Play all audios:
ABSTRACT Ethnicity has a significant role in shaping the composition of the gut microbiome, which has implications in human physiology. This study intends to investigate the gut microbiome
of Bengali people as well as several indigenous ethnicities (Chakma, Marma, Khyang, and Tripura) residing in the Chittagong Hill Tracts areas of Bangladesh. Following fecal sample collection
from each population, part of the bacterial 16 s rRNA gene was amplified and sequenced using Illumina NovaSeq platform. Our findings indicated that Bangladeshi gut microbiota have a
distinct diversity profile when compared to other countries. We also found out that Bangladeshi indigenous communities had a higher _Firmicutes_ to _Bacteroidetes_ ratio than the Bengali
population. The investigation revealed an unclassified bacterium that was differentially abundant in Bengali samples while the genus _Alistipes_ was found to be prevalent in Chakma samples.
Further research on these bacteria might help understand diseases associated with these populations. Also, the current small sample-sized pilot study hindered the comprehensive understanding
of the gut microbial diversity of the Bangladeshi population and its potential health implications. However, our study will help establish a basic understanding of the gut microbiome of the
Bangladeshi population. SIMILAR CONTENT BEING VIEWED BY OTHERS ELUCIDATING THE GUT MICROBIOME ALTERATIONS OF TRIBAL COMMUNITY OF ARUNACHAL PRADESH: PERSPECTIVES ON THEIR LIFESTYLE OR FOOD
HABITS Article Open access 31 October 2022 COMPARATIVE ANALYSIS OF GUT MICROBIOTA IN FREE RANGE AND HOUSE FED YAKS FROM LINZHOU COUNTY Article Open access 24 April 2025 GUT MICROBIOMES OF
AGROPASTORAL CHILDREN FROM THE ADADLE REGION OF ETHIOPIA REFLECT THEIR UNIQUE DIETARY HABITS Article Open access 01 December 2023 INTRODUCTION The human gut microbiome which is composed of
an enormous collection of microorganisms plays a crucial role in modulating various physiological processes, metabolic diseases, and immunological dysregulation. The composition of the gut
microbiome is influenced by a multitude of factors, including diet, lifestyle, environment, genetics, and ethnicity1. They forge a symbiotic relationship with the host in the
gastrointestinal tract, where they are monitored by the innate immune system using pattern recognition receptors such as toll-like receptors and Nucleotide-binding oligomerization
domain-containing protein (NOD)-like receptors2. A number of these bacteria are involved in regulating host metabolism by producing metabolites such as folate, indoles, secondary bile acids,
trimethylamine-N-oxide, neurotransmitters (e.g., serotonin, gamma amino butyric acid), and short-chain fatty acids3. Alteration in the host–symbiont relationships in the gut has been
observed in diseases like obesity, type 2 diabetes mellitus, non-alcoholic liver disease, cardio-metabolic diseases, and so on4. Since different ethnic groups have been reported to have
variations in genetics, food habits, lifestyle, their alterations can be greatly influenced by ethnicity. As a result, some ethnic groups are predisposed to certain metabolic diseases, while
others are inclined to others5. For example, high-risk variants of _NOD2_ are less prevalent in African Americans which makes their microbiome less prone to Crohn’s Disease6,7. Similarly,
the _lactase_ gene varies in different populations8. In the Japanese population higher abundance of _Bifidobacterium_ has been observed due to their characteristic _LCT_ genotypes9,10.
Hence, gut microbiome studies on ethnic groups can be helpful in identifying prognostic microbial biomarkers for various diseases. The genetic makeup, dietary patterns, and geographic
regions shape the gut microbiome of various ethnicities in a distinct pattern11,12,13,14. Previously, a number of gut microbiome studies in South-East Asia have been conducted. Gut
microbiomes of populations living in India and China which are geographically close to Bangladesh, showed an abundance of _Prevotella_ and _Bacteroidetes_ respectively5. In India, gut
microbiomes from the Adi, Apatani, and Nyshi tribes of Arunachal Pradesh (a state in Northeastern India) demonstrated the predominance of _Firmicutes_, _Bacteroidates_, _Actinobacteria_, and
_Proteobacteria_. On the other hand, Mongoloid and Proto-Australoid tribes of India were dominated by _Prevotella_15,16. However, studies from ethnic groups in Bangladesh are still not
available. Bangladeshi ethnic groups have diverse dietary habits. Bengali food customs stem from their agrarian culture, which is defined by a profusion of rice, fish, and different
vegetables. Dishes like biryani and kebabs are examples of the historical effects of Mughal and British dynasties on the cuisine. Seasonal fruit preparations, street snacks like shingara and
samosas, and traditional cakes called “Pitha” are all common17. Bangladeshi tribal people use forest resources to eat diversely. They feed on ‘bans koral,’ bamboo shoots from _Melocanna
baccifera_ and _Bambusa tulda_ during the rainy season. Fresh or dried wild plants commonly accompany staple grain recipes. Acidic leaves are often eaten as salad or chutney. Woody
perennials including _Albizia procera_, wild mango, and _Daemonorops jenkinsianus_ are vegetables for these ethnic groups. Lentinus, Shizophyllum, and Jew’s Ear mushrooms from decomposing
wood are eaten. Wild banana inflorescences and leaf sheath soft cores are also edible. During food shortages, banana core cooked with rice or bran is crucial18. Bangladesh is a country
inhabited by many different ethnicities. Bengalis are the largest among them. Other major groups are Chakma, Marma, Khyang, and Tripura with distinguishable food habits and lifestyles. The
Chittagong Hill Tracts in the southeastern parts of Bangladesh are home to the Chakma, Marma, Khyang, and Tripura people. Bengali people (Bengali language speakers) have Indian ancestry with
Proto-Australoid, Caucasoid, and Mongoloid genetics19,20,21. On the other hand, Chakma, Marma, Khyang, and Tripura people (Tibeto-Burman language speakers) have predominance of
Tibeto-Burman genetics. Tibeto-Burman populations originated from northwestern China and moved to the South. They have interbred with numerous southern tribes for the past 2600 years,
creating specific genetic traits among southern Tibeto-Burman populations22,23. Hence, Tibeto-Burman speakers from Northeast India are more distinctive than the ones living in the
Himalayas23. In Bangladesh, the Tibeto-Burman populations (e.g., Chakma, Marma, Khyang, Tripura etc.) have higher similarity with Northeast Indian Tibeto-Burman but they contain more
mainland Indian ancestry24. In this study, we investigated the gut microbiomes of Bengali, Chakma, Marma, Khyang, and Tripura populations in order to determine whether Bangladeshi
Tibeto-Burman populations differ from Bengali people in terms of their gut microbiomes. METHODS SAMPLE COLLECTION Prior to sample collection, written informed consent was obtained from all
participants. Ethical approval and necessary permits were obtained from the National Institute of Biotechnology Ethical Review Committee, Bangladesh (NIBERC2022-01). All ethical regulations
relevant to human research participants were followed. Supplementary Data 1 contains detailed sample information regarding age, gender, and cohort. All of the indigenous volunteers (Chakma,
Marma, Khyang, and Tripura) in this study reside in the Rangamati district (22°37’60 N 92°12’0E) of the Chittagong Hill Tracts region. Bengali samples were collected from Dhaka Division
(23.9536° N, 90.1495° E). Based on ethnicity, the participants were divided into five groups (Bengali, Chakma, Marma, Khyang, and Tripura). A total of 55 individuals were sampled, of which
13 were Bengali, 15 were Chakma, 10 were Khyang, 6 were Marma, and 11 were Tripura. Fecal samples of the participants were collected using sterile stool collection tubes. The samples were
then transported to the National Institute of Biotechnology using Icebox and subsequently stored at −80 °C temperature. Fecal DNA extraction was executed using the PureLink™ Microbiome DNA
Purification Kit (Catalog number: A29790). Specialized beads were used along with a combination of heat, chemical, and mechanical disruption to lyse the microorganisms efficiently.
Precipitation with a proprietary cleaning buffer removed inhibitors. After that, the samples were placed in spin columns, and the DNA attached to the column was washed once before elution.
DNA concentration and purity were estimated by Thermo Scientific NanoDrop 2000/2000c and then stored at −20 °C. AMPLICON GENERATION AND LIBRARY PREPARATION FOR SEQUENCING Amplicons were
generated using the 341 F (5’-CCTAYGGGRBGCASCAG-3’) and 806 R (5’-GGACTACNNGGGTATCTAAT-3’) primers that targeted the V3-V4 region of the bacterial 16 S rRNA gene. All PCR reactions were
performed with 15 μL of Phusion® High-Fidelity PCR Master Mix (New England Biolabs), 0.2 μM of forward and reverse primers, and around 10 ng template DNA. For thermal cycling, initial
denaturation at 98 °C for 1 min was followed by 30 cycles of denaturation at 98 °C for 10 s, annealing at 50 °C for 30 s, and elongation at 72 °C for 30 s. Final elongation was carried out
for 5 min at 72 °C. TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina, USA) was used as per the manufacturer’s protocol for sequencing library preparation, and index codes were appended.
The quality of the library was evaluated by employing the [email protected] Fluorometer (Thermo Scientific) and the Agilent Bioanalyzer 2100 system. Finally, the library was sequenced on an
Illumina NovaSeq platform, resulting in 250 bp paired-end reads. DATA PRE-PROCESSING AND QUALITY CONTROL The paired end sequences were converted into QIIME2 format for data pre-processing,
quality control, taxonomic assignment, differential abundance identification, functional analysis using QIIME2 platform (version 2021.4.0)25. More specifically, the data pre-processing of
paired end sequences were performed using the DADA2 plug-in within QIIME226. DADA2 filtered noisy reads, performed error correction in marginal sequences, removed chimeras and singletons,
joined denoised paired-end reads, and also de-replicated the filtered reads. The features produced by DADA2 are denoted as amplicon sequence variants. TAXONOMIC ASSIGNMENT For taxonomic
assignment, a pre-trained classifier based on the Naive Bayes machine-learning model was implemented. This model was trained on 99% sequence similarity of Greengenes 13_8 data. This
classifier was then deployed for taxonomy assignment of amplicon sequence variants. DIVERSITY ANALYSIS Several diversity metrics in QIIME2 require a rooted phylogenetic tree generated from
the amplicon sequence variants of the sampled data. A reference-based fragment insertion method, using q2-fragment-insertion tool, was applied to construct the rooted tree for this purpose.
Greengenes 13_8 data was used as a reference database in the q2-fragment-insertion tool27,28. The sequencing depth of the samples was 3525 to observe the richness. This phenomenon was
checked with the alpha rarefaction curve generated by the q2-diversity tool. The microbiome within and between samples was calculated by the core-metric-phylogenetic method of the
q2-diversity tool. This method computes several alpha (Observed features, Shanon diversity, Faith’s phylogenetic diversity, Pielou evenness) and beta (Jaccard distance, Bray–Curtis distance,
unweighted UniFrac distance, and weighted UniFrac distance) diversity metrics altogether29. Based on each beta diversity metric, this command also performed principal coordinates analysis
(PCoA)30. To visualize the PCoA plots for every beta diversity metric, EMPeror visualization tool was utilized to generate the figures31. Several statistical tests were conducted during
diversity analysis such as Kruskal–Wallis H test, two-way ANOVA, paired _t_-test and PERMANOVA test32,33,34,35. The boxplot() function in R was used to draw boxplots based on alpha diversity
values. This function follows the 1.5 IQR method for detecting outliers which are placed above and below the whiskers on the boxplots. Here, only one sample was identified as outlier
(Sample ID: BBT19). The result including the outlier has been provided in Supplementary Fig. 1. After removal of the outlier, the number of samples used for analysis were reduced from 55 –
54. DIFFERENTIAL ABUNDANCE TEST To classify the features that were differentially abundant across various sample groups the analysis of the composition of microbiomes (ANCOM) method was
applied by the q2-composition tool36. This statistical framework was deployed at the genus level. The minimum sample size for each feature was set to 27 (half of the total samples) because
ANCOM fails to manage false discovery rates at sample sizes <10 as well as to remove very lowly abundant features37. For linear discriminant analysis by the LEfSe, the feature table was
collapsed at the genus level and the minimum sample size was chosen at 2738. This tool first performs the non-parametric Kruskal-Wallis (KW) sum-rank test to identify the features which had
significant differential abundance across different metadata categories. Finally, LEfSe applies linear discriminant analysis to compute the effect size of each differentially abundant
feature and plot the linear discriminant analysis score in the log10 scale. Result of both ANCOM and LEfSe analysis conducted for all samples including the previously mentioned outliers has
been provided in Supplementary Fig. 2. FUNCTIONAL ANALYSIS BURRITO, an interactive visualization web server (http://elbo-spice.cs.tau.ac.il/shiny/burrito/), was utilized to explore the
taxa-function relationship within the samples of the study39. To acquire gene contents and functional annotations, this tool adopts the PICRUSt and KEGG Orthology databases,
respectively40,41. At first, features with a sample size of <27 were filtered from the original feature table to remove very low abundant taxon. The q2-vsearch tool was employed for
closed-reference clustering of retrieved features at 97% identity based on greengenes 97% OTU IDs as reference 42,43. Thus the acquired OTU table was then converted to the appropriate table
format for BURRITO input. COMPARATIVE ANALYSIS WITH TROPICAL AND SUBTROPICAL COUNTRIES Since Bangladesh is a tropical country, we compared the gut microbiome of Bangladeshi samples with
several tropical and subtropical countries to see if there is any similarity between them. To do this we took sequence data from publicly available 16 s rRNA amplicon sequence data from the
NCBI Sequence Read Archive and MG-RAST databases. Only healthy / control samples were taken from the selected datasets. Countries included in the comparative analysis were Australia, Egypt,
India, Indonesia, Malaysia, Mexico, Thailand, and Vietnam. Information about the samples taken from NCBI and MG-RAST has been presented in Supplementary Data 2 and Supplementary Data 3
respectively. The samples downloaded from MG-RAST were processed using q2-deblur while the samples downloaded from NCBI were processed using q2-dada234,44. This is because the samples were
taken from different regions and were prepared and sequenced using different methods. Amplicon sequencing using the Illumina MiSeq technology was done on Indian samples, encompassing the
V3-V4 region of bacterial 16 S rDNA. On the other hand, the V4 region of bacterial 16 S ribosomal RNA genes from Malawi, Amerindians, and the United States was amplified and sequenced using
an Illumina HiSeq 2000. The V1-V3 region of the 16 S ribosomal RNA (rRNA) gene of the Mongolian samples was amplified and sequenced using pyrosequencing on a Roche GS FLX. The samples were
merged with the qiime feature-table merge method. All the samples were rarefied to the same depth (3525). These foreign samples (_n_ = 181) were then compared with all Bangladeshi samples
(_n_ = 55) using the q2-diversity tool via Alpha diversity and Beta Diversity analysis. Afterwards, a phylogenetic rooted tree was generated by the sepp fragment insertion approach. This
rooted phylogeny was used to create a Unweighted Pair Group Method with Arithmetic Mean tree based on the Unweighted UniFrac metric by beta-rarefaction command of the q2-diversity tool45.
REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. RESULTS BENGALI POPULATION POSSESSED LOWER
_FIRMICUTES_ TO _BACTEROIDETES_ RATIO COMPARED TO INDIGENOUS GROUPS Among the 19 classified phyla, _Firmicutes_ and _Bacteroidetes_ were the most prevalent (48% and 34% of the total) in the
gut microbiome of all Bangladeshi populations. Other phyla with higher abundance were Proteobacteria (14%), Actinobacteria (3%), Tenericutes (0.5%), etc (Supplementary Fig. 3).
Interestingly, 100% of taxa from the _Bacteroidetes_ phylum belonged to the _Bacteroidia_ class. On the other hand, considering _Firmicutes_ phylum, 87% of features are _Clostridia_ class,
11% _Bacilli_, and 2% are _Erysipelotrichi_ (Supplementary Fig. 4). In Supplementary Data 4, the taxonomic classification of each feature id is documented, along with the confidence value.
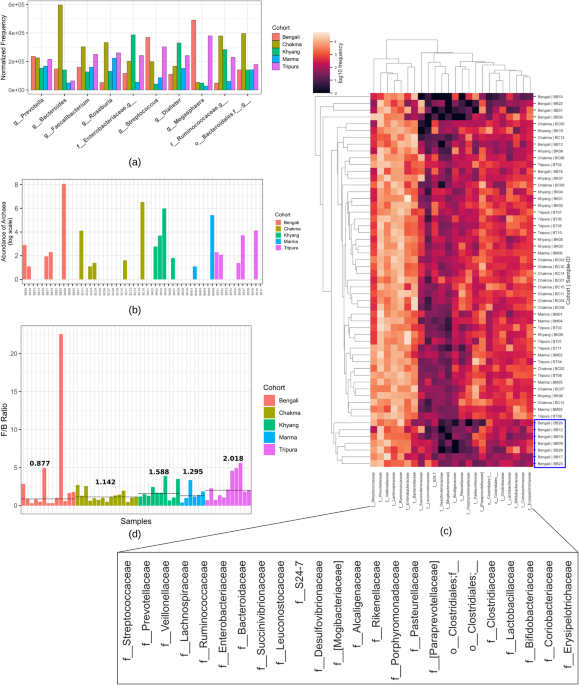
The normalized abundance of the top ten genera across different cohorts is represented in cpm (counts per million) (Fig. 1a). The _Prevotella_ genus abundance was relatively similar in all
cohorts. Nevertheless, the prevalence of the _Bacteroides_ genus was drastically higher in the Chakma population and very low in Marma and Tripura samples. Moreover, Chakma tribal group also
contained _Faecalibacterium_, _Roseburia_, and uncharacterized genera from the _Ruminococcaceae_ family and _Bacteroidales_ order in a relatively higher amount. On the other hand,
_Streptococcus_ and _Megasphaera_ genera were highly abundant in the Bengali and Tripura populations and the Marma cohort contained these genera in relatively lower quantities. The Khyang
group contained the genus _Dialister_ and uncharacterized genera from _Enterobacteriaceae_ and _Ruminococcaceae_ families. About 0.05% amplicon sequence variants were classified as archaea
of the _Methanobacteriaceae_ family, of which only 0.1% were from the _Methanosphaera_ genus, and the rest of the features belonged to the _Methanobrevibacter_ genus (Fig. 1b). Twenty-four
families of features were found as core features and the frequency heatmap of these features revealed that there were two main clusters, one with higher frequency and another with lower
frequency (Fig. 1c). The _Streptococcaceae_, _Prevotellaceae_, _Veillonellaceae_, _Lachnospiraceae_, _Ruminococcaceae_, _Enterobacteriaceae_, and _Bacteroidaceae_ families were in the higher
frequency cluster. Most of the samples from the Bengali population clustered together and contained these higher frequency microbial families in relatively lower amounts. The median value
of the _Firmicutes_ to _Bacteroidetes_ ratio was highest (2.018) in the case of Tripura samples, while the Bengali population had the lowest (0.877) median ratio (Fig. 1d). All other cohorts
(Chakma, Khyang, Tripura) had a median _Firmicutes_ to _Bacteroidetes_ ratio of more than one. THE BENGALI POPULATION SHOWED INFERIOR MICROBIOME RICHNESS THAN OTHER ETHNIC COHORTS Alpha
diversity depicts the diversity within the sample. Alpha diversity is measured via Shannon diversity, Observed features, Faith pd and Pielou evenness parameters. Shannon diversity calculates
both the number of species in a community and their relative abundance. The median of Shannon diversity was relatively lower in the Bengali group compared to others (Fig. 2). For ethnicity
based cohorts, Kruskal-Wallis test for all groups had a _p-_value of 0.0033 (Supplementary Data 5). Observed features counted the number of distinct features present in the cohorts (Fig. 2).
Chakma, Marma, Khyang and Tripura populations had higher species richness than the Bengalis (_p-_value = 0.0003) (Supplementary Data 6). Faith pd incorporated information of the
evolutionary relationships between different bacterial species. Bengali samples had lower Fatih _p_-values than indigenous populations (Fig. 3). However, the difference in Faith pd score was
statistically insignificant (_p_-value = 0.3816) (Supplementary Data 7). Pielou evenness measured how evenly different species’ abundances were distributed within a community (Fig. 3).
There was no significant difference in Pielou evenness profile between indigenous populations (_p-_value = 0.0631) (Supplementary Data 8). PRESENCE AND ABSENCE OF VARIOUS SPECIES MADE
BENGALI MICROBIOME DISTINCT The PCoA plot revealed that the Bengali cohort was distinct from others in terms of Jaccard distance based on ethnicity (Fig. 4b) Jaccard distance is a binary
distance matrix that only considers whether a taxon is present or not. In other metrics, such as Bray–Curtis, weighted and unweighted UniFrac distances calculate presence and relative
abundances of microbial taxa but no separated clusters were observed there (Figs. 4a, c, and d). _ALISTIPES_ GENUS IS DIFFERENTIALLY ABUNDANT IN CHAKMA POPULATION ANCOM analysis revealed the
microbial community compositions between the various Bangladeshi ethnic groups. It used a log-ratio test to compare the relative abundance using non-parametric methods. Here, only the
Bengali vs. Non-Bengali and the Chakma vs. Non-Chakma comparisons demonstrated differentially abundant taxa between the groups. In terms of ANCOM, the other groups (Marma, Khyang, and
Tripura) did not reveal any differentially abundant bacteria. In the Bengali group, an unclassified bacteria showed a negative centered log ratio (clr) with higher W score (Fig. 5a).
Whereas, the Chakma population depicted abundance for _Alistipes_ and _Odoribacter_ (Fig. 5b). Another tool, LEfSe implemented statistical methods to identify microbial features that were
differentially abundant. LEfSe unveiled differentially abundant genus _Paraprevotella_, _Lactonifactor, Barnesiella, Bacteroides_, _Alistipes_, and _Ruminococcus_ within the Chakma
population (Fig. 5c). The results from ANCOM and LEfSe analysis complemented each other as they both unveiled that the genus _Alistipes_ is differentially abundant in the Chakma population.
BENGALI GUT MICROBIOME CONTRIBUTED TO MARKEDLY DIVERSE BIOLOGICAL PATHWAYS Relationships between microbiome composition and its effect on different biological functions were also explored in
this study. Among all metadata categories, only the Bengali, Chakma, and Marma groups showed differential enrichment of function based on their gut microbiome composition (Fig. 6). All
differentially abundant functions along with related contributing taxa and BH (Benjamini and Hochberg) FDR-adjusted _p_-values are tabulated in Supplementary Data 9. Thirty-three pathways
were differentially abundant in Bengali samples. Several of them were highly enriched in Bengali samples such as peptidases, Ribosomes, Purine, and Pyrimidine metabolism (Fig. 6a). The genus
_Bacteroides_, _Blautia_, _Collinsella_, _Coprococcus_, _Dorea_, _Parabacteroides_, SMB53, and _Slackia_ were the top significantly contributing taxa in the enrichment of these functions.
Chakma samples seemed to be enriched with histidine metabolism, arginine biosynthesis, and transcription machinery (Fig. 6b). But, in the case of the Marma samples histidine metabolism,
arginine biosynthesis functions were in relatively lower abundance than other Non-Marma samples, and transcription machinery function was in higher frequency (Fig. 6c). For both categories,
the genus _Bacteroides_, _Blautia_, and _Parabacteroides_ were the most contributing taxa for function abundance. Detailed statistics for significant pathways and taxa contributions for each
pathway are documented in Supplementary Data 10. BANGLADESHI GUT MICROBIOME HAS A DISTINCT COMPOSITION COMPARED TO TROPICAL AND SUBTROPICAL POPULATIONS When compared with the gut microbiome
of several other countries, the Bangladeshi samples formed a vividly distinct cluster based on bacterial composition and abundance (Fig. 7a, b). Cluster for Bray–Curtis and Jaccard distance
indicates that bacterial composition and their abundances are unique for Bangladeshi populations. Their species richness was higher than other countries (Fig. 7f). The unweighted UniFrac,
which incorporates phylogenetic information, demonstrated that most of the samples from the Bangladeshi population clustered closer to the Indian samples (Fig. 7c). All numerical source data
of Fig. 7 can be found in Supplementary Data 11a–h. Furthermore, the Unweighted Pair Group Method with Arithmetic Mean based tree showed that most of the indigenous samples of Bangladesh
have clustered with different Indian indigenous groups while the Bangladeshi samples as a whole were scattered across different phylogenetic clades (Fig. 8). DISCUSSION The human gut
microbiome is an integral part of human growth and development and it is highly influenced by ethnicity14,46. Diverse ethnic populations reside in Bangladesh. The majority of the population
is Bengali whereas large indigenous communities such as the Chakma, Marma, Tripura, and Khyang live in Chittagong Hill Tracts region of the country. The eating habits of these communities
substantially differ from Bengali people. Their gut microbiomes, which may have an impact on important public health issues, can be influenced by their genetic make-up, geographic location,
and lifestyle. In this study, we conducted a pilot survey to shed light of these areas. Here, we have performed a 16 s rRNA gene amplicon sequencing to identify the gut microbiome
composition. To execute this, we have collected fecal samples from Bengali, Chakma, Marma, Khyang, and Tripura populations. After collecting the microbial DNA from fecal samples, parts of
the 16 S rRNA genes of the bacterial species were amplified. The amplified products were then sequenced. The sequence data was then processed and analyzed to draw interpretations. After all
the analysis, we found that at the phylum level, Bengali population has lower _Firmicutes_ to _Bacteroidetes_ ratio compared to indigenous groups. Among _Firmicutes_ phylum, _Dialister_ and
_Faecalibacterium_ are highly abundant in Bangladeshi microbiomes (Fig. 1a). On the other hand, _Prevotella_, a member of the _Bacteroidetes_ phylum, was distributed evenly across each
cohort. People from the Indian subcontinent usually have _Prevotella_ abundance in their gut47. Here, we have explored the alpha and beta diversity of the gut microbiome. Alpha diversity
depicted within-group diversity and beta diversity showed different diversity ratios between the groups. The alpha diversity of the Bengali population was lower compared to others according
to the Shannon diversity and species richness (Observed features) (Fig. 2). The overall species evenness was enriched for indigenous populations (Fig. 3). Based on the phylogenetic diversity
of the gut microbes, the Bengali and the people of the Chittagong Hill Tracts were found not to be very distant. However, our findings indicate that the overall gut microbial population and
abundance is unique for Bangladesh when compared with international datasets. However, most of the indigenous samples from Bangladesh (Chakma, Marma, Khyang, Tripura) clustered separately
with a common origin with the Indian indigenous population whereas Bengali samples were spread across various clades (Fig. 8). Some bacterial species were found to be abundant in indigenous
groups that were not enriched in Bengali samples or vice versa. To identify this differential abundance, we employed several statistical approaches using LEfSe and ANCOM. Both of these tools
identified an unclassified bacteria, which can be studied further, that is significantly abundant in Bengali samples whereas Chakma samples had enriched levels of _Alistipes_ species.
_Alistipes_ is a newly discovered genus under _Bacteroidetes_ phylum. The gut microbial pathways play a critical role in health and diseases48. In Bengali samples DNA repair system,
pyrimidine and purine metabolism, lipopolysaccharide (LPS) biosynthesis, peptidases, ribosome, etc functions were enriched. On the other hand, Chakma samples were enriched with histidine
metabolism and arginine biosynthesis (Fig. 6b) whereas in the Marma group abundance of these pathways were lower (Fig. 6c). Furthermore, Bangladeshi gut microbes possessed a distinctive
diversity profile. Further research is needed to investigate the specific differences in responses and underlying mechanisms associated with distinct gut microbiota profiles. LIMITATIONS OF
THE STUDY The primary limitation of the study is small sample size, which exhibits significant variability among individuals sampled. With a sample size of _n_ = 55 divided into five ethnic
groups, two genders, and three large age groups (20–40, 40–60, and 60–80 years), the representativeness for each combination is low. The gut microbiome (GM) is well-known to be highly
influenced by factors such as sex, age, current health status and so on. In our study, the representativeness of each of these groups is limited. A higher sample size would certainly enhance
the credibility of the differences between these groups and also help draw more meaningful conclusions. However, the primary goal of our study was to explore and characterize the gut
microbiome of Bangladeshi population for the first time and thus establish the baseline data for further gut microbiome research in Bangladesh. During the course of the study, as a secondary
goal, we also wanted to see if there is any difference in the gut microbiome of the various ethnicities. Since our main priority was to characterize the Bangladeshi population as a whole,
the difference between various subgroups based on age, sex, and health status was given less importance. We want to reiterate that the number of participants in the current study was not
sufficient to provide a comprehensive representation of the overall gut microbial diversity within neither the Bangladeshi population as a whole nor the ethnicities studied (Bengali, Chakma,
Marma, Khyang, and Tripura). As a result, the findings from this study may lack statistical power and robustness, leading to potential biases and limited generalizability of the results.
Moreover, the small sample size may hinder the identification of subtle microbial variations that could be crucial in understanding complex interactions between the gut microbiota and
various health conditions. Consequently, caution must be exercised when interpreting the results of this gut microbiome study due to its limited sample size. Further research with larger
cohorts is absolutely necessary to draw more definitive and reliable conclusions about the role of gut microbiota in the Bangladeshi population. In conclusion, the current study indicates
that the indigenous gut microbiome was more diverse and distinct from the Bengali population. Our study will help establish the baseline data for the gut microbiome of the Bangladeshi
population. DATA AVAILABILITY All newly generated 16rRNA amplicon sequencing data used in this study can be freely accessed via NCBI BioProject number PRJNA876782. Source data have been
included in the supplementary data files. All other data are available from the corresponding author on reasonable request. CODE AVAILABILITY Code can be accessed from the corresponding
author on reasonable request. Versions of software used to process the current dataset: QIIME2 (version 2021.4.0). REFERENCES * Bull, M. J. & Plummer, N. T. Part 1: The human gut
microbiome in health and disease. _Integr. Med. Clin. J._ 13, 17–22 (2014). Google Scholar * Chassaing, B., Kumar, M., Baker, M. T., Singh, V. & Vijay-Kumar, M. Mammalian gut immunity.
_Biomed. J._ 37, 246–258 (2014). Article PubMed Google Scholar * Cani, P. D. Human gut microbiome: hopes, threats and promises. _Gut_ 67, 1716–1725 (2018). Article CAS PubMed Google
Scholar * Fan, Y. & Pedersen, O. Gut microbiota in human metabolic health and disease. _Nat. Rev. Microbiol._ 19, 55–71 (2021). Article CAS PubMed Google Scholar * Gupta, V. K.,
Paul, S. & Dutta, C. Geography, ethnicity or subsistence-specific variations in human microbiome composition and diversity. _Front. Microbiol._ 8, 1162 (2017). Article PubMed PubMed
Central Google Scholar * Adeyanju, O. et al. Common NOD2 risk variants in African Americans with Crohn’s disease are due exclusively to recent caucasian admixture. _Inflamm. Bowel Dis._
18, 2357–2359 (2012). Article PubMed Google Scholar * Lauro, M. L., Burch, J. M. & Grimes, C. L. The effect of NOD2 on the microbiota in Crohn’s disease. _Curr. Opin. Biotechnol._ 40,
97–102 (2016). Article CAS PubMed PubMed Central Google Scholar * Charati, H. et al. The evolutionary genetics of lactase persistence in seven ethnic groups across the Iranian plateau.
_Hum. Genomics_ 13, 7 (2019). Article CAS PubMed PubMed Central Google Scholar * Hall, A. B., Tolonen, A. C. & Xavier, R. J. Human genetic variation and the gut microbiome in
disease. _Nat. Rev. Genet._ 18, 690–699 (2017). Article CAS PubMed Google Scholar * Kato, K. et al. Association between functional lactase variants and a high abundance of
bifidobacterium in the gut of healthy Japanese people. _PloS One_ 13, e0206189 (2018). Article PubMed PubMed Central Google Scholar * Huang, T., Shu, Y. & Cai, Y.-D. Genetic
differences among ethnic groups. _BMC Genomics_ 16, 1093 (2015). Article PubMed PubMed Central Google Scholar * Bennett, G., Bardon, L. A. & Gibney, E. R. A comparison of dietary
patterns and factors influencing food choice among ethnic groups living in one locality: a systematic review. _Nutrients_ 14, 941 (2022). Article PubMed PubMed Central Google Scholar *
Manica, A., Prugnolle, F. & Balloux, F. Geography is a better determinant of human genetic differentiation than ethnicity. _Hum. Genet._ 118, 366–371 (2005). Article PubMed PubMed
Central Google Scholar * Dwiyanto, J. et al. Ethnicity influences the gut microbiota of individuals sharing a geographical location: a cross-sectional study from a middle-income country.
_Sci. Rep._ 11, 2618 (2021). Article CAS PubMed PubMed Central Google Scholar * Dehingia, M. et al. Gut bacterial diversity of the tribes of India and comparison with the worldwide
data. _Sci. Rep._ 5, 18563 (2015). Article CAS PubMed PubMed Central Google Scholar * Hazarika, P., Chattopadhyay, I., Umpo, M., Choudhury, Y. & Sharma, I. Elucidating the gut
microbiome alterations of tribal community of Arunachal Pradesh: perspectives on their lifestyle or food habits. _Sci. Rep._ 12, 18296 (2022). Article CAS PubMed PubMed Central Google
Scholar * Alam, S. M. N. & Naser, M. N. Chapter 2—Role of traditional foods of Bangladesh in reaching-out of nutrition. In _Nutritional and Health Aspects of Food in South Asian
Countries_ (eds. Prakash, J., Waisundara, V. & Prakash, V.) 217–235 (Academic Press, 2020) https://doi.org/10.1016/B978-0-12-820011-7.00025-3. * Haque, M. M. Dietary practice among
mainstream Bengali population and ethnic communities in Bangladesh. _Arch. Nutr. Food Sci_. 1, 5–7 (2020). * Chakraborty, R. et al. Gene differentiation among ten endogamous groups of West
Bengal, India. _Am. J. Phys. Anthropol._ 71, 295–309 (1986). Article CAS PubMed Google Scholar * Saha, N. Blood genetic markers in Bengali muslims of Bangladesh. _Hum. Hered._ 37, 86–93
(1987). Article CAS PubMed Google Scholar * Hasan, M. M. et al. Phylogenetic and forensic studies of the Bangladeshi population using next-generation powerPlex® Y23 STR marker system.
_Int. J. Leg. Med._ 130, 1493–1495 (2016). Article Google Scholar * Wen, B. et al. Analyses of genetic structure of Tibeto-Burman populations reveals sex-biased admixture in Southern
Tibeto-Burmans. _Am. J. Hum. Genet._ 74, 856–865 (2004). Article CAS PubMed PubMed Central Google Scholar * Gayden, T. et al. Genetic insights into the origins of Tibeto-Burman
populations in the himalayas. _J. Hum. Genet._ 54, 216–223 (2009). Article CAS PubMed Google Scholar * Gazi, N. N. et al. Genetic structure of Tibeto-Burman populations of Bangladesh:
evaluating the gene flow along the sides of Bay-of-Bengal. _PLoS One_ 8, e75064 (2013). Article CAS PubMed PubMed Central Google Scholar * Bolyen, E. et al. Reproducible, interactive,
scalable and extensible microbiome data science using QIIME 2. _Nat. Biotechnol._ 37, 852–857 (2019). Article CAS PubMed PubMed Central Google Scholar * Callahan, B. J. et al. DADA2:
high-resolution sample inference from Illumina amplicon data. _Nat. Methods_ 13, 581–583 (2016). Article CAS PubMed PubMed Central Google Scholar * Janssen, S. et al. Phylogenetic
placement of exact amplicon sequences improves associations with clinical information. _mSystems_ 3, e00021–18 (2018). Article CAS PubMed PubMed Central Google Scholar * Matsen, F. A.,
Hoffman, N. G., Gallagher, A. & Stamatakis, A. A format for phylogenetic placements. _PLoS One_7, e31009 (2012). Article CAS PubMed PubMed Central Google Scholar * Weiss, S. et al.
Normalization and microbial differential abundance strategies depend upon data characteristics. _Microbiome_ 5, 27 (2017). Article PubMed PubMed Central Google Scholar * Halko, N.,
Martinsson, P.-G., Shkolnisky, Y. & Tygert, M. An algorithm for the principal component analysis of large data sets. _SIAM J. Sci. Comput._ 33, 2580–2594 (2011). Article Google Scholar
* Vázquez-Baeza, Y., Pirrung, M., Gonzalez, A. & Knight, R. EMPeror: a tool for visualizing high-throughput microbial community data. _GigaScience_ 2, 16 (2013). Article PubMed
PubMed Central Google Scholar * Kruskal, W. H. & Wallis, W. A. Use of ranks in one-criterion variance analysis. _J. Am. Stat. Assoc._ 47, 583–621 (1952). Article Google Scholar *
McKinney, W. Data Structures for Statistical Computing in Python. _Proc. 9th Python Sci. Conf._ 56–61 (2010). * Bokulich, N. A. et al. q2-longitudinal: longitudinal and paired-sample
analyses of microbiome data. _mSystems_ 3, e00219–18 (2018). Article PubMed PubMed Central Google Scholar * Anderson, M. J. A new method for non-parametric multivariate analysis of
variance. _Austral Ecol._ 26, 32–46 (2001). Google Scholar * Mandal, S. et al. Analysis of composition of microbiomes: a novel method for studying microbial composition. _Microb. Ecol.
Health Dis._ 26, 27663 (2015). PubMed Google Scholar * Lin, H. & Peddada, S. D. Analysis of microbial compositions: a review of normalization and differential abundance analysis. _NPJ
Biofilms. Microbiomes._ 6, 1–13 (2020). Article Google Scholar * Segata, N. et al. Metagenomic biomarker discovery and explanation. _Genome Biol._ 12, R60 (2011). Article PubMed PubMed
Central Google Scholar * McNally, C. P., Eng, A., Noecker, C., Gagne-Maynard, W. C. & Borenstein, E. BURRITO: An interactive multi-omic tool for visualizing taxa–function relationships
in microbiome data. _Front. Microbiol._ 9, 365 (2018). Article PubMed PubMed Central Google Scholar * Langille, M. G. I. et al. Predictive functional profiling of microbial communities
using 16S rRNA marker gene sequences. _Nat. Biotechnol._ 31, 814–821 (2013). Article CAS PubMed PubMed Central Google Scholar * Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M.
& Tanabe, M. KEGG as a reference resource for gene and protein annotation. _Nucleic Acids Res._ 44, D457–D462 (2016). Article CAS PubMed Google Scholar * DeSantis, T. Z. et al.
Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. _Appl. Environ. Microbiol._ 72, 5069–5072 (2006). Article CAS PubMed PubMed Central Google Scholar
* Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: a versatile open source tool for metagenomics. _PeerJ_ 4, e2584 (2016). Article PubMed PubMed Central Google
Scholar * Amir, A. et al. Deblur rapidly resolves single-nucleotide community sequence patterns. _mSystems_ 2, e00191–16 (2017). Article PubMed PubMed Central Google Scholar * Letunic,
I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. _Nucleic Acids Res._ 44, W242–W245 (2016). Article CAS
PubMed PubMed Central Google Scholar * Guinane, C. M. & Cotter, P. D. Role of the gut microbiota in health and chronic gastrointestinal disease: understanding a hidden metabolic
organ. _Ther. Adv. Gastroenterol._ 6, 295–308 (2013). Article Google Scholar * Prasoodanan, P. K. V. et al. Western and non-western gut microbiomes reveal new roles of prevotella in
carbohydrate metabolism and mouth–gut axis. _NPJ Biofilms. Microbiomes._ 7, 1–17 (2021). Article Google Scholar * Hou, K. et al. Microbiota in health and diseases. _Signal Transduct.
Target. Ther._ 7, 1–28 (2022). Google Scholar Download references ACKNOWLEDGEMENTS This study was funded under the “Establishment of National Gene Bank Project”, National Institute of
Biotechnology (NIB), Ministry of Science and Technology, Government of the People’s Republic of Bangladesh. We would like to thank Md. Amjad Hossain, Ritaren Chakma, Tina Tripura, Barshan
Chakma, and Suborna Dash for their assistance with sample and metadata collection. AUTHOR INFORMATION Author notes * These authors contributed equally: Ishtiaque Ahammad, Arittra
Bhattacharjee, Zeshan Mahmud Chowdhury, Anisur Rahman, and Mohammad Uzzal Hossain. AUTHORS AND AFFILIATIONS * Bioinformatics Division, National Institute of Biotechnology, Ganakbari,
Ashulia, Savar, Dhaka, 1349, Bangladesh Ishtiaque Ahammad, Arittra Bhattacharjee, Zeshan Mahmud Chowdhury, Anisur Rahman & Mohammad Uzzal Hossain * Rangamati Medical College, Hospital
Road, Rangamati-4500, Rangamati, Bangladesh Gourab Dewan & Shiny Talukder * Molecular Biotechnology Division, National Institute of Biotechnology, Ganakbari, Ashulia, Savar, Dhaka, 1349,
Bangladesh Keshob Chandra Das & Md Salimullah * Department of Biochemistry and Microbiology, North South University, Bashundhara, Dhaka, 1229, Bangladesh Chaman Ara Keya Authors *
Ishtiaque Ahammad View author publications You can also search for this author inPubMed Google Scholar * Arittra Bhattacharjee View author publications You can also search for this author
inPubMed Google Scholar * Zeshan Mahmud Chowdhury View author publications You can also search for this author inPubMed Google Scholar * Anisur Rahman View author publications You can also
search for this author inPubMed Google Scholar * Mohammad Uzzal Hossain View author publications You can also search for this author inPubMed Google Scholar * Gourab Dewan View author
publications You can also search for this author inPubMed Google Scholar * Shiny Talukder View author publications You can also search for this author inPubMed Google Scholar * Keshob
Chandra Das View author publications You can also search for this author inPubMed Google Scholar * Chaman Ara Keya View author publications You can also search for this author inPubMed
Google Scholar * Md Salimullah View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Conceptualization: Mohammad Uzzal Hossain, Keshob Chandra
Das, Chaman Ara Keya, Md Salimullah, Analysis: Ishtiaque Ahammad, Arittra Bhattacharjee, Zeshan Mahmud Chowdhury, Anisur Rahman, Resources: Gourab Dewan, Shiny Talukder, Keshob Chandra Das,
Md Salimullah. Writing—Original Draft: Ishtiaque Ahammad, Arittra Bhattacharjee, Zeshan Mahmud Chowdhury, Anisur Rahman, Writing—Review & Editing: Ishtiaque Ahammad, Arittra
Bhattacharjee, Zeshan Mahmud Chowdhury, Anisur Rahman, Mohammad Uzzal Hossain, Chaman Ara Keya, Md Salimullah. Supervision: Keshob Chandra Das, Chaman Ara Keya, Md Salimullah. Funding
acquisition: Md Salimullah. CORRESPONDING AUTHOR Correspondence to Md Salimullah. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW
INFORMATION _Communications Biology_ thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Kevin Theis and Tobias Goris. ADDITIONAL
INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY
INFORMATION DESCRIPTION OF SUPPLEMENTARY MATERIALS SUPPLEMENTARY DATA 1-11 REPORTING SUMMARY RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution
4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and
the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s
Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not
permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ahammad, I., Bhattacharjee, A., Chowdhury, Z.M. _et al._ Gut microbiome composition
reveals the distinctiveness between the Bengali people and the Indigenous ethnicities in Bangladesh. _Commun Biol_ 7, 500 (2024). https://doi.org/10.1038/s42003-024-06191-9 Download
citation * Received: 30 September 2022 * Accepted: 15 April 2024 * Published: 25 April 2024 * DOI: https://doi.org/10.1038/s42003-024-06191-9 SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative