of OS and subsequent histopathological evaluation was completed
to confirm the diagnosis. All study dogs underwent amputation of the affected limb and samples were collected for single-cell RNA sequencing processing within 30 min. The amputated limb was
then submitted to the Colorado State University Veterinary Diagnostic Laboratory where representative samples of the tumor were processed for histopathologic confirmation of the clinical
diagnosis. The osteosarcoma subcategorization presented in Table 1 was based on pathology reports for each sample and confirmed by a second veterinary pathologist. All studies were approved
by the Colorado State University (CSU) Institutional Animal Care and Use Committee and the CSU Clinical Review Board. We have complied with all relevant ethical regulations for animal use
and all dog owners provided informed consent prior to sample collection. SAMPLE PREPARATION The amputated limb was dissected to the level of the tumor and a stainless-steel Michele trephine
biopsy needle was used to obtain 3–5 biopsy cores from the tumor, targeting areas where there was an obvious mass effect and/or lysis of cortical bone. The biopsy cores were then washed with
phosphate buffered saline (PBS), minced using a scalpel, and digested with collagenase type II (250 U/mL) in Hanks’ Balanced Salt Solution (HBSS) for 45 min at 37 °C with agitation (Thermo
Fisher Scientific Inc.). Samples were passed through a 70-µm cell strainer, washed with PBS, then centrifuged for 5 min at 400 rcf. Each of the separately collected biopsies were inspected
to ensure the presence of viable cells (as determined using trypan blue exclusion; Thermo Fisher Scientific Inc.), then all samples with detectable live cells were pooled into 4-mL HBSS. To
enrich for live cells and remove debris, the pooled cell suspension was layered onto 3-mL Ficoll Paque (Cytiva; Marlborough, MA), and centrifuged for 30 minutes at 400 rcf with acceleration
at 9 and brake at 0. Following density centrifugation, the cell interface layer was collected, washed one time with PBS, and resuspended in 10-mL of Ammonium-Chloride-Potassium lysis buffer
for 3–7 min at room temperature. To remove small debris and platelets, a final wash at 100 rcf for 15 min was completed. Cells were resuspended in 0.04% molecular grade BSA (Sigma-Aldrich;
St. Louis, MO) in PBS, confirmed to have a viability greater than 90% (as determined using trypan blue exclusion), and transported to a Chromium iX instrument (10x Genomics; Pleasanton, CA)
for cell capture. All samples were captured within 30 min of preparation. LIBRARY PREPARATION AND SEQUENCING Single cells were isolated and tagged with molecular barcodes using a Chromium iX
instrument with a target of 5000 cells per sample. Two of the six dogs (dogs 1 and 2) had two samples processed each with a 5000-cell target, for a total target of 10,000 cells for dogs 1
and 2. Single cells were processed using a Chromium Next GEM Single Cell 3ʹ v3.1 Kit and a standard Illumina library dual index library construction kit (10x Genomics). Sample quality was
analyzed using a LabChip (PerkinElmer; Waltham, MA) and submitted for sequencing on an Illumina NovaSeq 6000 sequencer (Novogene Corporation; Sacramento, CA) with a target of 100,000 150 bp
paired-end reads per cell. READ MAPPING AND QUANTIFICATION A Cell Ranger analysis pipeline (version 6.1.2, 10× Genomics) was utilized to process raw FASTQ sequencing data, align reads to the
canine genome, and generate a count matrix. The default settings were used when running “cellranger count” and aligned to a CanFam3.1 reference (Ensembl release 104) prepared as previously
described21. DATA FILTERING AND INTEGRATION For each sample, the count matrix was imported into R using the Read10X() function then converted to a Seurat object using the
CreateSeuratObject() function18. To estimate the number of dead/poor quality cells, the percentage of reads mapping to mitochondrial chromosomes per cell (“percent.MT”) was calculated using
PercentageFeatureSet() to count all reads mapped to features with the prefix “MT-“. Each object was filtered to only retain cells which met the following requirements: 200 < nFeature_RNA
< 5500, percent.mt < 12.5, and 100 < nCount_RNA < 75000. Next, DoubletFinder, was used to identify and remove putative cell doublets59. After completing quality control filtering
on each sample, all samples were normalized using SCTransform (Pearson residuals of regularized negative binomial regression) then integrated into one object using Seurat’s alignment
workflow48. The alignment workflow consisted of (1) identification of variable features in each sample, (2) scaling data for variable features in each sample, (3) identification and
filtering of conserved variable features between samples (“anchors”) using canonical correlation analysis (2000 integration anchors used), and (4) pairwise integration of the samples. During
the data scaling step, we used the “percent.MT” value as latent variable in a linear regression framework to minimize the impact of mitochondrial reads on dimension reduction and
integration60. Following data integration, the dataset was inspected and three low quality clusters (defined by low UMI counts) were identified and removed from the dataset. The filtered
dataset was then divided into a list of count matrices by sample and Seurat’s alignment workflow was repeated, as the selection of variable features is potentially altered with removal of
cells. Ideal clustering parameters (res = 0.8, dims = 45, n.neighbors = 40, min.dist = 0.35) were determined using the R package clustree61. Dimension reduction and visualization was
completed, and the data were projected using 2-dimensional, non-linear uniform manifold approximation and projection (UMAP) plots. SUBCLUSTERING ANALYSIS For each major cell type, we subset
the integrated dataset onto the population of interest to exclude all additional cells. The subset dataset was then divided into a list of count matrices by sample and Seurat’s alignment
workflow was repeated as described above. In the process of repeating the integration new variable features were identified which can enhance the ability to detect rare cell types through
unsupervised clustering of the reintegrated dataset. The integration, dimension reduction, and clustering parameters were as follows; tumor/stroma: integration anchors = 3000, res = 0.5,
dims = 40, n.neighbors = 50, min.dist = 0.5, T cell: integration anchors = 2500, res = 0.6, dims = 40, n.neighbors = 50, min.dist = 0.3, dendritic cells: integration anchors = 2000, res =
0.3, dims = 35, n.neighbors = 50, min.dist = 0.3, and macrophage/osteoclasts: integration anchors = 2500, res = 0.6, dims = 40, n.neighbors = 40, min.dist = 0.25. During subclustering
analysis additional low-quality clusters (low UMI or heterotypic doublets) were filtered out. Specifically, 3 (tumor/stroma), 1 (T cell), 0 (dendritic cells), and 1 (macrophage/osteoclasts)
cluster(s) were removed from each major subset. CELL CLASSIFICATION High level cell type annotations were established using unsupervised clustering results, gene set enrichment analysis, and
manual annotation based on the literature for human cell type markers62. Briefly, features used to identify major cell populations included _CD3E_/_CD5_/_CD7_ for T cells,
_CTSK_/_ACP5_/_ATP6V0D2_ for osteoclasts, _CD68_/_AIF1_/_MRC1_ for macrophages, _S100A12_/_CD4_/_CXCL8_ for neutrophils, _COL1A1_/_ALPL_/_FAP_ for tumor/fibroblasts, _FLT3_/_CD1C_/_DNASE1L3_
for dendritic cells, _MS4A1_/_JCHAIN_ for B cells, _ESAM_/_PLVAP_/_CD34_ for endothelial cells, _TOP2A_/_H1-5_/_MKI67_ for cycling cells, and _FCER1A_/_GATA2_/_MS4A2_ for mast cells. In
addition to the use of canonical markers, singleR and reference mapping to a canine leukocyte atlas was completed to provide support for immune cell classifications21,63. Further
high-resolution cell identification was completed through subclustering analysis on cells within each major population (Tumor/fibroblast, macrophage/monocyte, osteoclast, dendritic cell, and
T cell). Cell type gene signatures, as determined in this dataset using the FindAllMarkers() function (Wilcoxon Rank Sum test), can be found in Supplementary Data 1. In addition to the full
gene signatures, we provide curated short cell type gene signatures in Supplementary Data 2 and Supplementary Table 1. FEATURE VISUALIZATION Feature expression was visualized using violin
plots, feature plots, and dot plots. Selected features were chosen based on prior biological knowledge and classification of features as statistically significant using the FindAllMarkers()
function. Y-axis scales for violin plots within a figure are on fixed scales. Feature plots show normalized expression for each feature on variable scales. For all feature plots, gray/light
purple coloration indicates low expression and dark purple coloration indicates high expression. Dot plots use scaled expression data which depicts deviation from the average value for a
gene across the cells being sampled. DIFFERENTIAL GENE EXPRESSION ANALYSIS Differential gene expression (DGE) analysis was completed using pseudobulk conversion followed by a DESeq2
pipeline64. Prior to running DESeq2, low abundance features, defined as features with less than 10 raw counts across all cells sampled, were filtered out. Features that had an adjusted _P_
value of less than 0.05 (as determined using a Benjamini and Hochberg correction method) and a log2(fold change) greater than 0.58 were considered to be statistically significant65. GENE SET
ENRICHMENT ANALYSIS When completing follow-up gene set enrichment analysis (GSEA) on the gene lists generated from DGE analysis, the significantly upregulated and downregulated features
were processed separately. The upregulated and downregulated gene lists were used as input for evaluation using the clusterProfiler and msigdbr R packages to infer pathway activity66,67.
Terms which reached an adjusted _P_ value of 0.05 or lower (Benjamini and Hochberg correction method) were discussed as significantly enriched. In addition to using GSEA following DGE
analysis we also used the R package singleseqgset to complete GSEA on cell type clusters. The analysis used a competitive gene set enrichment test which was based on a Correlation Adjusted
MEan RAnk gene set test68. The log2(fold change) and mean expression for every feature within each cell type was calculated and used to complete GSEA. _P_ values were corrected for multiple
comparisons using a false discovery rate (FDR) method and corrected _P_ values were filtered to only retain terms in which at least one cell type had a value less than 0.05. The enrichment
values were scaled, and the top pathways (weighted by _P_ value) were plotted using a heatmap. COPY NUMBER VARIATION ANALYSIS Copy number variation (CNV) analysis was completed using CopyKAT
on cells that contained more than 2000 unique molecular identifiers (UMIs)14. Briefly, the approach segmented the human genome into 220-kb variable genomic bins to establish a genome-wide
copy number profile for each single cell at an approximate resolution of 5 Mb. Eash sample was run individually with a known normal cell population consisting of osteoclasts, neutrophils,
macrophages, and T cells when inferring CNV status. Individual cell classifications were extracted from the CopyKat output files, transferred to the integrated dataset, and CNV status was
visualized in the UMAP space. This approach was only used to infer if a cell was aneuploid or diploid. Individual chromosomal mutations were not evaluated due to incompatibilities of the
software across species. To supplement the CopyKAT analysis we also applied the inferCNV algorithm to the dataset which yielded similar results in terms of aneuploid and diploid
classifications15. Briefly, the inferCNV approach was run with an ordered gene file generated using the Ensembl CanFam3.1.104 gene transfer format (.gtf) file that was used to build the
reference used for alignment of the single-cell data. An infercnv object was then created using the canine gene positions with endothelial cells and macrophages selected as the “normal”
reference populations. InferCNV was then run using the default settings with the recommended “cutoff” argument set to 0.1. REGULON ACTIVITY The python implementation of Single-Cell
Regulatory Network Inference and Clustering (pySCENIC) was used to infer activity of gene regulatory networks within cell types69,70. To complete this analysis the gene symbols were
converted from canine to human using the convert_orthologs() function from the orthogene R package71. During conversion, genes that had duplicate mappings in either canine or human
annotations were dropped from the matrix and excluded from downstream analysis. The count matrices with converted gene symbols were loaded into SCANPY and a standard pySCENIC workflow with
default settings was followed72. The regulatory feather files used in the analysis were obtained from https://resources.aertslab.org/cistarget/, with file names being
hg38__refseq-r80__10kb_up_and_down_tss.mc9nr.feather hg38__refseq-r80__500bp_up_and_100bp_down_tss.mc9nr.feather. After predicting regulon activity with pySCENIC, regulon specificity scores
(rss) were calculated using AUCell and the rss values were used to infer regulon activity in the cell types analyzed73. CELL-CELL INTERACTION INFERENCE ANALYSIS The R package, CellChat, was
used to make inferences about cell-cell interactions within the tumor microenvironment35. Using a list of known human receptor-ligand pairs provided through CellChat, we calculated the
interaction scores (strength and weight) which represent the probability of two cells interacting. Analysis was initially completed on the final fully annotated dataset with 41 cell types
and secondarily completed on a subset of only the immune cells (exclusion of the major cell types: osteoblast, cycling osteoblast, endothelial, and fibroblast). Prior to analysis the raw
count matrices were extracted and the gene symbols were converted from canine to human as described in the “Regulon activity” section. Broadly the analysis evaluated the interactivity
between ligand expressing cells (senders/outgoing signals) and receptor expressing cells (receivers/incoming signals). Inferences regarding potential interactivity were then made based on
the law of mass action using the average expression values of receptors and ligands within cell types. Statistical enrichment of interaction networks was determined using permutation testing
and an adjusted _P_ value < 0.05 was used to determine significance. Following identification of enriched cell-cell interaction networks, we further classified each enriched pathway as
“immune specific”, “immune related”, and “non-immune” based on reported expression patterns of receptors and ligands (Supplementary Table 2). Analysis of the immune cell subset involved
further classifying a subset of interaction networks (PD-L1, CD80, and CD86) as immune regulatory. HUMAN OS HOMOLOGY ANALYSIS Six treatment-naive human OS samples were obtained from the NCBI
GEO database accession GSE16245412. The count matrices reported from the previous study were loaded in as Seurat objects and were filtered using the same parameters as used to preprocess
the 6 canine OS tumor samples. The human dataset was annotated using low resolution unsupervised clustering while referencing the primary article in an attempt to recreate the original
annotations. Prior to integrating data across species, the raw count matrices from each dataset were extracted and canine gene symbols were converted from canine to human as described in the
“Regulon activity” section. Following conversion of gene symbols, the 12 OS samples (6 human and 6 canine) were integrated into one object using the same Seurat alignment workflow described
above, with the exception that 3000 variable features were selected as anchors. SCTransform normalized counts were then used to complete hierarchical clustering using the hclust() function
with method set to “complete”. Subsequent cell type gene signatures were established using FindAllMarkers() and DGE analysis contrasting cell types within each species was completed within
the respective dataset. The resulting cell type gene signatures were used to calculate a Jaccard similarity index, whereas the adjusted _P_ values were assigned a sign (+/−) based on the
log2(fold change) then the signed _P_ values were used to generate scatter plots. STATISTICS AND REPRODUCIBILITY Raw data from a total of 8 canine scRNA-seq osteosarcoma samples were
generated in this study. Two of the 8 samples were technical replicates, which were considered as one sample when completing computational analysis to retain a total of 6 biological
replicates. Biological replicates were used for pseudobulk differential gene expression analysis, while cellular replicates were used for all other analysis completed in this study. Detailed
descriptions of the statistical analyses and significance thresholds used in this study are provided in the respective methods section. REPORTING SUMMARY Further information on research
design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY Raw sequencing data are available on the NCBI Gene Expression Omnibus database under
the accession number GSE252470. The annotated dataset is available for browsing at the UCSC Cell Browser (https://cells.ucsc.edu/?ds=canine-os-atlas)74, and the processed data (Seurat v4.3.0
RDS objects) are available on Zenodo (https://doi.org/10.5281/zenodo.10666968)75. Results of all differential gene expression analysis and cell type gene signatures are provided in
Supplementary Data files. CODE AVAILABILITY A project specific GitHub page containing all analysis code and software versions used to analyze the data presented in this manuscript is
available at https://github.com/dyammons/canine_osteosarcoma_atlas75. Any additional data requests can be made by contacting a corresponding author. REFERENCES * Link, M. P. et al. The
effect of adjuvant chemotherapy on relapse-free survival in patients with osteosarcoma of the extremity. _N. Engl. J. Med._ 314, 1600–1606 (1986). Article CAS PubMed Google Scholar *
Tarone, L. et al. Naturally occurring cancers in pet dogs as pre-clinical models for cancer immunotherapy. _Cancer Immunol. Immunother._ 68, 1839–1853 (2019). Article PubMed Google Scholar
* Dow, S. A role for dogs in advancing cancer immunotherapy research. _Front. Immunol._ 10, 2935 (2020). Article PubMed PubMed Central Google Scholar * Schiffman, J. D. & Breen, M.
Comparative oncology: what dogs and other species can teach us about humans with cancer. _Philos. Trans. R. Soc. B Biol. Sci._ 370, 20140231 (2015). Article Google Scholar * Wu, C. C. et
al. Immuno-genomic landscape of osteosarcoma. _Nat. Commun._ 11, 1008 (2020). Article CAS PubMed PubMed Central Google Scholar * Cao, J., Chow, L. & Dow, S. Strategies to overcome
myeloid cell induced immune suppression in the tumor microenvironment. _Front. Oncol._ 13, 1116016 (2023). Article CAS PubMed PubMed Central Google Scholar * Roma-Rodrigues, C., Mendes,
R., Baptista, P. V. & Fernandes, A. R. Targeting tumor microenvironment for cancer therapy. _Int. J. Mol. Sci._ 20, 840 (2019). Article CAS PubMed PubMed Central Google Scholar *
Withers, S. S. et al. Association of macrophage and lymphocyte infiltration with outcome in canine osteosarcoma. _Vet. Comp. Oncol._ 17, 49–60 (2019). Article CAS PubMed Google Scholar *
Gomez-Brouchet, A., et al. CD163-positive tumor-associated macrophages and CD8-positive cytotoxic lymphocytes are powerful diagnostic markers for the therapeutic stratification of
osteosarcoma patients: An immunohistochemical analysis of the biopsies from the French OS2006 phase 3 t. [Internet]. 6. Available from:
https://www.tandfonline.com/doi/abs/10.1080/2162402X.2017.1331193 (2017). * Buddingh, E. P. et al. Tumor-Infiltrating Macrophages Are Associated with Metastasis Suppression in High-Grade
Osteosarcoma: A Rationale for Treatment with Macrophage Activating AgentsImpact of Macrophages on Osteosarcoma Metastases. _Clin. Cancer Res._ 17, 2110–2119 (2011). Article CAS PubMed
Google Scholar * Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. _Nat. Commun._ 8, 14049 (2017). Article CAS PubMed PubMed Central Google
Scholar * Liu, Y. et al. Single-cell transcriptomics reveals the complexity of the tumor microenvironment of treatment-naive osteosarcoma. _Front. Oncol._ 11, 709210 (2021). Article CAS
PubMed PubMed Central Google Scholar * Zhou, Y. et al. Single-cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma. _Nat.
Commun._ 11, 6322 (2020). Article CAS PubMed PubMed Central Google Scholar * Gao, R. et al. Delineating copy number and clonal substructure in human tumors from single-cell
transcriptomes. _Nat. Biotechnol._ 39, 599–608 (2021). Article CAS PubMed PubMed Central Google Scholar * inferCNV of the Trinity CTAT Project.
https://github.com/broadinstitute/inferCNV. * Zhang, H. et al. A novel molecular classification method for osteosarcoma based on tumor cell differentiation trajectories. _Bone Res._ 11, 1
(2023). Article PubMed PubMed Central Google Scholar * Wang, X. et al. Reinvestigation of classic T cell subsets and identification of novel cell subpopulations by single-cell RNA
sequencing. _J. Immunol._ 208, 396–406 (2022). Article CAS PubMed Google Scholar * Hao, Y. et al. Integrated analysis of multimodal single-cell data. _Cell_ 184, 3573–3587 (2021).
Article CAS PubMed PubMed Central Google Scholar * Tabib, T. et al. Myofibroblast transcriptome indicates SFRP 2hi fibroblast progenitors in systemic sclerosis skin. _Nat. Commun._ 12,
4384 (2021). Article CAS PubMed PubMed Central Google Scholar * Chung, W. et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer.
_Nat. Commun._ 8, 1–12 (2017). Article Google Scholar * Ammons D. T., et al. A single-cell RNA sequencing atlas of circulating leukocytes from healthy and osteosarcoma affected dogs. Front
Immunol. 14, 1162700 (2023). * Chtanova, T. et al. T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide
help for B cells. _J. Immunol._ 173, 68–78 (2004). Article CAS PubMed Google Scholar * Kim, C. H. et al. Unique gene expression program of human germinal center T helper cells. _Blood_
104, 1952–1960 (2004). Article CAS PubMed Google Scholar * Magen, A., et al. Intratumoral dendritic cell–CD4+ T helper cell niches enable CD8+ T cell differentiation following PD-1
blockade in hepatocellular carcinoma. _Nat Med_. 29, 1389–1399 (2023). * Ukita, M. et al. CXCL13-producing CD4+ T cells accumulate in the early phase of tertiary lymphoid structures in
ovarian cancer. _JCI Insight_ 7, e157215 (2022). Article PubMed PubMed Central Google Scholar * Yang, M. et al. CXCL13 shapes immunoactive tumor microenvironment and enhances the
efficacy of PD-1 checkpoint blockade in high-grade serous ovarian cancer. _J. Immunother. Cancer_ 9, e001136 (2021). Article PubMed PubMed Central Google Scholar * Bausch-Fluck, D. et
al. The in silico human surfaceome. _Proc. Natl Acad. Sci._ 115, E10988–E10997 (2018). Article CAS PubMed PubMed Central Google Scholar * See, P. et al. Mapping the human DC lineage
through the integration of high-dimensional techniques. _Science_ 356, eaag3009 (2017). Article PubMed PubMed Central Google Scholar * Collin, M. & Bigley, V. Human dendritic cell
subsets: an update. _Immunology_ 154, 3–20 (2018). Article CAS PubMed PubMed Central Google Scholar * Maier, B. et al. A conserved dendritic-cell regulatory program limits antitumour
immunity. _Nature_ 580, 257–262 (2020). Article CAS PubMed PubMed Central Google Scholar * Li, J. et al. Mature dendritic cells enriched in immunoregulatory molecules (mregDCs): A novel
population in the tumour microenvironment and immunotherapy target. _Clin. Transl. Med._ 13, e1199 (2023). Article CAS PubMed PubMed Central Google Scholar * Bao, M. & Liu, Y. J.
Regulation of TLR7/9 signaling in plasmacytoid dendritic cells. _Protein Cell_ 4, 40–52 (2013). Article CAS PubMed Google Scholar * Sawai, C. M. et al. Transcription factor Runx2
controls the development and migration of plasmacytoid dendritic cells. _J. Exp. Med._ 210, 2151–2159 (2013). Article CAS PubMed PubMed Central Google Scholar * Cheng, S. et al. A
pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. _Cell_ 184, 792–809 (2021). Article CAS PubMed Google Scholar * Jin, S. et al. Inference and analysis of
cell-cell communication using CellChat. _Nat. Commun._ 12, 1088 (2021). Article CAS PubMed PubMed Central Google Scholar * Ma, R. Y., Black, A., Qian, B. Z. Macrophage diversity in
cancer revisited in the era of single-cell omics. _Trends Immunol._ 43, 546–563 (2022). * Rzepecka, A., Żmigrodzka, M., Witkowska-Piłaszewicz, O., Cywińska, A. & Winnicka, A. CD4 and
MHCII phenotypic variability of peripheral blood monocytes in dogs. _PLoS One_ 14, e0219214 (2019). Article CAS PubMed PubMed Central Google Scholar * Jablonski, K. A. et al. Novel
markers to delineate murine M1 and M2 macrophages. _PLoS One_ 10, e0145342 (2015). Article PubMed PubMed Central Google Scholar * Mosser, D. M. & Edwards, J. P. Exploring the full
spectrum of macrophage activation. _Nat. Rev. Immunol._ 8, 958–969 (2008). Article CAS PubMed PubMed Central Google Scholar * Eisinger, S. et al. Targeting a scavenger receptor on
tumor-associated macrophages activates tumor cell killing by natural killer cells. _Proc. Natl Acad. Sci._ 117, 32005–32016 (2020). Article CAS PubMed PubMed Central Google Scholar *
Omata, Y. et al. Interspecies single‐cell RNA‐seq analysis reveals the novel trajectory of osteoclast differentiation and therapeutic targets. _JBMR_ 6, e10631 (2022). CAS Google Scholar *
Dougall, W. C. et al. RANK is essential for osteoclast and lymph node development. _Genes Dev._ 13, 2412–2424 (1999). Article CAS PubMed PubMed Central Google Scholar * Takayanagi, H.
et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. _Dev. Cell_ 3, 889–901 (2002). Article CAS
PubMed Google Scholar * Zhu, L. et al. A Zeb1/MtCK1 metabolic axis controls osteoclast activation and skeletal remodeling. _EMBO J._ 42, e111148 (2023). Article CAS PubMed PubMed
Central Google Scholar * De Frutos, C. A. et al. Snail1 controls bone mass by regulating Runx2 and VDR expression during osteoblast differentiation. _EMBO J._ 28, 686–696 (2009). Article
PubMed PubMed Central Google Scholar * Koirala, P. et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. _Sci. Rep._ 6, 30093
(2016). Article CAS PubMed PubMed Central Google Scholar * Ashley, J. W. et al. Genetic ablation of CD68 results in mice with increased bone and dysfunctional osteoclasts. _PLoS One_ 6,
e25838 (2011). Article CAS PubMed PubMed Central Google Scholar * Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using
regularized negative binomial regression. _Genome Biol._ 20, 1–15 (2019). Article Google Scholar * Pombo Antunes, A. R. et al. Single-cell profiling of myeloid cells in glioblastoma across
species and disease stage reveals macrophage competition and specialization. _Nat. Neurosci._ 24, 595–610 (2021). Article CAS PubMed Google Scholar * Mannheimer, J. D. et al.
Transcriptional profiling of canine osteosarcoma identifies prognostic gene expression signatures with translational value for humans. _Commun. Biol._ 6, 856 (2023). Article CAS PubMed
PubMed Central Google Scholar * Musumeci, A., Lutz, K., Winheim, E. & Krug, A. B. What makes a pDC: recent advances in understanding plasmacytoid DC development and heterogeneity.
_Front Immunol._ 10, 1222 (2019). Article CAS PubMed PubMed Central Google Scholar * Li, Y., Qi, X., Liu, B. & Huang, H. The STAT5–GATA2 pathway is critical in basophil and mast
cell differentiation and maintenance. _J. Immunol._ 194, 4328–4338 (2015). Article CAS PubMed Google Scholar * Li, Y. et al. GATA2 regulates mast cell identity and responsiveness to
antigenic stimulation by promoting chromatin remodeling at super-enhancers. _Nat. Commun._ 12, 494 (2021). Article CAS PubMed PubMed Central Google Scholar * Ohmori, S. et al. GATA2 and
PU. 1 Collaborate To Activate the Expression of the Mouse Ms4a2 Gene, Encoding FcεRI β, through Distinct Mechanisms. _Mol. Cell Biol._ 39, e00314–e00319 (2019). Article CAS PubMed PubMed
Central Google Scholar * Newman, A. M. et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. _Nat. Biotechnol._ 37, 773–782 (2019). Article CAS
PubMed PubMed Central Google Scholar * Li, T. et al. TIMER2. 0 for analysis of tumor-infiltrating immune cells. _Nucleic Acids Res_ 48, W509–W514 (2020). Article CAS PubMed PubMed
Central Google Scholar * Liu, W. et al. Characterizing the tumor microenvironment at the single-cell level reveals a novel immune evasion mechanism in osteosarcoma. _Bone Res._ 11, 4
(2023). Article CAS PubMed PubMed Central Google Scholar * Luo, Z. W., Liu, P. P., Wang, Z. X., Chen, C. Y. & Xie, H. Macrophages in osteosarcoma immune microenvironment:
implications for immunotherapy. _Front. Oncol._ 10, 586580 (2020). Article PubMed PubMed Central Google Scholar * McGinnis, C. S., Murrow, L. M. & Gartner, Z. J. DoubletFinder:
doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. _Cell Syst._ 8, 329–337 (2019). Article CAS PubMed PubMed Central Google Scholar * Butler, A.,
Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. _Nat. Biotechnol._ 36, 411–420
(2018). Article CAS PubMed PubMed Central Google Scholar * Zappia, L. & Oshlack, A. Clustering trees: a visualization for evaluating clusterings at multiple resolutions.
_Gigascience_ 7, giy083 (2018). Article PubMed PubMed Central Google Scholar * Thomas, D. D., Lacinski, R. A., Lindsey, B. A. Single-cell RNA-seq reveals intratumoral heterogeneity in
osteosarcoma patients: A review. _J. Bone Oncol._ 39, 100475 (2023). * Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage.
_Nat. Immunol._ 20, 163–172 (2019). Article CAS PubMed PubMed Central Google Scholar * Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for
RNA-seq data with DESeq2. _Genome Biol._ 15, 1–21 (2014). Article Google Scholar * Squair, J. W. et al. Confronting false discoveries in single-cell differential expression. _Nat. Commun._
12, 5692 (2021). Article CAS PubMed PubMed Central Google Scholar * Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. _Innovation_ 2, 100141
(2021). CAS PubMed PubMed Central Google Scholar * Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles.
_Proc. Natl Acad. Sci._ 102, 15545–15550 (2005). Article CAS PubMed PubMed Central Google Scholar * Wu, D. & Smyth, G. K. Camera: a competitive gene set test accounting for
inter-gene correlation. _Nucleic Acids Res._ 40, e133–e133 (2012). Article CAS PubMed PubMed Central Google Scholar * Aibar, S. et al. SCENIC: single-cell regulatory network inference
and clustering. _Nat. Methods_ 14, 1083–1086 (2017). Article CAS PubMed PubMed Central Google Scholar * Van de Sande, B. et al. A scalable SCENIC workflow for single-cell gene
regulatory network analysis. _Nat. Protoc._ 15, 2247–2276 (2020). Article PubMed Google Scholar * Schilder, B. M. & Skene, N. G. orthogene: an R package for easy mapping of
orthologous genes across hundreds of species. _Bioconductor_ https://doi.org/10.18129/B9.bioc.orthogene (2022). * Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell
gene expression data analysis. _Genome Biol._ 19, 1–5 (2018). Article Google Scholar * Suo, S. et al. Revealing the critical regulators of cell identity in the mouse cell atlas. _Cell
Rep._ 25, 1436–1445 (2018). Article CAS PubMed PubMed Central Google Scholar * Speir, M. L. et al. UCSC Cell Browser: visualize your single-cell data. _Bioinformatics_ 37, 4578–4580
(2021). Article CAS PubMed PubMed Central Google Scholar * Ammons, D. Canine osteosarcoma single-cell RNA sequencing reference dataset: analysis code and processed data for publication,
https://doi.org/10.5281/zenodo.10666968 (2024). Download references ACKNOWLEDGEMENTS This project was supported by grants from the National Institutes of Health (NIH): U01 CA224182 (to
S.D.) and T32 OD012201 (to D.T.A.), the Boettcher Foundation Webb-Waring Biomedical Research Award (to D.P.R.), and the Shipley Family Foundation (to S.D.).The authors would like to
acknowledge Lynelle Lopez, Kara Hall, and Allister Aradi for their assistance with management of clinical cases and sample collection. This work utilized the Alpine high performance
computing resource at the University of Colorado Boulder. Alpine is jointly funded by the University of Colorado Boulder, the University of Colorado Anschutz, and Colorado State University.
Data storage was supported by the University of Colorado Boulder ‘PetaLibrary. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Microbiology, Immunology, and Pathology, College of
Veterinary Medicine and Biomedical Sciences, Colorado State University, Fort Collins, CO, USA Dylan T. Ammons, Kathryn E. Cronise, Daniel P. Regan & Steven Dow * Flint Animal Cancer
Center, Department of Clinical Sciences, College of Veterinary Medicine and Biomedical Sciences, Colorado State University, Fort Collins, CO, USA Leone S. Hopkins, Jade Kurihara, Daniel P.
Regan & Steven Dow Authors * Dylan T. Ammons View author publications You can also search for this author inPubMed Google Scholar * Leone S. Hopkins View author publications You can also
search for this author inPubMed Google Scholar * Kathryn E. Cronise View author publications You can also search for this author inPubMed Google Scholar * Jade Kurihara View author
publications You can also search for this author inPubMed Google Scholar * Daniel P. Regan View author publications You can also search for this author inPubMed Google Scholar * Steven Dow
View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Conception and design: D.T.A., S.D. Experimentation and data acquisition: D.T.A., J.K.,
D.P.R. Data analysis and manuscript drafting: D.T.A., L.S.H., K.E.C., D.P.R., S.D. Final approval of completed manuscript: All authors. CORRESPONDING AUTHORS Correspondence to Dylan T.
Ammons or Steven Dow. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Communications Biology_ thanks Samir Amin and
the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: George Inglis and Johannes Stortz. A peer review file is available.
ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION PEER
REVIEW FILE SUPPLEMENTARY INFORMATION DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 SUPPLEMENTARY DATA 3 SUPPLEMENTARY DATA 4 SUPPLEMENTARY DATA 5
SUPPLEMENTARY DATA 6 SUPPLEMENTARY DATA 7 SUPPLEMENTARY DATA 8 REPORTING SUMMARY RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0
International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the
source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative
Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ammons, D.T., Hopkins, L.S., Cronise, K.E. _et al._ Single-cell RNA sequencing
reveals the cellular and molecular heterogeneity of treatment-naïve primary osteosarcoma in dogs. _Commun Biol_ 7, 496 (2024). https://doi.org/10.1038/s42003-024-06182-w Download citation *
Received: 06 November 2023 * Accepted: 10 April 2024 * Published: 24 April 2024 * DOI: https://doi.org/10.1038/s42003-024-06182-w SHARE THIS ARTICLE Anyone you share the following link with
will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative