Play all audios:
ABSTRACT DNA methylation marks have recently been used to build models known as epigenetic clocks, which predict calendar age. As methylation of cytosine promotes C-to-T mutations, we
hypothesized that the methylation changes observed with age should reflect the accrual of somatic mutations, and the two should yield analogous aging estimates. In an analysis of multimodal
data from 9,331 human individuals, we found that CpG mutations indeed coincide with changes in methylation, not only at the mutated site but with pervasive remodeling of the methylome out to
±10 kilobases. This one-to-many mapping allows mutation-based predictions of age that agree with epigenetic clocks, including which individuals are aging more rapidly or slowly than
expected. Moreover, genomic loci where mutations accumulate with age also tend to have methylation patterns that are especially predictive of age. These results suggest a close coupling
between the accumulation of sporadic somatic mutations and the widespread changes in methylation observed over the course of life. Access through your institution Buy or subscribe This is a
preview of subscription content, access via your institution ACCESS OPTIONS Access through your institution Access Nature and 54 other Nature Portfolio journals Get Nature+, our best-value
online-access subscription $29.99 / 30 days cancel any time Learn more Subscribe to this journal Receive 12 digital issues and online access to articles $119.00 per year only $9.92 per issue
Learn more Buy this article * Purchase on SpringerLink * Instant access to full article PDF Buy now Prices may be subject to local taxes which are calculated during checkout ADDITIONAL
ACCESS OPTIONS: * Log in * Learn about institutional subscriptions * Read our FAQs * Contact customer support SIMILAR CONTENT BEING VIEWED BY OTHERS MAKING SENSE OF THE AGEING METHYLOME
Article 02 May 2022 DISENTANGLING AGE-DEPENDENT DNA METHYLATION: DETERMINISTIC, STOCHASTIC, AND NONLINEAR Article Open access 28 April 2021 UNIVERSAL DNA METHYLATION AGE ACROSS MAMMALIAN
TISSUES Article Open access 10 August 2023 DATA AVAILABILITY All data analyzed were from The Cancer Genome Atlas Pan-Can cohort34,35,36 (http://xena.ucsc.edu/) and the Pan-Cancer Analysis of
Whole Genomes48 (https://xenabrowser.net/datapages/?hub=https://pcawg.xenahubs.net:443). Data can be accessed from the provided links and are described further in the respective
publications (https://doi.org/10.1038/ng.2764, https://doi.org/10.1038/s41586-020-1969-6)35,37. Data to replicate the figures in this manuscript can be found on figshare (‘Somatic mutation
as an explanation for epigenetic aging (Koch et al. 2024)’, https://figshare.com/projects/Somatic_mutation_as_an_explanation_for_epigenetic_aging_Koch_et_al_2024_/224232)75. The panel of
normal and gnomAD resources used for filtering the somatic mutation calls can be accessed by downloading Mutect2 (https://gatk.broadinstitute.org/hc/en-us/articles/360037593851-Mutect2). A
file containing Illumina 450k array CpG locations and characteristics can be accessed on the Illumina website
(https://webdata.illumina.com/downloads/productfiles/humanmethylation450/humanmethylation450_15017482_v1-2.csv). The hg19 genome annotation can be accessed through the University of
California, Santa Cruz, website (https://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/cpgIslandExt.txt.gz). CODE AVAILABILITY All custom algorithms and analysis code are in the GitHub
repository at https://github.com/zanekoch/MutationsAndMethylationAging/. REFERENCES * Szilard, L. On the nature of the aging process. _Proc. Natl Acad. Sci. USA_ 45, 30–45 (1959). Article
CAS PubMed PubMed Central Google Scholar * Cagan, A. et al. Somatic mutation rates scale with lifespan across mammals. _Nature_ 604, 517–524 (2022). Article CAS PubMed PubMed Central
Google Scholar * Alexandrov, L. B. et al. Clock-like mutational processes in human somatic cells. _Nat. Genet._ 47, 1402–1407 (2015). Article CAS PubMed PubMed Central Google Scholar
* Moore, L. et al. The mutational landscape of human somatic and germline cells. _Nature_ 597, 381–386 (2021). Article CAS PubMed Google Scholar * Jaiswal, S. & Ebert, B. L. Clonal
hematopoiesis in human aging and disease. _Science_ 366, eaan4673 (2019). Article CAS PubMed PubMed Central Google Scholar * Lodato, M. A. et al. Aging and neurodegeneration are
associated with increased mutations in single human neurons. _Science_ 359, 555–559 (2018). Article CAS PubMed Google Scholar * Bae, T. et al. Analysis of somatic mutations in 131 human
brains reveals aging-associated hypermutability. _Science_ 377, 511–517 (2022). Article CAS PubMed PubMed Central Google Scholar * Stratton, M. R., Campbell, P. J. & Futreal, P. A.
The cancer genome. _Nature_ 458, 719–724 (2009). Article CAS PubMed PubMed Central Google Scholar * Blagosklonny, M. V. DNA- and telomere-damage does not limit lifespan: evidence from
rapamycin. _Aging (Albany NY)_ 13, 3167–3175 (2021). Article CAS PubMed Google Scholar * López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of
aging. _Cell_ 153, 1194–1217 (2013). Article PubMed PubMed Central Google Scholar * Moore, L. D., Le, T. & Fan, G. DNA methylation and its basic function. _Neuropsychopharmacology_
38, 23–38 (2013). Article CAS PubMed Google Scholar * Li, E., Beard, C. & Jaenisch, R. Role for DNA methylation in genomic imprinting. _Nature_ 366, 362–365 (1993). Article CAS
PubMed Google Scholar * Deaton, A. M. & Bird, A. CpG islands and the regulation of transcription. _Genes Dev._ 25, 1010–1022 (2011). Article CAS PubMed PubMed Central Google
Scholar * Ehrlich, M. et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. _Nucleic Acids Res._ 10, 2709–2721 (1982). Article CAS
PubMed PubMed Central Google Scholar * Jabbari, K. & Bernardi, G. Cytosine methylation and CpG, TpG (CpA) and TpA frequencies. _Gene_ 333, 143–149 (2004). Article CAS PubMed Google
Scholar * Meaney, M. J. & Szyf, M. Environmental programming of stress responses through DNA methylation: life at the interface between a dynamic environment and a fixed genome.
_Dialogues Clin. Neurosci._ 7, 103–123 (2005). Article PubMed PubMed Central Google Scholar * Hannum, G. et al. Genome-wide methylation profiles reveal quantitative views of human aging
rates. _Mol. Cell_ 49, 359–367 (2013). Article CAS PubMed Google Scholar * Horvath, S. DNA methylation age of human tissues and cell types. _Genome Biol._ 14, R115 (2013). Article
PubMed PubMed Central Google Scholar * McCrory, C. et al. GrimAge outperforms other epigenetic clocks in the prediction of age-related clinical phenotypes and all-cause mortality. _J.
Gerontol. A Biol. Sci. Med. Sci._ 76, 741–749 (2021). Article PubMed Google Scholar * Lu, A. T. et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. _Aging (Albany
NY)_ 11, 303–327 (2019). Article CAS PubMed Google Scholar * Levine, M. E. et al. An epigenetic biomarker of aging for lifespan and healthspan. _Aging (Albany NY)_ 10, 573–591 (2018).
Article PubMed Google Scholar * Li, A., Koch, Z. & Ideker, T. Epigenetic aging: biological age prediction and informing a mechanistic theory of aging. _J. Intern. Med._ 292, 733–744
(2022). Article PubMed Google Scholar * Yang, J.-H. et al. Loss of epigenetic information as a cause of mammalian aging. _Cell_ 186, 305–326 (2023). Article CAS PubMed PubMed Central
Google Scholar * de Magalhães, J. P. Ageing as a software design flaw. _Genome Biol._ 24, 51 (2023). Article PubMed PubMed Central Google Scholar * López-León, M. & Goya, R. G. The
emerging view of aging as a reversible epigenetic process. _Gerontology_ 63, 426–431 (2017). Article PubMed Google Scholar * Ito, S. et al. Tet proteins can convert 5-methylcytosine to
5-formylcytosine and 5-carboxylcytosine. _Science_ 333, 1300–1303 (2011). Article CAS PubMed PubMed Central Google Scholar * Wang, M. et al. Identification of DNA motifs that regulate
DNA methylation. _Nucleic Acids Res._ 47, 6753–6768 (2019). Article CAS PubMed PubMed Central Google Scholar * Nachun, D. et al. Clonal hematopoiesis associated with epigenetic aging
and clinical outcomes. _Aging Cell_ 20, e13366 (2021). Article CAS PubMed PubMed Central Google Scholar * Gibbs, J. R. et al. Abundant quantitative trait loci exist for DNA methylation
and gene expression in human brain. _PLoS Genet._ 6, e1000952 (2010). Article PubMed PubMed Central Google Scholar * McCartney, D. L. et al. Genome-wide association studies identify 137
genetic loci for DNA methylation biomarkers of aging. _Genome Biol._ 22, 194 (2021). Article CAS PubMed PubMed Central Google Scholar * Youk, J., An, Y., Park, S., Lee, J.-K. & Ju,
Y. S. The genome-wide landscape of C:G > T:A polymorphism at the CpG contexts in the human population. _BMC Genomics_ 21, 270 (2020). Article CAS PubMed PubMed Central Google Scholar
* Ju, Y. S. et al. Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. _Nature_ 543, 714–718 (2017). Article CAS PubMed PubMed Central Google Scholar *
Duncan, B. K. & Miller, J. H. Mutagenic deamination of cytosine residues in DNA. _Nature_ 287, 560–561 (1980). Article CAS PubMed Google Scholar * Ellrott, K. et al. Scalable open
science approach for mutation calling of tumor exomes using multiple genomic pipelines. _Cell Syst._ 6, 271–281 (2018). Article CAS PubMed PubMed Central Google Scholar * Cancer Genome
Atlas Research Network et al. The Cancer Genome Atlas Pan-Cancer analysis project. _Nat. Genet._ 45, 1113–1120 (2013). Article PubMed Central Google Scholar * Liu, J. et al. An integrated
TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. _Cell_ 173, 400–416 (2018). Article CAS PubMed PubMed Central Google Scholar * ICGC/TCGA
Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. _Nature_ 578, 82–93 (2020). Article Google Scholar * Bibikova, M. et al. High density DNA methylation
array with single CpG site resolution. _Genomics_ 98, 288–295 (2011). Article CAS PubMed Google Scholar * Liu, X. et al. Metallothionein 2A (MT2A) controls cell proliferation and liver
metastasis by controlling the MST1/LATS2/YAP1 signaling pathway in colorectal cancer. _Cancer Cell Int._ 22, 205 (2022). Article CAS PubMed PubMed Central Google Scholar * Si, M. &
Lang, J. The roles of metallothioneins in carcinogenesis. _J. Hematol. Oncol._ 11, 107 (2018). Article PubMed PubMed Central Google Scholar * Fu, J. et al. Metallothionein 1G functions
as a tumor suppressor in thyroid cancer through modulating the PI3K/Akt signaling pathway. _BMC Cancer_ 13, 462 (2013). Article PubMed PubMed Central Google Scholar * Tong, M. et al.
Evaluation of MT family isoforms as potential biomarker for predicting progression and prognosis in gastric cancer. _Biomed Res. Int._ 2019, 2957821 (2019). Article PubMed PubMed Central
Google Scholar * Pinney, S. E. Mammalian non-CpG methylation: stem cells and beyond. _Biology (Basel)_ 3, 739–751 (2014). PubMed Google Scholar * Mathelier, A. et al. Cis-regulatory
somatic mutations and gene-expression alteration in B-cell lymphomas. _Genome Biol._ 16, 84 (2015). Article PubMed PubMed Central Google Scholar * Luo, X. et al. Effects of DNA
methylation on TFs in human embryonic stem cells. _Front. Genet._ 12, 639461 (2021). Article CAS PubMed PubMed Central Google Scholar * Wang, M., Ngo, V. & Wang, W. Deciphering the
genetic code of DNA methylation. _Brief. Bioinform._ 22, bbaa424 (2021). Article PubMed PubMed Central Google Scholar * Villicaña, S. & Bell, J. T. Genetic impacts on DNA
methylation: research findings and future perspectives. _Genome Biol._ 22, 127 (2021). Article PubMed PubMed Central Google Scholar * Russo, G. et al. DNA damage and repair modify DNA
methylation and chromatin domain of the targeted locus: mechanism of allele methylation polymorphism. _Sci. Rep._ 6, 33222 (2016). Article CAS PubMed PubMed Central Google Scholar *
Morano, A. et al. Targeted DNA methylation by homology-directed repair in mammalian cells. Transcription reshapes methylation on the repaired gene. _Nucleic Acids Res._ 42, 804–821 (2014).
Article CAS PubMed Google Scholar * Allen, B., Pezone, A., Porcellini, A., Muller, M. T. & Masternak, M. M. Non-homologous end joining induced alterations in DNA methylation: a
source of permanent epigenetic change. _Oncotarget_ 8, 40359–40372 (2017). Article PubMed PubMed Central Google Scholar * Pagès-Gallego, M. et al. Direct detection of 8-oxo-dG using
nanopore sequencing. Preprint at _bioRxiv_ https://doi.org/10.1101/2024.05.17.594638 (2024). * Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and
adult fibroblast cultures by defined factors. _Cell_ 126, 663–676 (2006). Article CAS PubMed Google Scholar * Gill, D. et al. Multi-omic rejuvenation of human cells by maturation phase
transient reprogramming. _eLife_ 11, e71624 (2022). Article CAS PubMed PubMed Central Google Scholar * Ocampo, A. et al. In vivo amelioration of age-associated hallmarks by partial
reprogramming. _Cell_ 167, 1719–1733 (2016). Article CAS PubMed PubMed Central Google Scholar * Martincorena, I. et al. Tumor evolution. High burden and pervasive positive selection of
somatic mutations in normal human skin. _Science_ 348, 880–886 (2015). Article CAS PubMed PubMed Central Google Scholar * Martincorena, I. & Campbell, P. J. Somatic mutation in
cancer and normal cells. _Science_ 349, 1483–1489 (2015). Article CAS PubMed Google Scholar * Li, R. et al. A body map of somatic mutagenesis in morphologically normal human tissues.
_Nature_ 597, 398–403 (2021). Article CAS PubMed Google Scholar * Chen, Y. et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450
microarray. _Epigenetics_ 8, 203–209 (2013). Article CAS PubMed PubMed Central Google Scholar * Jaffe, A. E. & Irizarry, R. A. Accounting for cellular heterogeneity is critical in
epigenome-wide association studies. _Genome Biol._ 15, R31 (2014). Article PubMed PubMed Central Google Scholar * Tomusiak, A. et al. Development of an epigenetic clock resistant to
changes in immune cell composition. _Commun. Biol._ 7, 934 (2024). Article CAS PubMed PubMed Central Google Scholar * Wang, T. et al. Quantitative translation of dog-to-human aging by
conserved remodeling of the DNA methylome. _Cell Syst._ 11, 176–185 (2020). Article PubMed PubMed Central Google Scholar * Lu, A. T. et al. Universal DNA methylation age across mammalian
tissues. _Nat. Aging_ 3, 1144–1166 (2023). Article CAS PubMed PubMed Central Google Scholar * Rozenblit, M. et al. Evidence of accelerated epigenetic aging of breast tissues in
patients with breast cancer is driven by CpGs associated with polycomb-related genes. _Clin. Epigenetics_ 14, 30 (2022). Article CAS PubMed PubMed Central Google Scholar * Moqri, M. et
al. PRC2-AgeIndex as a universal biomarker of aging and rejuvenation. _Nat. Commun._ 15, 5956 (2024). Article CAS PubMed PubMed Central Google Scholar * Van Egeren, D. et al.
Reconstructing the lineage histories and differentiation trajectories of individual cancer cells in myeloproliferative neoplasms. _Cell Stem Cell_ 28, 514–523 (2021). Article PubMed PubMed
Central Google Scholar * Ferrall-Fairbanks, M. C. et al. Progenitor hierarchy of chronic myelomonocytic leukemia identifies inflammatory monocytic-biased trajectory linked to worse
outcomes. _Blood Cancer Discov._ 3, 536–553 (2022). Article CAS PubMed PubMed Central Google Scholar * McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for
analyzing next-generation DNA sequencing data. _Genome Res._ 20, 1297–1303 (2010). Article CAS PubMed PubMed Central Google Scholar * Nassar, L. R. et al. The UCSC Genome Browser
database: 2023 update. _Nucleic Acids Res._ 51, D1188–D1195 (2023). Article CAS PubMed Google Scholar * Raney, B. J. et al. Track data hubs enable visualization of user-defined
genome-wide annotations on the UCSC Genome Browser. _Bioinformatics_ 30, 1003–1005 (2014). Article CAS PubMed Google Scholar * Kent, W. J. et al. The human genome browser at UCSC.
_Genome Res._ 12, 996–1006 (2002). Article CAS PubMed PubMed Central Google Scholar * Tang, G., Cho, M. & Wang, X. OncoDB: an interactive online database for analysis of gene
expression and viral infection in cancer. _Nucleic Acids Res._ 50, D1334–D1339 (2022). Article CAS PubMed Google Scholar * Chen, T. & Guestrin, C. XGBoost: a scalable tree boosting
system. In _Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining_ 785–794 (Association for Computing Machinery, 2016). * Alexandrov, L. B. et
al. The repertoire of mutational signatures in human cancer. _Nature_ 578, 94–101 (2020). Article CAS PubMed PubMed Central Google Scholar * Pedregosa, F. et al. Scikit-learn: machine
learning in Python. _J. Mach. Learn. Res._ 12, 2825–2830 (2011). Google Scholar * Koch, Z. Zip of all data. _figshare_ https://doi.org/10.6084/m9.figshare.27270468.v1 (2024). Download
references ACKNOWLEDGEMENTS This study was funded by the National Institutes of Health under awards U54 CA274502 (T.I.), P41 GM103504 (T.I.) and R01AG059416 (S.C.). S.C. and D.E. also
receive support from The Sequoia Center for Research on Aging, California Pacific Medical Center Research Institute. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Program in Bioinformatics
and Systems Biology, University of California, San Diego, La Jolla, CA, USA Zane Koch, Adam Li & Trey Ideker * California Pacific Medical Center Research Institute, San Francisco, CA,
USA Daniel S. Evans & Steven Cummings * Department of Epidemiology and Biostatistics, University of California, San Francisco, San Francisco, CA, USA Daniel S. Evans & Steven
Cummings * Department of Medicine, University of California, San Diego, La Jolla, CA, USA Trey Ideker Authors * Zane Koch View author publications You can also search for this author
inPubMed Google Scholar * Adam Li View author publications You can also search for this author inPubMed Google Scholar * Daniel S. Evans View author publications You can also search for this
author inPubMed Google Scholar * Steven Cummings View author publications You can also search for this author inPubMed Google Scholar * Trey Ideker View author publications You can also
search for this author inPubMed Google Scholar CONTRIBUTIONS Z.K. designed the study, carried out the primary data analyses and wrote the manuscript. A.L. and D.S.E. assisted with data
analysis and study design considerations. T.I. and S.C. designed the study and wrote the manuscript. CORRESPONDING AUTHORS Correspondence to Steven Cummings or Trey Ideker. ETHICS
DECLARATIONS COMPETING INTERESTS T.I. is a cofounder of Serinus and Data4Cure, is on their scientific advisory boards and has an equity interest in both companies. T.I. is on the scientific
advisory board of IDEAYA Biosciences and has an equity interest. The terms of these arrangements have been reviewed and approved by the University of California, San Diego, in accordance
with its conflict of interest policies. The other authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Aging_ thanks Wolfgang Wagner and the other, anonymous,
reviewer(s) for their contribution to the peer review of this work. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published
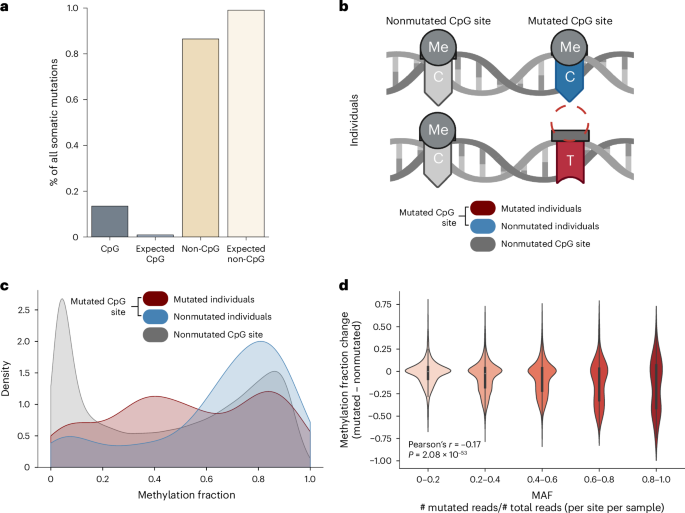
maps and institutional affiliations. EXTENDED DATA EXTENDED DATA FIG. 1 LINKS AMONG CPG MUTATIONS, METHYLOME REMODELING, AND AGING. A) Various mutational processes affect the genome. Here,
we show that some of these mutations associate with an aberrant DNA methylation pattern at both the mutated site and at numerous neighboring CpGs. B) An individual’s DNA mutation profile and
DNA methylation profile make similar predictions of their calendar age and rate of aging. Panel A created with BioRender.com. EXTENDED DATA FIG. 2 SUPPLEMENTAL CHARACTERIZATION OF CPG
MUTATIONS. A) The distribution of methylation fraction values of each CpG site in the TCGA and PCAWG datasets separately (TCGA = 273,202 and PCAWG = 326,749 CpG sites) in each sample (TCGA =
8,680 and PCAWG = 651 samples). B) The CpG density (number of CpGs per base pair) in the 50 and 125 base pairs surrounding each of the CpG sites in (A). The central line of the inner
boxplot represents the median, the edges of the box the interquartile range (IQR), and the whiskers 1.5-times the IQR. C) Violin plots of the distribution of mean methylation fraction of
non-mutated individuals at the same mutated CpG sites as in Fig. 1d (n = 8,037 sites), stratified by CpG mutation type. D) As in (C), but the distribution of CpG density in the 125 bp
surrounding each CpG site. E) Pie chart showing the proportion of CpG mutations (n = 467,079 mutations) that result in specific mutated nucleotides. Note that 5’-CpG-3’ sites are
palindromic, corresponding to a 3’-GpC-5’ sequence on the opposite strand; thus, mutation of the C residue is equivalent to mutation of the complementary G residue. For simplicity, we refer
to all CpG mutations by the status of the C residue. F) Violin plot showing the mean methylation fraction across all PCAWG samples, considering CpG sites where a mutation has occurred in at
least one sample (left, n = 1,137 CpG sites), CpG sites where no mutation has occurred in any sample (middle, n = 325,614 CpG sites), and all measured CpG sites (right, n = 326,751).
Significant difference of distribution (p ≤ 3.03 × 10–50) is marked with (***) and non-significant (p > 0.05) with (n.s.), based on a two-sided Mann-Whitney test. G) Methylation fraction
at the same mutated CpG sites as Fig. 1d (n = 8,037 sites). CpG sites are binned into five groups based on MAF, with violin plots summarizing the distribution of methylation fraction within
each group. Vertical bars inside each violin represent the interquartile range. Two-sided p value calculated based on the exact distribution of Pearson’s r modeled as a beta function.
EXTENDED DATA FIG. 3 MAGNITUDE OF METHYLATION CHANGE NEAR SOMATIC MUTATIONS BY TISSUE AND GENOMIC CONTEXT. A) Boxplots of the distribution of ΔMF10kb values for mutated (red) versus random
control (n = 260,000, blue) sites for each tissue type separately (n = 813, 144, and 1,643 mutated sites from Pancreas, Brain, and Ovary tissues, respectively). P value shown for a two-sided
Mann-Whitney test for a difference in median methylation fraction between the mutated and non-mutated random control loci. P value shown for a two-sided Mann-Whitney test for a difference
in median absolute deviation (MAD) of ΔMF10kb between the mutated and non-mutated random control loci. The central line represents the median, the edges of the box the interquartile range
(IQR), and the whiskers 1.5-times the IQR. B) A histogram of the median methylation fraction across comparison sites within ±10 kb of mutated (n = 2,600, red) and random control sites (n =
260,000, blue). Mutated sites are the same as Fig. 3b. Random control sites have been selected as before, with the additional criteria of having a methylation profile matched to that of the
matched samples at mutated sites (as measured by the median methylation fraction of comparison sites, Methods). P value shown for a two-sided Mann-Whitney test for a difference in median
methylation fraction between the mutated and random control loci. C) Probability distribution of ΔMF10kb values for mutated (red) versus random control (blue) sites. Mutated and random sites
are the same as (B). P value calculated as in (A). D) Line plot depicting the fold enrichment for mutated over non-mutated random control sites as a function of ΔMF10kb, for the same sites
as Fig. 3b. Sites are stratified depending on whether the site is a CpG and/or falls within a CpG island (n = 419 CpG-non-CGI, 21 CpG-CGI, 2,120 non-CpG-non-CGI, and 39 non-CpG-CGI sites).
Fold enrichment is the ratio of the probability of observing a given ΔMF10kb for mutated sites versus non-mutated random control sites. ΔMF10kb is divided into equally spaced bins from –0.4
to 0.4. E) Barchart showing the fold-enrichment of mutated sites with the most extreme methylation changes (absolute ΔMF10kb | Z-score | > 1.96, n = 401 mutated sites) in various genomic
regions, compared to all other mutated sites (n = 2,199 mutated sites). P values were calculated using a two-sided Fisher exact test. The categories ‘Upstream gene’ and ‘Downstream gene’
refer to variants located within 1 kb of the 5’ transcription start site and the 3’ transcription stop site, respectively, but outside the gene itself. F) As in (E), but comparing the
mutated sites with the most extreme gains of methylation (Z-score of ΔMF10kb > 1) to those with the most extreme losses of methylation (Z-score of ΔMF10kb < –1). G) Boxplot of the
ΔMF10kb value as a function of the mutated allele frequency (MAF). Same sites and samples as Fig. 3e (n = 3,880 mutated loci. The Pearson correlation is shown for the association of MAF with
ΔMF10kb and the absolute value of ΔMF10kb. Two-sided p values were calculated based on the exact distribution of Pearson’s r modeled as a beta function. The central line represents the
median, the edges of the box the interquartile range (IQR), the whiskers 1.5-times the IQR, and the points all ΔMF10kb value outside of these ranges. EXTENDED DATA FIG. 4 MUTATION-ASSOCIATED
METHYLATION CHANGE IN NORMAL TISSUES. A) Probability distribution of ΔMF1kb values for mutated (red) versus random control (blue) sites. Includes n = 463 mutated sites (n = 146 samples)
with MAF ≤ 0.15, ≥10 matched individuals (individuals of same tissue type within ± 10 years of age), and ≥1 measured CpG within the window. Random control sites include n = 46,300
non-mutated sites (n = 146 samples, Methods). P value shown for a two-sided Mann-Whitney test for a difference in median absolute deviation (MAD) of ΔMF1kb between the mutated and
non-mutated random control loci. B) Line plot depicting the fold enrichment for mutated over non-mutated sites as a function of ΔMF1kb. Fold enrichment is the ratio of the probability of
observing a given ΔMF1kb for mutated sites versus the probability of that ΔMF1kb for non-mutated control sites. ΔMF1kb is divided into equally spaced bins from –0.45 to 0.45. C) Absolute
ΔMF1kb as the window center is moved away from the mutated site (n = 463, red). This quantity is also shown for non-mutated random control sites (n = 46,300, blue) (Methods). Points indicate
the mean value and error bars denote the 95% confidence interval. A significant difference in distribution of absolute ΔMF1kb values (two-sided t-test) is marked (**, p ≤ .01), (*, p ≤
.05). Other comparisons are non-significant (n.s., p > 0.05). EXTENDED DATA FIG. 5 SUPPLEMENTAL AGE PREDICTION ACCURACY. A) Bar plot indicating the correlation of chronological age with
the age predictions of mutation clocks (left) or methylation clocks (right). Correlations are shown across all tumor tissues (n = 1,601) and in each of five TCGA tumor tissues individually:
LGG (Brain), GBM (Brain-2), SARC (Bone), KIRP (Kidney), and THCA (Thyroid). B) As in (A) but for age predictions using samples from normal (that is non-cancerous) tissues (n = 40
individuals). C) Heatmap indicating the pairwise consistencies (Pearson correlation) among the mutation age in normal tissue, mutation age in tumor tissue, and chronological age. Data shown
for n = 22 individuals with mutations measured in both normal and tumor tissues (the same individuals as from panel B with the exception of 11 colon samples and 7 liver samples as these were
not available in the tumor samples). D) As in (c), but comparing predictions from methylation clocks. E) Scatter plot of human individuals, showing age predictions from the mutation model
versus their chronological age. Shared area denotes the 95% confidence interval of the line of best fit. Includes 40 individuals from four normal tissues (Methods). A two-sided p value was
calculated based on the exact distribution of Pearson’s r modeled as a beta function. F) Similar to panel (B) but showing age predictions from the methylation rather than mutation model. G)
Violin plots of the methylation age residual versus mutation age residual (Methods). Plots include the same individuals as in panels (B,C). Pearson r refers to the correlation between
methylation age residual and mutation age residual, controlling for chronological age (that is, partial correlation, p = 1.76 × 10–3). The central line of the inner boxplot represents the
median, the edges of the box the interquartile range (IQR), the whiskers 1.5-times the IQR, and the points all the methylation age residual values. Statistics calculated as in (E). EXTENDED
DATA FIG. 6 PERFORMANCE COMPARISON TO PREVIOUS EPIGENETIC CLOCKS. A) Pearson r between predicted and chronological age for Hannum, Horvath, and PhenoAge clocks across the same samples as
Fig. 4b (n = 1,601). Predictions were done using the subset of features from each clock that existed in our methylation data after quality control (66%, 63%, and 61% of CpG sites from the
Hannum, Horvath, and PhenoAge clocks, respectively). The performance of this study’s methylation clock is not shown as it is inherently fit to the TCGA dataset in 5-fold CV. B) Pearson r
between predicted and chronological age for Hannum, Horvath, and PhenoAge clocks after re-fitting (Methods). Same samples as (A). The performance of the methylation clock trained in this
study (‘This study’) is shown for reference. EXTENDED DATA FIG. 7 MUTATION AGE PREDICTION WITHOUT WHOLE-GENOME FEATURES. A) Correlation of chronological versus predicted age, shown for
mutation or methylation clocks built without whole-genome features (n = 1,601 individuals). Correlations are shown across all tissues and in each of five TCGA tissues individually: LGG
(Brain), GBM (Brain-2), SARC (Bone), KIRP (Kidney), and THCA (Thyroid). B) As in (A) but for age predictions using samples from normal (that is non-cancerous) tissues (n = 40). C) The
methylation age residual is plotted versus the mutation age residual, using clocks without whole-genome features (Methods). Violin plots summarize the same samples as in panel (A). Pearson r
refers to the correlation between methylation age residual and mutation age residual, controlling for chronological age (that is, partial correlation, p = 6.66 × 10–105). The central line
of the inner boxplot represents the median, the edges of the box the interquartile range (IQR), and the whiskers 1.5-times the IQR. A two-sided p value was calculated based on the exact
distribution of Pearson’s r modeled as a beta function. D) Similar to (C), but for the samples in (B). The central line of the inner boxplot represents the median, the edges of the box the
interquartile range (IQR), the whiskers 1.5-times the IQR, and the points all the methylation age residual values. Statistics calculated as in (C). SUPPLEMENTARY INFORMATION REPORTING
SUMMARY RIGHTS AND PERMISSIONS Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or
other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Koch, Z., Li, A., Evans, D.S. _et al._ Somatic mutation as an explanation for epigenetic aging. _Nat Aging_ 5, 709–719 (2025).
https://doi.org/10.1038/s43587-024-00794-x Download citation * Received: 08 December 2023 * Accepted: 12 December 2024 * Published: 13 January 2025 * Issue Date: April 2025 * DOI:
https://doi.org/10.1038/s43587-024-00794-x SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative