Play all audios:
ABSTRACT Access to carbocyclic C-nucleosides (CC-Ns) is currently restricted. The few methods available to make CC-Ns suffer from long syntheses and poor modularity, hindering the
examination of potentially important chemical space. Here we report an approach to CC-Ns which uses an asymmetric Suzuki-Miyaura type reaction as the key C-C bond forming step. After
coupling the densely functionalized racemic bicyclic allyl chloride and heterocyclic boronic acids, the trisubstituted cyclopentenyl core is elaborated to RNA analogues via a
hydroborylation-homologation-oxidation sequence. We demonstrate that the approach can be used to produce a variety of enantiomerically enriched CC-Ns, including a carbocyclic derivative of
Showdomycin. SIMILAR CONTENT BEING VIEWED BY OTHERS AN ENANTIOSELECTIVE AND MODULAR PLATFORM FOR C4ʹ-MODIFIED NUCLEOSIDE ANALOGUE SYNTHESIS ENABLED BY INTRAMOLECULAR _TRANS_-ACETALIZATIONS
Article Open access 17 August 2024 SYNTHESIS OF (+)-RIBOSTAMYCIN BY CATALYTIC, ENANTIOSELECTIVE HYDROAMINATION OF BENZENE Article 26 May 2022 AN ENGINEERED ALDOLASE ENABLES THE BIOCATALYTIC
SYNTHESIS OF 2′-FUNCTIONALIZED NUCLEOSIDE ANALOGUES Article Open access 05 November 2024 INTRODUCTION Nucleosides and their analogs are widely studied antiviral and anticancer agents1,2,3.
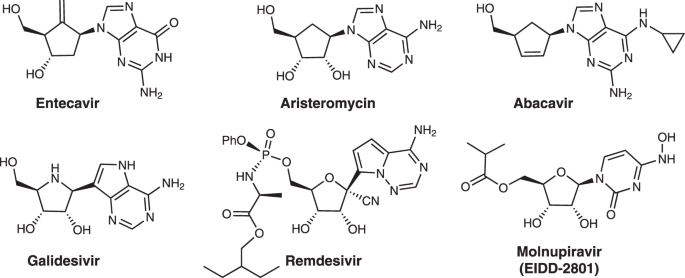
Carbocyclic nucleoside analogs show a broad spectrum of antiviral activity and are known to exhibit enhanced flexibility, lipophilicity and metabolic stability4,5,6. For instance, Entecavir
is clinically used to treat hepatitis B;7,8 Aristeromycin and Abacavir are active against HIV9,10. Additionally, nucleoside analog Galidesivir is a potent inhibitor of Ebola and Zika
viruses11,12. There has been much recent interest in using nucleoside in the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-COV-2); Remdesivir13,14 and Molnupiravir15,16
demonstrate activity against SARS-COV-2 and have been approved for high-risk patients with severe symptoms (Fig. 1). The synthesis and bioactivities of N-nucleosides, C-nucleosides, and
carbocyclic N-nucleosides (Fig. 2A) are extensively studied17,18,19,20,21. Nucleophilic addition to carbohydrate-derived oxocarbeniums is widely used to access N- and C-nucleosides20.
Strategies for the synthesis of carbocyclic nucleosides revolve around construction of the carbocycle and the mode of nucleobase addition18, for example Pd-catalyzed allylic amination
followed by late-stage construction of the nucleobase allows access to carbocyclic N-nucleoside derivatives19. Carbocyclic C-nucleosides (CC-Ns) are rare, and this is almost certainly due to
difficulties in their synthesis (Fig. 2A)22,23,24,25. Few methods for the synthesis of CC-Ns are known, and these methods tend to be long and non-modular. For instance, the synthesis of a
potential Alzheimer’s disease drug by Merck is reported in 17 steps from D-Ribose (Fig. 2B)23. Here, we report a cross-coupling approach to CC-Ns which uses an asymmetric Suzuki-Miyaura type
reaction as the key C–C bond forming step followed by hydroborylation-homologation-oxidation strategy to access the RNA analogs. STRATEGY Using a cross-coupling strategy to solve the CC-N
synthesis problem may be ideal as it could provide a general and modular route to access many derivatives. With the development of a catalytic asymmetric cross-coupling strategy in mind, we
envisaged that coupling heteroaryl boronic acids 2 with a suitably functionalized halide such as racemic 1 could provide enantioenriched 3. Allyl halide 1 is racemic, but is
pseudo-symmetrical about the allyl halide unit, which would allow a successful desymmetrizing reaction to set the absolute and relative stereochemistry of the three contiguous stereocenters
on the cyclopentene core while simultaneously forming the key C–C bond. These cyclopentenes may be suitable precursors to CC-Ns (Fig. 2C) if modification of 3 by alkene functionalization
could produce RNA analogs. Our approach relies on a catalytic asymmetric Suzuki–Miyaura coupling (SMC) reaction to make the C–C bond26,27,28,29,30,31. However, CC-Ns that resemble other
nucleoside derivatives (c.f. Fig. 1 for example), and that we naively suspect would be more biologically interesting, would feature rather elaborate nucleobase units capable of
H-bonding/acid-base interactions. While we felt that relatively simple boronic acids (Fig. 3A) may be suitable for the asymmetric cross-coupling strategy to CC-Ns, the use of these coupling
partners would give relatively simple (although almost unknown) products. Accessing more typical nucleobases would require using functionalized heterocyclic boronic acids (Fig. 3B) that are
significantly more complex than have been used in comparable asymmetric transformations. At the outset of this project it was not at all clear from the literature if appropriately complex
boronic acid derivatives would undergo SCM reactions, what impact they would have on enantioselectivity over using benzene-derived boronic acids, or if they would even be stable32,33. The
incompatibility of complex heterocycles with transition-metal catalyzed reactions (particularly asymmetric transformations) remains a major challenge for a number of reasons, including
catalyst poisoning, undesired reactivity patterns, and proto-demetallation (or deborylation)34,35,36,37,38,39. The use of complex boronic acids early in the sequence would also require that
the installed nucleobase units were compatible with the chemistry used to convert the cyclopentene core 3 to CC-Ns. Another strategy to potentially access CC-Ns involves using relatively
less complex boronic acids (Fig. 3C) which are designed so that late-stage modifications would reveal or allow construction of heterobase units. While this approach involves more steps than
Strategy B, it has significant advantages in terms of how easy it would be to perform each step in the sequence. This would likely make Strategy C more suitable for the production of
libraries of related compounds, as having to heavily optimize each individual reaction for each target molecule would be unwelcome. Despite this approach’s relative length, it would still
compare favorably to known approaches to CC-Ns involving long, non-modular sequences starting from the chiral pool (c.f. Fig. 2B). RESULTS AND DISCUSSION We first targeted relatively simple
CC-Ns derived from the addition of six-membered rings; benzenes, pyridines and pyrimidines. Previous reports of asymmetric SMC conditions, extensively explored variables such as solvent and
ligand28, and using these conditions addition of PhB(OH)2 to racemic bicycle 1 gave 3A as a single diastereoisomer in 91% yield with very high enantioselectivity (95% ee, Table 1, Entry 1).
The absolute configuration was assigned by analogy to previous work28. The efficiency and selectivity of this reaction, combined with previous observations that a wide variety of
benzene-derived nucleophiles are generally well tolerated in related reactions, encouraged us not to dwell on simple all carbon nucleophiles and to instead explore nitrogen containing
six-membered rings. When addition of pyridines and pyrimidines was attempted no desired product was observed (Table 1, entries 2–4). We chose to next look at addition of 2-halo-pyridyl
boronic acids, which have two advantages over simple pyridines, (i) that the presence of the halogen at the 2-position moderates the Lewis-basicity of the heterocycle, and (ii) that the
halogen can either be used as a handle in further functionalization reactions or simply removed to reveal the parent pyridine32,34,35,36,37,38,39. Addition of 2-chloro-5-pyridyl boronic acid
2E to allyl chloride 1 provided ~60% conversion to the desired product, and 3B was isolated in 50% yield and 95% ee (Table 1, Entry 5). The reaction could be improved by adding water as
co-solvent which led to the full consumption of starting material and 3B could be isolated as a single diastereoisomer in 70% yield and 94% ee (Table 1, Entry 6). Similar reactivity was
observed with 2-fluoro-5-pyridine boronic acid 2 F providing 3C in 90% yield and 92% ee (Table 1, Entry 7). REACTION SCOPE A variety of 2-halo-pyridyl boronic acid isomers were then used to
give products 3B–G as shown in Fig. 4. The reaction was conducted at 50 °C for 2-halo-pyridyl-4-boronic acids, and 3D and 3E were isolated in 85% yield and 90% ee, and 72% yield and 93% ee
respectively. 2-chloro-pyridyl-6-boronic acid provided 3 F in 80% yield and 96% ee. Ortho-substitution of these pyridines such as 2-chloro-pyridyl-3-boronic acid,
2,6-difluoropyridyl-3-boronic acid and 2,6-difluoropyridyl-3-boronic acid gave very poor results, but we did find we could use 2-fluoro-pyridyl-3-boronic acid to obtain 3 G in 32% yield and
85% ee. The use of 5-membered _O_- and _N_-containing heterocyclic nucleophiles was then explored, with 2- and 3-furan boronic acids providing 3 H (80%, 97% ee) and 3 L (81%, 97% ee).
Pyrrole derived boronic acids similarly undergo highly efficient reactions to give highly enantioenriched (>94% ee) products, but 3J and 3K are a bit sensitive to decomposition and were
isolated in 55% and 65% yield, respectively. Encouraged by these results, we decided to explore more heavily functionalized 5- and 6-membered heterocycles. In the few examples we examined,
di-halo-substituted pyridines performed poorly in reactions at 60 °C, we are unsure why but by simply doing the reaction at room temperature 3L and 3M were found to give a single
diastereoisomer of product in >80% yield, and reasonable levels of enantioselectivity (80% ee). A 2-chloro-aminopyridine derived boronic acid, which features both hydrogen-bond donating
and accepting moieties, was also used and gave 3N in 79% yield (80% ee). Similarly, we were able to add 2-cyano pyrrol-5-boronic acid to give 3O (48%, 94% ee). We note that this 2-cyano
pyrrole moiety is important because it is used in the late-stage construction of the pyrrolo-triazine nucleobase found in Remdesivir, and nucleobase which is otherwise difficult to make
(Fig. 4)40. Boronic acids featuring more than one heteroatom are a further step away from benzene rings to more elaborate cross coupling partners that are unexplored in asymmetric catalysis
and have less well understood reactivity patterns in the context of asymmetric cross-coupling reactions; 2-chloro-pyrimidine-5-boronic acid was successfully employed and 3P was isolated in
70% yield (94% ee). The ability to run reactions on scale is tremendously important to any multi-step synthetic sequence and so we investigated the synthesis of 3H and 3K on a 5 mmol scale
(~1 g). We found these could repeatedly make these products with consistent yields and enantioselectivities, and so these reactions – with challenging nucleophiles – should allow them to be
used in the synthesis of complex products such as CC-Ns. SYNTHESIS OF CARBOCYCLIC C-NUCLEOSIDES In order to produce carbocyclic ribose analogs from 3 a 5’-hydroxymethyl group must be added
to the 4’ carbon in the ring (ribose numbering). From the carbocyclic alkene in 3 both the regiochemistry (attachment at the 4’ position) and stereochemistry (_cis_ or _trans_ relative to
the nucleobase) must be controlled with the _cis_ or β-stereochemistry desired as it mimics the stereochemistry most often found in natural nucleosides. After considerable exploratory work,
including examining metal catalyzed direct carbonylation and carboxylation, hydrozirconation followed by trapping and photochemical approaches, we were able to identify a useful 3-step
hydroborylation-homologation-oxidation sequence in order to add the hydroxymethyl group with complete stereo- and regio-chemical control in roughly 50% yield (Fig. 5) – however it does
involve the use of fairly reactive species and so it would not be expected to be compatible with all functional groups. The homologation first uses Wilkinson’s catalyst (2.5 mol%) with
pinacol borane to give boronic ester 4 as a single isomer. The presence of cesium carbonate is crucial for the suppression of competitive and undesired alkene reduction. Matteson
homologation using _n_-BuLi and CH2ICl was then used to convert the secondary boronic ester 4 to primary ester 5, and subsequent hydrogen peroxide oxidation furnished alcohol 6 as a key CC-N
precursor (Fig. 5). The route successfully furnished phenyl (6A), fluoropyridines (6C, 6E, 6L), furan (6H) and pyrrole (6J and 6K) derivatives in high yields. However, most chloropyridines
(except 6B) and the cyano-pyrrole decomposed during attempted hydroborylation. The hydroborylation-homologation-oxidation sequence was successfully performed on 4 mmol scales to give 6H and
6K. By performing a final acetonide deprotection under standard acidic conditions (Fig. 6), a series of phenyl, pyridine and furan derived CC-Ns (7A–7C, 7E, 7H, and 7K) was isolated in high
yields. Encouraged by the success in obtaining a collection of CC-Ns bearing simple nucleobase moieties we became interested in the problem of how to make more elaborate CC-Ns. We started
preparing some complex heterocyclic boronic acids and exploring their synthesis and compatibility with the SMC reaction, (for example see SI; See supplemantry data for (a) Attepmts toward
complex boronic acids. (b) Detailed synthetic schemes and procedures) Overall, we found that a wide selection of what we imagined to be suitably complex boronic acids (and corresponding
esters) were difficult to prepare and not very stable. In the absence of a specific target molecule that would provide the motivation to overcome stability problems we decided to focus our
efforts on nucleobase 9, featured in Remdesivir (Fig. 1). These pyrrolo-triazines contain 4 nitrogen atoms, and were additionally attractive because boronic ester 9D had been reported and
seemed easy to prepare25. Our first attempts at making suitable boronic acid derivatives were based on the above success with 2-halo-substitution, and we chose to examine the synthesis of 9A
and 9B. While a halogen-lithium exchange approach was unsuccessful, presumably due to incompatibility with nBuLi, we were able to make boronic esters using Ir-catalyzed C-H
borylation41,42,43. However, borylation to give 9A-C was complicated because mixture of regioisomers were obtained which proved difficult to separate. Eventually, we found that 9D and 9E
could be prepared as single isomers and these were examined in the asymmetric cross-coupling reaction. Repeated attempts to add 9D gave poor conversion, however when using 9E (Fig. 7) we
obtained 3Q in 40% yield and 93% ee when doubling the normal catalyst loading. The pyrrolo-triazine carbocycle 3Q underwent hydroborylation to 4Q, but attempts to homologate this boronic
ester to add the hydroxymethyl group were unsuccessful and gave decomposition products, almost certainly due to incompatibility of Matteson homologation using _n_-BuLi and CH2ICl with the
rich array of nitrogen atoms in 4Q. We were able to access carbocyclic triol 10 however by oxidation followed by trifloroacetic acid mediated global deprotection to give the TFA salt in 43%
yield over 3 steps (Fig. 7). Overall, the synthesis of complex boronic acids featuring nucleobases was not only difficult, but they were more often than not either inherently unstable or
incompatible with the relatively mild conditions of the asymmetric SMC reaction. Most of the complex boronic acids led to protodeborylation and it was difficult to see any obvious
correlation between heterocycle structure and boronic acid stability/reactivity. As the approach (Fig. 3: Strategy 2) of adding highly complex boronic acids proved to be difficult and would
likely require extensive optimization in cases where it was viable, and the addition of the 5’ hydroxyl methyl group also appeared challenging in the presence of complex heterocycles we
decided to pursue the alternative approach (Fig. 3: Strategy 3) of adding relatively simple boronic acids which could later be transformed into more elaborate units. We envisaged that the
oxidative cleavage of furan could provide carboxylic acid moiety primed for the construction of complex heterocycles. Alcohol 6H was first TBS-protected and then oxidative cleavage of furan
moiety was accomplished by the combination of ruthenium chloride (10 mol%) and sodium periodate. The resulting carboxylic acid 12 was isolated in 65% yield over 2 steps (Fig. 8). As a simple
proof of concept of this approach we chose to make the benzoimidazole derived carbocyclic C-nucleoside 13 which was accomplished via in situ generation of the corresponding acid chloride,
and addition of 1,2-diaminobenzene to form the amide. Acetic acid mediated cyclization and global deprotection gave CC-N 11 in 52% isolated yield from 12 (Fig. 8). We note that carboxylic
acids can readily be transformed into a whole range of structures and so it seems very likely that intermediate 1244 could also be used to access many other complex CC-Ns. Showdomycin is a
C-nucleoside with antiviral, antibacterial and antitumor properties22,45,46. We chose to produce a carbocyclic analog of Showdomycin in order to showcase our approach to enantiomerically
enriched CC-N derivatives of natural products with important biological activity (Fig. 9). After hydroborylation-homologation-oxidation sequence of 3K (94% ee) to 6K (Fig. 5, 62% yield over
three steps), the alcohol was subjected to TBS protection followed by pyrrole Boc-deprotection to give 14 (65% yield over 2 steps). Pyridinium chlorochromate (PCC) oxidation of the pyrrole
group followed by deprotection with trifluoroacetic acid gave carbocyclic Showdomycin analog 16 in 53% isolated yield over 2 steps (Fig. 9). CONCLUSIONS We present new synthetic strategies
for the synthesis of carbocyclic C-nucleosides. The approaches use a key asymmetric Suzuki-Miyaura-type coupling reaction followed by late-stage addition of the hydroxymethyl group to give
ribose analogs. Using relatively simple boronic acids nucleophiles Strategy A provides a route to simple CC-N’s featuring benzene derivatives or heterocycles featuring a single heteroatom in
the ‘nucleobase’ moiety. We identified two strategies (B and C, Fig. 3) which could produce more complex CC-Ns. While strategy B would provide direct access to complex CC-N’s by using
complex boronic acid derivatives, these nucleophiles are difficult to prepare, and if they are compatible with the sequence then it seems very likely that optimization of many of the
individual steps would be required to prepare each substrate (Fig. 7). We have demonstrated that strategy C is capable of producing CC-Ns featuring more complex nucleobases including the
carbocyclic analog of Showdomycin, a biologically active natural product. Strategy C involves the addition of relatively less complex boronic acids followed by late-stage modification, and
therefore requires more steps than strategy B but we would recommend this approach to anyone seeking to produce more complex CC-N’s in the future (although there may of course be shorter
alternative routes that can be devised to access a particular target); carboxylic acid 12 can obviously be converted into a wide range of structures and there are many relatively simple
heterocycles available which would be expected to be compatible with our sequence and could very likely be converted into complex CC-N targets. METHODS GENERAL PROCEDURE FOR ENANTIOSELECTIVE
SUZUKI–MIYAURA COUPLING [Rh(COD)OH]2 (4.6 mg, 0.010 mmol, 2.5 mol%) and (S)-Segphos (14.6 mg, 0.026 mmol, 6.0 mol%) were added to a 7 mL dram vial, sealed with a rubber septum under an
argon atmosphere, dissolved in THF (0.80 mL) and stirred at 60 °C. After 30 min, a solution (or suspension) of boronic acid (0.80 mmol, 2.0 equiv) and allylic chloride (62 µL, 0.40 mmol, 1.0
equiv) in THF (0.8 mL) and H2O (0.2 mL) was added via syringe and the flask was rinsed with THF (0.4 mL). Lastly, CsOH (50 wt% aq. solution, 70 µL, 0.40 mmol, 1.00 equiv) was added and the
resulting mixture was then stirred at indicted temperature for the period of time indicated. The mixture was then cooled to room temperature and diluted with Et2O (2 mL) before passing
through a plug of SiO2. The plug was washed with an additional 10 mL of Et2O and the solvents were removed in vacuo. Purification by flash chromatography afforded the desired product 3.
GENERAL PROCEDURE FOR SYNTHESIS OF 7A–L [Rh(PPh3)3Cl] (23.1 mg, 0.025 mmol, 2.5 mol%) and Cs2CO3 (325.8 mg, 1.0 mmol, 1 equiv) were added to a flame dried 25 mL round bottom flask, sealed
with a rubber septum under an argon atmosphere, dissolved in THF (0.5 mL) and stirred at room temperature. After 5 min, a solution (or suspension) of 3 (1 mmol, 1.0 equiv) in THF (3 mL) was
added via syringe and the flask was rinsed with THF (0.5 mL). Pinacolborane (0.29 mL, 2 mmol, 2.0 equiv) was added dropwise to the reaction mixture at 50 °C and stirred for 16 h at 50 °C.
The mixture was then cooled to room temperature and diluted with Et2O (5 mL) before passing through a plug of silica. The plug was washed with an additional 10 mL of Et2O and the solvents
were removed in vacuo. The crude product boronic acid pinacol ester 4 was charged to the next step without further purification. In a flamed dried flask under nitrogen, _n_-butyllithium (2.5
M in hexanes, 0.75 mL, 1.88 mmol, 2.5 equiv) was added dropwise to a solution of boronic acid pinacol ester 4 obtained above (0.75 mmol, 1.0 equiv) and iodochloromethane (165 µL, 2.25 mmol,
3 equiv) in THF (3.5 mL) at −78 °C. The reaction mixture was slowly allowed to warm to ambient temperature over 12 h. The mixture was diluted with Et2O (5 mL) and the organic layer was
washed with an aq. sat. solution of NH4Cl (2 × 3 mL) and dried over Na2SO4, filtered, and the solvent was removed in vacuo. The crude product boronic acid pinacol ester 5 was charged to the
next step. 30%wt. H2O2 (1 mL) was added to a solution of boronic acid pinacol ester 5 obtained above (0.75 mmol, 1.0 equiv) and 2 N aq. NaOH (1.5 ml) in THF (4 mL) at 0 °C. The reaction
mixture was slowly allowed to warm to ambient temperature. After 3 h, the mixture was diluted with Et2O (7 mL). The organic layer was separated and the aqueous layer was extracted with Et2O
(2 × 5 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and the solvent was removed in vacuo. Purification by flash chromatography afforded alcohol 6. A solution
of alcohol 6 (0.2 mmol) in AcOH (0.5 mL) and H2O (0.5 mL) was stirred at 50 °C for 16 h. The reaction mixture was concentrated under reduced pressure and purification by flash chromatography
afforded compound 7. DATA AVAILABILITY The authors declare that supporting information containing experimental procedures, compound synthesis and characterization, and supporting discussion
are available within the paper and its supplementary data files (Supplementary Methods and Supplementary Spectra). REFERENCES * Sofia, M. J. Nucleosides and Nucleotides for the Treatment of
Viral Diseases. In _Annual Reports in Medicinal Chemistry_. Vol. 49 Ch. 221, 221–247 (Elsevier, 2014). * Singh, S., Bhattarai, D., Veeraswamy, G., Choi, Y. & Lee, K. Nucleosides with
modified sugar ring: synthesis and biological activities. _Curr. Org. Chem._ 20, 856–897 (2016). Article CAS Google Scholar * Pruijssers, A. J. & Denison, M. R. Nucleoside analogues
for the treatment of coronavirus infections. _Curr. Opin. Virol._ 35, 57–62 (2019). Article PubMed PubMed Central CAS Google Scholar * Agrofoglio, L. A. & Challand, S. R. Biological
activity of carbocyclic nucleosides. In _Acyclic, Carbocyclic L-Nucleosides_. Ch. 5, 256–284 (Springer, 1998). * Chu, D. & Rao, J. Carbocyclic compounds and methods for treating
emerging diseases, including Influenza and Venezuela Equine Encephalitis virus. WO2008124157A1 (2008). * Wang, J., Rawal, R. K. & Chu, C. K. Recent Advances in Carbocyclic Nucleosides:
Synthesis and Biological Activity. In _Medicinal Chemistry of Nucleic Acids_. 1–100 (Wiley, 2011). * Zahler, R. & Slusarchyk, W. A. _Hydroxymethyl (methylenecyclopentyl) Purines And
Pyrimidines_. US5206244A (1993). * Dimou, E., Papadimitropoulos, V. & Hadziyannis, S. J. The role of entecavir in the treatment of chronic hepatitis B. _Ther. Clin. Risk Manag._ 3,
1077–1086 (2007). PubMed PubMed Central CAS Google Scholar * Hervey, P. S. & Perry, C. M. Abacavir: a review of its clinical potential in patients with HIV infection. _Drugs_ 60,
447–479 (2000). Article PubMed CAS Google Scholar * Yoon, J. S. et al. Design, synthesis, and anti-RNA virus activity of 6’-fluorinated-aristeromycin analogues. _J. Med. Chem._ 62,
6346–6362 (2019). Article PubMed PubMed Central CAS Google Scholar * Warren, T. K. et al. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430.
_Nature_ 508, 402–405 (2014). Article PubMed PubMed Central CAS Google Scholar * Lim, S. Y. et al. A direct-acting antiviral drug abrogates viremia in Zika virus-infected rhesus
macaques. _Sci. Transl. Med._ 12, eaau9135 (2020). * Eastman, R. T. et al. Remdesivir: a review of its discovery and development leading to emergency use authorization for treatment of
COVID-19. _ACS Cent. Sci._ 6, 672–683 (2020). Article PubMed PubMed Central CAS Google Scholar * Yin, W. et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from
SARS-CoV-2 by remdesivir. _Science_ 368, 1499–1504 (2020). Article PubMed PubMed Central CAS Google Scholar * Kabinger, F. et al. Mechanism of molnupiravir-induced SARS-CoV-2
mutagenesis. _Nat. Struct. Mol. Biol._ 28, 740–746 (2021). Article PubMed PubMed Central CAS Google Scholar * Zhao, Y., He, G. & Huang, W. A novel model of molnupiravir against
SARS-CoV-2 replication: accumulated RNA mutations to induce error catastrophe. _Signal Transduct. Target Ther._ 6, 410 (2021). Article PubMed PubMed Central CAS Google Scholar *
Agrofoglio, L. et al. Synthesis of carbocyclic nucleosides. _Tetrahedron_ 50, 10611–10670 (1994). Article CAS Google Scholar * Vorbrüggen, H. & Ruh-Pohlenz, C. Synthesis of
Nucleosides. In _Organic Reactions_. 1–630 (Wiley, 2004). * Boutureira, O., Matheu, M. I., Diaz, Y. & Castillon, S. Advances in the enantioselective synthesis of carbocyclic nucleosides.
_Chem. Soc. Rev._ 42, 5056–5072 (2013). Article PubMed CAS Google Scholar * Merino, P. _Chemical Synthesis of Nucleoside Analogues_ (Wiley, 2013). * Ojeda-Porras, A. C., Roy, V. &
Agrofoglio, L. A. Chemical approaches to carbocyclic nucleosides. _Chem. Rec._ 22, e202100307 (2022). Article PubMed CAS Google Scholar * Just, G. & Kim, S. C-nucleosides and related
compounds: synthesis of the carbocyclic analogues of D, L-pyrazofurin a (pyrazomycin) and showdomycin. _Tetrahedron Lett._ 17, 1063–1066 (1976). Article Google Scholar * Woa, E. P. A. D.,
Converso, A., Fraley, M. E. & Hartingh, J. _Ahcy Hydrolase Inhibitors For Treatment Of Hyper Homocysteinemia_. WO2009108546A1 (2009). * Maier, L. et al. New carbocyclic nucleosides:
synthesis of carbocyclic pseudoisocytidine and its analogs. _Tetrahedron Lett._ 55, 3713–3716 (2014). Article CAS Google Scholar * Maier, L. et al. Diastereoselective flexible synthesis
of carbocyclic C-nucleosides. _J. Org. Chem._ 82, 3382–3402 (2017). Article PubMed CAS Google Scholar * Sidera, M. & Fletcher, S. P. Rhodium-catalysed asymmetric allylic arylation of
racemic halides with arylboronic acids. _Nat. Chem._ 7, 935–939 (2015). Article PubMed CAS Google Scholar * Goetzke, F. W., Dijk, L. & Fletcher, S. P. Catalytic Asymmetric
Suzuki–Miyaura Couplings. In _PATAI’S Chemistry of Functional Groups_. 1–54 (Wiley, 2019). * Goetzke, F. W., Mortimore, M. & Fletcher, S. P. Enantio- and diastereoselective
suzuki-miyaura coupling with racemic bicycles. _Angew. Chem. Int. Ed._ 58, 12128–12132 (2019). Article CAS Google Scholar * Gonzalez, J., van Dijk, L., Goetzke, F. W. & Fletcher, S.
P. Highly enantioselective rhodium-catalyzed cross-coupling of boronic acids and racemic allyl halides. _Nat. Protoc._ 14, 2972–2985 (2019). Article PubMed CAS Google Scholar * Fletcher,
S. P. & Goetzke, F. W. Additions to racemates: a strategy for developing asymmetric cross-coupling reactions. _Synlett_ 32, 1816–1825 (2021). Article Google Scholar * Hedouin, G.,
Hazra, S., Gallou, F. & Handa, S. The catalytic formation of atropisomers and stereocenters via asymmetric suzuki–miyaura couplings. _ACS Catal._ 12, 4918–4937 (2022). Article CAS
Google Scholar * Schafer, P., Palacin, T., Sidera, M. & Fletcher, S. P. Asymmetric Suzuki-Miyaura coupling of heterocycles via Rhodium-catalysed allylic arylation of racemates. _Nat.
Commun._ 8, 15762 (2017). Article PubMed PubMed Central Google Scholar * Schafer, P., Sidera, M., Palacin, T. & Fletcher, S. P. Asymmetric cross-coupling of alkyl, alkenyl and
(hetero)aryl nucleophiles with racemic allyl halides. _Chem. Commun._ 53, 12499–12511 (2017). Article CAS Google Scholar * Knapp, D. M., Gillis, E. P. & Burke, M. D. A general
solution for unstable boronic acids: slow-release cross-coupling from air-stable MIDA boronates. _J. Am. Chem. Soc._ 131, 6961–6963 (2009). Article PubMed PubMed Central CAS Google
Scholar * Cox, P. A., Leach, A. G., Campbell, A. D. & Lloyd-Jones, G. C. Protodeboronation of heteroaromatic, vinyl, and cyclopropyl boronic acids: pH-rate profiles, autocatalysis, and
disproportionation. _J. Am. Chem. Soc._ 138, 9145–9157 (2016). Article PubMed CAS Google Scholar * Blakemore, D. C. et al. Organic synthesis provides opportunities to transform drug
discovery. _Nat. Chem._ 10, 383–394 (2018). Article PubMed CAS Google Scholar * Bostrom, J., Brown, D. G., Young, R. J. & Keseru, G. M. Expanding the medicinal chemistry synthetic
toolbox. _Nat. Rev. Drug Discov._ 17, 709–727 (2018). Article PubMed Google Scholar * Cook, X. A. F., de Gombert, A., McKnight, J., Pantaine, L. R. E. & Willis, M. C. The 2-pyridyl
problem: challenging nucleophiles in cross-coupling arylations. _Angew. Chem. Int. Ed._ 60, 11068–11091 (2021). Article CAS Google Scholar * Hayes, H. L. D. et al. Protodeboronation of
(hetero)arylboronic esters: direct versus prehydrolytic pathways and self-/auto-catalysis. _J. Am. Chem. Soc._ 143, 14814–14826 (2021). Article PubMed CAS Google Scholar * Paymode, D. J.
et al. Expanding access to remdesivir via an improved pyrrolotriazine synthesis: supply centered synthesis. _Org. Lett._ 22, 7656–7661 (2020). Article PubMed PubMed Central CAS Google
Scholar * Harrisson, P., Morris, J., Marder, T. B. & Steel, P. G. Microwave-accelerated iridium-catalyzed borylation of aromatic C-H bonds. _Org. Lett._ 11, 3586–3589 (2009). Article
PubMed CAS Google Scholar * Klecka, M., Pohl, R., Klepetarova, B. & Hocek, M. Direct C-H borylation and C-H arylation of pyrrolo[2,3-d]pyrimidines: synthesis of 6,8-disubstituted
7-deazapurines. _Org. Biomol. Chem._ 7, 866–868 (2009). Article PubMed CAS Google Scholar * Larsen, M. A. & Hartwig, J. F. Iridium-catalyzed C-H borylation of heteroarenes: scope,
regioselectivity, application to late-stage functionalization, and mechanism. _J. Am. Chem. Soc._ 136, 4287–4299 (2014). Article PubMed CAS Google Scholar * Tanaka, M., Yoshioka, M.
& Sakai, K. Practical enzymatic procedure for the synthesis of (–)-aristeromycin. _J. Chem. Soc. Chem. Commun_. 19, 1454–1455 (1992). * Banwell, M. G., Hungerford, N. L. & Armitt, D.
J. Syntheses of Showdomycin andits anomer Using N-(triisopropylsilyl)pyrrole as a synthetic equivalent for the maleimide C3-anion. _Synthesis,_ 12, 1837–1843 (2003). * Renner, J. et al. The
synthesis and biological evaluation of two analogues of the C-riboside Showdomycin. _Australian Journal of Chemistry_ 58, 86–93 (2005). Download references ACKNOWLEDGEMENTS The authors
thank the UK Engineering and Physical Sciences Research Council (EP/V015087/1) for financial support. S.M. is grateful to The European Union’s Horizon 2020 research and innovation programe
for the Marie Skłodowska-Curie fellowship (890680) for funding. For the purpose of Open Access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript
(AAM) version arising from this submission. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Chemistry, Chemistry Research Laboratory, University of Oxford, Oxford, OX1 3TA, UK
Sourabh Mishra, Florian C. T. Modicom, Conor L. Dean & Stephen P. Fletcher Authors * Sourabh Mishra View author publications You can also search for this author inPubMed Google Scholar *
Florian C. T. Modicom View author publications You can also search for this author inPubMed Google Scholar * Conor L. Dean View author publications You can also search for this author
inPubMed Google Scholar * Stephen P. Fletcher View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS S.P.F conceived the project and guided the
research. S.M., F.C.T.M., and C.L.D. performed the experimental work. All the authors analyzed the data and planned the experiments. S.M. and S.P.F. wrote the manuscript. All authors have
given approval to the final version of the manuscript. CORRESPONDING AUTHOR Correspondence to Stephen P. Fletcher. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing
interests. PEER REVIEW PEER REVIEW INFORMATION _Communications Chemistry_ thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. ADDITIONAL INFORMATION
PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION
DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License,
which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a
link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license,
unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory
regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Mishra, S., Modicom, F.C.T., Dean, C.L. _et al._ Catalytic asymmetric synthesis of
carbocyclic C-nucleosides. _Commun Chem_ 5, 154 (2022). https://doi.org/10.1038/s42004-022-00773-6 Download citation * Received: 21 August 2022 * Accepted: 07 November 2022 * Published: 19
November 2022 * DOI: https://doi.org/10.1038/s42004-022-00773-6 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative