Play all audios:
ABSTRACT Gliomas are the most common brain tumors affecting the central nervous system and are associated with a high mortality rate. DCF1 is a membrane protein that was previously found to
play a role in neural stem cell differentiation. In the present study, we found that overexpression of _dcf1_ significantly inhibited cell proliferation, migration and invasion and
dramatically promoted apoptosis in the glioblastoma U251 cell line. DCF1 deletion mutations in the functional region showed that the complete structure of DCF1 was necessary for apoptosis.
Furthermore, significantly lower tumorigenicity was observed in athymic nude mice by transplanting U251 cells overexpressing _dcf1_. To decode the apoptosis induced by _dcf1_, mitochondrial
structure and membrane potential in glioma cells were investigated and the results indicated obvious mitochondrial swelling, destruction of cristae and a significant decline in membrane
potential. Mechanismly, caspase-3 signaling was activated. Finally, endogenous _dcf1_ silence in U251 cells was investigated. Results showed a highly methylation at −1339 and −1322 position
at _dcf1_ promoter sequence, revealing the causal relationship between _dcf1_ gene and tumorigencicity. The present study identified a previously unknown cancer apoptosis mechanism involving
_dcf1_ overexpression and provided a novel approach to potentially treat glioma patients. SIMILAR CONTENT BEING VIEWED BY OTHERS CDC42EP3 PROMOTES GLIOMA PROGRESSION VIA REGULATION OF CCND1
Article Open access 01 April 2022 CARD16 RESTORES TUMORIGENESIS AND RESTRAINTS APOPTOSIS IN GLIOMA CELLS VIA FOXO1/TRAIL AXIS Article Open access 08 November 2024 MEX3A CONTRIBUTES TO
DEVELOPMENT AND PROGRESSION OF GLIOMA THROUGH REGULATING CELL PROLIFERATION AND CELL MIGRATION AND TARGETING CCL2 Article Open access 04 January 2021 INTRODUCTION Glioblastomas (GBMs) are
the most aggressive brain tumors in humans and have the worst prognoses, with a median survival time of approximately 14 months and are almost invariably fatal1. Due to the resistance of
tumor cells to conventional therapies, it is very difficult to treat GBMs2,3,4; however, the identification of genes or proteins that effectively inhibit tumor growth is expected to break
through this bottleneck5. Our previous study revealed that dendritic cell factor (_dcf1_), also known as _tmem59_, was involved in differentiation of neural stem cells (NSCs)6, since _dcf1_
silencing tends to differentiate NSCs into astrocytes7,8. We further found that GFAP, a marker of astrocytes, was significantly upregulated both in gene and protein arrays using _dcf1_
knockout mice, suggesting that there may be a lower expression level of _dcf1_ in glioma cells. Gene array analysis also showed that _dcf1_ expression in grade I and II gliomas was higher
than that in grade III and IV (Sup. 1a) and _dcf1_ expression was decreased in the U251 glioma cell line compared to normal astrocyte cells (Sup. 1b). At the same time, we detected _dcf1_
expression in normal mouse brain tissue and transplanted gliomas, with similar results (Sup. 1c–e). In view of these assumptions, we transfected _dcf1_ into the U251 glioma cell line and
identified an obvious tumor suppression not only in the cells but also in vivo. To study _dcf1_ membrane localization, we first performed immunofluorescence analysis using HeLa cells
overexpressing _dcf1_ and found that DCF1 was distributed outside the nucleus (Sup. 2a). Next, we constructed a _dcf1_ vector using the pEGFP-N2 vector for transfection of U251 glioma cells,
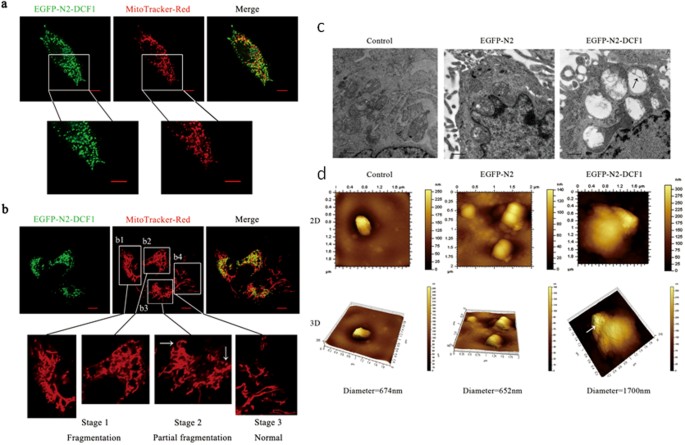
in which endogenous _dcf1_ expression was undetectable. The distribution of DCF1 in mitochondria was shown using the mitochondria-specific dye MitoTracker® Red CMXRos (Fig. 1a). A closer
examination revealed mitochondrial fragmentation or fission9 upon ectopic DCF1 expression (Fig. 1b). Interestingly, all DCF1-expressing mitochondria were ruptured (Fig. 1b1, b2), whereas
those not expressing DCF1 remained normal (Fig. 1b4). The same was true even in the same cell, non-expression of DCF1 retained normal mitochondrial structure (Fig. 1b3; white arrow on the
left), whereas DCF1 expression led to fragmentation of the mitochondrial structure (Fig. 1b3; white arrow on the right), suggesting a crucial step toward triggering cell apoptosis10,11. To
define the structural changes of mitochondria that are due to dcf1 expression, we observed mitochondria using transmission electron microscopy (TEM)12. Compared with controls, mitochondrial
swelling and cristae destruction were detected in cells overexpressing DCF1 (Fig. 1c; black arrow)13. Changes in mitochondrial morphology was further investigated using atomic force
microscopy (AFM), which showed that the mitochondria swelled to 1700 nm in cells overexpressing DCF1, which was more than twice that of controls (Fig. 1d), suggesting the role of DCF1 in
mitochondrial destruction. At the same time, we discovered many hollow spaces in the outer membrane of the mitochondria (indicated by white arrow), suggesting the opening of mitochondrial
permeability transition pores14. Following our observations of mitochondrial damage in U251 cells caused by DCF1 overexpression, we next examined the effects of DCF1 on cell viability
including proliferation, migration and invasion. The cell proliferation rate was evaluated using a CCK-8 Cell Counting Kit at 24, 36, 48, 60 and 72 h post-transfection15. In comparison to
control groups, exogenous DCF1 significantly inhibited growth of U251 cells at 24–72 h post-transfection (Fig. 2a). The same result was shown in the glioma cell line U87-MG, although not as
extensive as in U251 cells (Fig. 2b). However, there was no inhibition of DCF1 in other tumor cell lines, including MCF-7, H1299, K-562 and HEK293T, demonstrating specific inhibition of DCF1
in glioma cells. Considering the important characteristics of tumor cells, we next investigated the effects of DCF1 on cell migration and invasion16,17,18. When _dcf1_ was expressed in U251
cells, both cell migration and invasion were significantly decreased compared with control cells (Fig. 2c and 2e; Sup. 2b)19. To examine the functional region in DCF1, we examined the
entire protein sequence using the PSORT Nakai Server (psort.hgc.jp) and determined the existence of an N-terminal signal sequence, mitochondrial targeting sequential motif, a transmembrane
region and a cytoplasmic tail. Next, we designed and constructed 4 deletion mutants corresponding to these regions (Fig. 2d) and investigated the mitochondrial membrane potential20 following
transfection with full-length wild-type _dcf1_ and the 4 mutants in U251 cells. CCCP, a proton ionophore that disrupts membrane potential, was used as a positive control and the results
showed conclusively that only the complete structure of DCF1 reduced membrane potential21, whereas the other mutants did not have the same efficiency (Fig. 2f). Similar results were shown in
U87-MG cells (Sup 3a). Therefore, we investigated whether energy generation changed in such a state. As expected, ATP concentration in cells transfected with full-length _dcf1_ was
significantly reduced to approximately 30% of that in other mutants and controls (Fig. 2g), indicating the requirement of DCF1 integrity for biological function. Next, apoptosis of glioma
cells was investigated using fluorescence activated cell sorting. After _dcf1_ transfection for 48 h, 75.1% and 13.4% of cells were in the early and late stages of apoptosis, respectively,
compared with control cells (Fig. 3a). The TUNEL assay was also used to detect apoptosis in U251 cells and the results confirmed the occurrence of late apoptosis (Fig. 3b).Apoptosis was
further identified using TEM and the results showed DCF1 overexpression initiated a series of apoptotic phenomenon, such as cytoplasmic shrinkage, nuclear fragmentation, autophagosome
formation and clusters of vacuoles in the cytoplasm (Fig. 3c). To examine the apoptotic signal mechanism induced by DCF1, we evaluated expression changes in the apoptosis-related proteins
Bcl-2 and Bax22. Western blotting showed that DCF1 overexpression up-regulated pro-apoptotic protein Bax expression, whereas it down-regulated anti-apoptotic protein Bcl-2. Next, we
investigated whether DCF1 mediated the activation of caspase-3, an apoptosis-related cysteine peptidase. Western blotting revealed that Pro-caspase-3 was cleaved into two activated subunits,
suggesting that DCF1 induced glioma cell apoptosis via the Bcl-2 protein family and finally activated the caspase-3 signaling pathway (Fig. 4a). To elucidate the apoptotic mechanisms
induced by DCF1 at a systemic level, we proposed a signaling network based on the existing literature and our experiments (Fig. 4b), including known mitochondrial membrane permeability
transition pore proteins HTRA2, BIRC2, DIABLO and CYCS, as well as the apoptosis complex protein APAF123,24. Through activation of BAX, inhibition of BCL-2 and damage to mitochondria, DCF1
eventually leads to apoptosis by activating caspase-3 in an intrinsic apoptosis pathway. Assuming that tumorigenesis was caused by the downregulation of _dcf1_, we subcutaneously injected
U251 and U251-_dcf1_ cells into the right flank of nude mice to investigate its tumorigenicity25. After transplantation of U251 cells, tumors were visible at the transplant site with 75%
tumorigenicity, whereas nude mice transplanted with U251-_dcf1_ cells showed tumorigenicity of only 25% (Fig. 4c). More importantly, the difference in tumor size was significant, as the
average tumor volume was > 10-fold greater in U251 cells than that in U251-_dcf1_ cells (Fig. 4d). In addition, tumors in the control groups (U251 cells) had rich vessel distribution,
with increased proliferation ability compared with that in the experimental group (U251-_dcf1_ cells), in which the vessels appeared dark (Fig. 4d). Incidentally, when we evaluated GFAP
expression in primary glioma cells isolated from the tumors, the U251-_dcf1_ group showed less glioma content than the controls (Sup. 3b). An in vivo assay clearly showed that _dcf1_ indeed
contributed to glioma tumorigenicity and could serve as a potential therapeutic target for glioma treatment. Thanks of a lower expression of dcf1 in the U251 glioma cell, we finally argued
the causal relationship between dcf1 and tumorigenicity. We found by Bisulfite-modified genomic DNA analysis that there are 3 CpG sites (−1339, −1322, −1299) from −1350 to −1200 with a
complete methylation in _dcf1_ promoter in U251 cells (Fig. 5 A). To investigate the release from methylation –mediated repression, 5-Azacytidine, an inhibitor of DNA methyltransferaes was
used. Results show that expression of dcf1 at both protein and mRNA levels is in a concentration-dependent manner (Fig. 5 B and Fig. 5 C). Correspondingly, methylation mutation at −1339 and
−1322 was demonstrated a significant upregulation of dcf1 gene (Fig. 5 D). Further, to assess the effect of dcf1 demethylation on the tumor proliferation, CCK-8 assay was used to detect the
viability in cultured U251 glioma cells. The results showed that compared with controls, cells treated with 5-Azacytidine were significantly reduced in cell proliferation, powerfully
indicating inhibition of dcf1 to tumorigenicity (Fig. 5 E). Gliomas are the most lethal brain cancers. Despite great efforts to improve treatment, the survival time of patients remains 14–16
months after diagnosis26. It is therefore particularly important to find an effective treatment for gliomas. Recently, Dyn2 is as an effector to mediates glioma tumor growth and invasion26.
Silence of Dyn2 inhibited PDGFR alpha-stimulated phosphorylation of Akt and tumor growth and invasion. HES6, a transcription cofactor, was highly expressed in glioma samples. HES6 silencing
showed a reduction of cancer cell proliferation and migration27. Admittedly, it has been found that many genes are up- or down-regulated in tumorigenesis. Unfortunately, it has not so far
found a truly effective treatment of gliomas. In this study, we find that the overexpression of DCF1 significantly inhibits glioma cell proliferation, survivor and ultimately leads to
apoptosis, pushing a step forward in understanding the mechanism of tumorigenesis28. To better understand _dcf1_ functional mechanisms, we proposed a model based on our experiments and the
literature (Sup Fig. 4). In normal mitochondria with cristae integrity, cytochrome c is distributed between the outer and inner mitochondrial membranes29, AIF locates in the inner membrane
and Bax partly locates in the cytoplasm22,30. Mitochondrial membrane potential and ATP synthesis maintain normal, cellular physiological activity and procaspase-3 remains in a non-activated
state31. When DCF1 is overexpressed, it is embedded in the inner mitochondrial membrane, leading to destruction of the membrane structure, mitochondrial swelling, decreased mitochondrial
membrane potential and inhibited ATP synthesis. This extraneous interference disturbed mitochondrial homeostasis32, resulted in activating of BH-3 only protein10. Then activated BH-3 only
protein combined anti-apoptotic protein Bcl-2 and induced changes in Bcl-2 formation11,33. Bax then released from Bcl-2, transferred into the outer mitochondrial membrane34,35 and combined
with Bak in a dimeric complex comprising the mitochondrial permeability transition pore36, which altered mitochondrial outer membrane permeability and leaded to partial rupturing of the
outer membrane, resulting in mitochondrial swelling in a spherical shape37. Cytochrome c and AIF were released from the mitochondrial membrane interval29,38 and it further participated in
the formation of apoptosomes along with procaspase-9, dATP and APAF-1, which actives downstream caspase enzymes39. Procaspase-3 then becomes active caspase-340. On the other hand, after
release from the mitochondria, AIF enters the nucleus and induces nuclear shrinking and DNA fragmentation. METHODS All experiments were performed in accordance with guidelines and
regulations,Shanghai University Ethics Committee approved the experiments. PLASMID CONSTRUCTION Human _dcf1_ cDNA was amplified by PCR with _Pyrococcus furiosus_ DNA polymerase, then
subcloned into the _Eco_RI and _Bam_HI restriction sites in the pEGFP-N2 plasmid. For construction of the 4 mutants, the following primers were used: pEGFP-N2-DCF1 (sense,
5′-CGGAATTCATGGCGGCGCCGAAGGGGAG-3′; antisense, 5′-CGGGATCCGTAAAATTTCAGAATGAGCA-3′); pN-sig-DCF1 (sense, 5′-CCGGAATTCATGACCGCTTCGGCTGAAG-3′; antisense, 5′-CGGGATCCGTAAAATTTCAGAATGAGCA-3′);
pMito-DCF1(sense-1, 5′-C ATTAATAGTAATCAATTACGGGGTC-3′; antisense-1, 5′-GCTCTAGACCAGAGGCTCCCCTTCGGCG-3′; sense-2, 5′-GCTCTAGACTCCCGCCGCTGCTGCTGCT-3′; antisense-2,
5′-CGGGATCCGTAAAATTTCAGAATGAGCA-3′); and pTrans-DCF1 (sense-1, 5′-CGGAATTCATGGCGGCGCCGAAGGGGAG-3′; antisense-2, 5′-GCTCTAGAAGTTGTAGTTAAAATCCA-3′; sense-2, 5′-GCTCTAGAACAGCTGTGGAGCAGTAT-3′;
antisense-2, 5′-CGGGATCCGTAAAATTTCAGAATGAGCA-3′); pC-tail-DCF1 (sense, 5′-CGGAATTCATGGCGGCGCCGAAGGGGAG-3′; antisense, 5′-CGCGGATCCAGCAACAGTTGCACAACA-3′). CELL CULTURES The U251, U87 and
HEK293T cell lines were cultured in high-glucose DMEM supplemented with 10% fetal bovine serum (FBS), 1 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C in a humidified
atmosphere of 5% CO2. MCF-7, K-562 and H1299 cell lines were cultured in Roswell Park Memorial Institute 1640 medium supplemented with 10% FBS, 1 mM glutamine, 100 U/ml penicillin and 100
μg/ml streptomycin, at 37°C in a humidified atmosphere of 5% CO2. CELL TRANSFECTION U251 cells were plated in 24-well plates at a density of 1 × 105 cells/well and cultured at 37°C in an
atmosphere of 5% CO2 overnight for transfection. A total of l μg pEGFP-N2-DCF1 DNA per well was transfected into U251 cells using Lipofectamine 2000 according to the manufacturer's
protocol. MITOTRACKER RED STAINING Mitochondrial morphology was detected by MitoTracker Red staining. After transfection with pEGFP-N2-DCF1 for 48 h, the culture medium was removed and the
cells were stained with pre-warmed 1:1500 10% FBS–DMEM diluted MitoTracker® Red CMXRos dye (Invitrogen) at 37°C for 30 min in a humidified atmosphere of 5% CO2. Then, the loading solution
was replaced with fresh pre-warmed 10% FBS–DMEM. After staining, the cells were observed immediately by confocal laser microscopy. TEM OBSERVATION After transfection for 48 h, U251 cells
were harvested using a cell scraper and then washed 3 times with PBS. After centrifugation, cells were fixed with 1% glutaraldehyde at 4°C overnight and then fixed with 1% osmium tetroxide
at 4°C for 3 h. After dehydration and resin embedding, U251 cells were sectioned to a 70-nm thickness, stained with uranyl acetate and lead citrate and the ultrathin sectioned cells were
observed using TEM. MITOCHONDRIAL AFM OBSERVATION After transfection with plasmid pEGFP-N2 or recombinant plasmid pEGFP-N2-DCF1, mitochondria of U251 cells were isolated using the
Mitochondrial Protein Extraction Kit (KeyGen Biotech) using the manufacturer's protocol. Morphological analysis of U251 mitochondria was achieved using an ex situ Agilent 5500 AFM
system (Santa Clara, CA), with a scan rate in a tapping mode of 0.5–1 Hz and resonant frequency in the range 160–260 kHz. CELL PROLIFERATION The effect of _dcf1_ on cell proliferation was
assessed using the CCK-8 Cell Counting Kit. In brief, after seeding cells in 96-well plates for 24 h, the cells were transfected with plasmid pEGFP-N2 or recombinant plasmid pEGFP-N2-DCF1.
CCK-8 solution was added to each well at 12-h intervals. After the cells were incubated with CCK-8 solution for 30 min, the absorbance at 450 nm was measured using a microplate reader
(Bio-Rad).The cell viability was characterized by optical absorption and each experiment was performed in triplicate. CELL INVASION The effect of _dcf1_ on cell proliferation was assessed
using the BioCoat™ Matrigel™ Invasion Chamber (BD354480). After transfection of the 4 mutant cell lines with pEGFP-N2 and pEGFP-N2-DCF1 for 48 h, the cells were harvested and adjusted to a
density to 5 × 104/ml. After incubation in the invasion chamber for 22 h in a humidified atmosphere of 5% CO2, non-invaded cells were scrubbed with a cotton swab and transfected cells were
stained with crystal violetand observed using a Nikon Ti-S fluorescence microscopeat a magnification of 100×. JC-1 ASSAY The mitochondrial membrane potential was evaluated by using the JC-1
Mitochondrial Membrane Potential Detection Kit (Beyotime). In brief, before staining, 1:1000 diluted CCCP was added as a positive control for 30 min in a humidified atmosphere of 5% CO2.
After transfection for 48 h, cells were incubated with 1× JC-1 staining solution in 24-well plates for 20 min at 37°C and rinsed twice with 1× staining buffer. Finally, cells cultured in 10%
FBS/DMEM were detected using a Nikon Ti-S fluorescence microscope. CELLULAR ATP ASSAY ATP concentration was detected using the ATP Assay Kit (Beyotime). In brief, cells, including the 4
mutant strains, were cultured in 6-well plates after transfection with pEGFP-N2 or pEGFP-N2-DCF1 for 48 h, lysed and then centrifuged at 12,000 rpm for 10 min to isolate total protein. Next,
100 μl of supernatant was added to 100 μl of ATP detection solution and standard samples were generated. Luminescence was immediately detected using a plate reader (PerkinElmer). Protein
concentration was measured using the Bradford assay. FLOW CYTOMETRY After transfection with pEGFP-N2 or pEGFP-N2-DCF1 for 48 h, U251 cells were digested with trypsin (0.25% in D-Hanks
solution without EDTA) and harvested (1 × 106 cells) in 10% FBS–DMEM. Next, cells were washed twice with 2%-BSA–PBS, then resuspended in 500 μl of binding buffer, combined with 5 μl annexin
V-APC and gently mixed. Next, 5 μl of 7-AAD was added according to the protocol of the Annexin V-APC/7-AAD Apoptosis Detection Kit (KeyGen Biotech). Samples were cultured at room temperature
for 15 min before flow cytometry (Beckman). TUNEL ASSAY U251 cells were rinsed with PBS, fixed in 4% paraformaldehyde for 30 min at room temperature, permeabilized with 1% Triton X-100 for
5 min and blocked with 3% H202 at room temperature for 10 min. The samples were washed 3 times with PBS, 50 μl TdT enzyme reaction buffer was added to the slide and incubated at 37°C for 1 h
as described in the protocol of the TUNEL Apoptosis Detection Kit (KeyGEN Biotech). After removal of the reaction buffer, the sample was incubated with 50 μl DAPI for 5 min. Fluorescence at
543 nm was detected using the Zeiss LSM 710 confocal microscope. IMMUNOFLUORESCENCE U251 cells were rinsed with PBS, fixed in 4% paraformaldehyde for 10 min at room temperature and
permeabilized with 0.1% Triton X-100 for 10 min. The cells were blocked with 2% BSA–PBS at room temperature for 60 min and incubated with primary antibodies at 4°C overnight. After washing 3
times with PBS, cells were incubated with secondary antibodies for 50 min at 37°C and fluorescence at 488 nm (FITC) and 543 nm (TRITC) were detected using the Nikon Ti-S fluorescence
microscope. WESTERN BLOTTING At 48 h after transfection, the cells were washed twice with ice-cold PBS and protein was extracted according to the manufacturer's protocol (Beyotime).
Protein concentrations were detected using the BCA Protein Assay Kit (Beyotime). The total protein (30 μg) was separated by 12% SDS-PAGE and electroblotted onto nitrocellulose membranes,
which were blocked with 5% BSA–PBS for 1 h at room temperature and then incubated with β-actin antibody (1:1000; Santa Cruz), DCF1 (1:3000), Bcl-2 (1:2000, CST), Bax (1:2000, CST) and
procaspase-3 (1:2000, CST) overnight at 4°C. Next infrared dye 800-conjugated affinity-purified goat anti-rat IgG (Zemed, USA) and infrared dye 700-labeled goat anti-rabbit IgG (Zemed, USA)
secondary antibodies were added and incubated for 1 h at room temperature. Immunoblotting bands were detected and quantified using the LI-COR Odyssey infrared imaging system (simultaneous
two-color targeted analysis) and software (LI-COR). CELLULAR TUMORIGENICITY IN NUDE MICE U251 cells transfected with control plasmids or pEGFP-N2-DCF1 for 48 h, harvested by trypsin
digestion, then 107 cells were injected subcutaneously into the right flank of the nude mice (4 mice/treatment group). REFERENCES * Van Meir, E. G. et al. Exciting new advances in
neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin 60, 166–193 (2010). Article Google Scholar * Lawson, H. C. et al. Interstitial chemotherapy for malignant
gliomas: the Johns Hopkins experience. J Neurooncol 83, 61–70 (2007). Article CAS Google Scholar * Holland, E. C. Glioblastoma multiforme: the terminator. Proceedings of the National
Academy of Sciences of the United States of America 97, 6242–6244 (2000). Article CAS ADS Google Scholar * Chen, J. et al. A restricted cell population propagates glioblastoma growth
after chemotherapy. Nature 488, 522–526 (2012). Article CAS ADS Google Scholar * Eyler, C. E. et al. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide
synthase-2. Cell 146, 53–66 (2011). Article CAS Google Scholar * Wen, T., Gu, P. & Chen, F. Discovery of two novel functional genes from differentiation of neural stem cells in the
striatum of the fetal rat. Neuroscience letters 329, 101–105 (2002). Article CAS Google Scholar * Li, X. et al. MicroRNA-351 regulates TMEM 59 (DCF1) expression and mediates neural stem
cell morphogenesis. RNA biology 9, 292–301 (2012). Article CAS Google Scholar * Wang, L. et al. A novel function of dcf1 during the differentiation of neural stem cells in vitro. Cellular
and molecular neurobiology 28, 887–894 (2008). Article Google Scholar * Lin, H. Y. et al. Suppressor of cytokine signaling 6 (SOCS6) promotes mitochondrial fission via regulating DRP1
translocation. Cell death and differentiation 20, 139–153 (2013). Article CAS ADS Google Scholar * Sheridan, C. & Martin, S. J. Mitochondrial fission/fusion dynamics and apoptosis.
Mitochondrion 10, 640–648 (2010). Article CAS Google Scholar * Fulda, S., Galluzzi, L. & Kroemer, G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov 9, 447–464 (2010).
Article CAS Google Scholar * Liu, X. et al. A protein interaction network for the analysis of the neuronal differentiation of neural stem cells in response to titanium dioxide
nanoparticles. Biomaterials 31, 3063–3070 (2010). Article CAS Google Scholar * Sandoval, H. et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature 454, 232–235
(2008). Article CAS ADS Google Scholar * Sun, M. G. et al. Correlated three-dimensional light and electron microscopy reveals transformation of mitochondria during apoptosis. Nature cell
biology 9, 1057–1065 (2007). Article CAS Google Scholar * Li, Y. et al. Cholera toxin induces malignant glioma cell differentiation via the PKA/CREB pathway. Proceedings of the National
Academy of Sciences of the United States of America 104, 13438–13443 (2007). Article CAS ADS Google Scholar * Li, J. et al. Phosphorylation of ACAP1 by Akt regulates the
stimulation-dependent recycling of integrin beta1 to control cell migration. Dev Cell 9, 663–673 (2005). Article CAS Google Scholar * Friedl, P. & Wolf, K. Tumour-cell invasion and
migration: diversity and escape mechanisms. Nat Rev Cancer 3, 362–374 (2003). Article CAS Google Scholar * Zhang, K. et al. Slug enhances invasion ability of pancreatic cancer cells
through upregulation of matrix metalloproteinase-9 and actin cytoskeleton remodeling. Lab Invest 92, 1801 (2012). Article Google Scholar * Kleber, S. et al. Yes and PI3K bind CD95 to
signal invasion of glioblastoma. Cancer Cell 13, 235–248 (2008). Article CAS Google Scholar * Kanome, T. et al. Characterization of Jumping translocation breakpoint (JTB) gene product
isolated as a TGF-beta1-inducible clone involved in regulation of mitochondrial function, cell growth and cell death. Oncogene 26, 5991–6001 (2007). Article CAS Google Scholar * Brenner,
C. & Grimm, S. The permeability transition pore complex in cancer cell death. Oncogene 25, 4744–4756 (2006). Article CAS Google Scholar * Chipuk, J. E. et al. Mechanism of apoptosis
induction by inhibition of the anti-apoptotic BCL-2 proteins. Proceedings of the National Academy of Sciences of the United States of America 105, 20327–20332 (2008). Article CAS ADS
Google Scholar * Kinnally, K. W. & Antonsson, B. A tale of two mitochondrial channels, MAC and PTP, in apoptosis. Apoptosis: an international journal on programmed cell death 12,
857–868 (2007). Article CAS Google Scholar * Spencer, S. L. & Sorger, P. K. Measuring and modeling apoptosis in single cells. Cell 144, 926–939 (2011). Article CAS Google Scholar *
Huang, J. et al. Up-regulation of DLK1 as an imprinted gene could contribute to human hepatocellular carcinoma. Carcinogenesis 28, 1094–1103 (2007). Article CAS Google Scholar * Feng, H.
et al. Dynamin 2 mediates PDGFRalpha-SHP-2-promoted glioblastoma growth and invasion. Oncogene 31, 2691–2702 (2012). Article CAS Google Scholar * Haapa-Paananen, S. et al. HES6 gene is
selectively overexpressed in glioma and represents an important transcriptional regulator of glioma proliferation. Oncogene 31, 1299–1310 (2012). Article CAS Google Scholar * Lecoeur, H.
et al. HIV-1 Tat protein directly induces mitochondrial membrane permeabilization and inactivates cytochrome c oxidase. Cell Death Dis 3, e282 (2012). Article CAS Google Scholar * Chipuk,
J. E. et al. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 148, 988–1000 (2012). Article CAS Google Scholar * Benard, G. et
al. IBRDC2, an IBR-type E3 ubiquitin ligase, is a regulatory factor for Bax and apoptosis activation. The EMBO journal 29, 1458–1471 (2010). Article CAS Google Scholar * Li, Z. et al.
Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 141, 859–871 (2010). Article CAS Google Scholar * Nunnari, J. &
Suomalainen, A. Mitochondria: in sickness and in health. Cell 148, 1145–1159 (2012). Article CAS Google Scholar * Lomonosova, E. & Chinnadurai, G. BH3-only proteins in apoptosis and
beyond: an overview. Oncogene 27 SUPPL 1S2–19 (2008). Article CAS Google Scholar * Edlich, F. et al. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 145,
104–116 (2011). Article CAS Google Scholar * Dussmann, H. et al. Single-cell quantification of Bax activation and mathematical modelling suggest pore formation on minimal mitochondrial
Bax accumulation. Cell death and differentiation 17, 278–290 (2010). Article CAS Google Scholar * Soriano, M. E. & Scorrano, L. Traveling Bax and forth from mitochondria to control
apoptosis. Cell 145, 15–17 (2011). Article CAS Google Scholar * Yip, K. W. & Reed, J. C. Bcl-2 family proteins and cancer. Oncogene 27, 6398–6406 (2008). Article CAS Google Scholar
* Henry-Mowatt, J., Dive, C., Martinou, J. C. & James, D. Role of mitochondrial membrane permeabilization in apoptosis and cancer. Oncogene 23, 2850–2860 (2004). Article CAS Google
Scholar * Fulda, S. & Debatin, K. M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 25, 4798–4811 (2006). Article CAS Google Scholar * Rasola, A.
& Bernardi, P. Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium 50, 222–233 (2011). Article CAS Google Scholar Download references
ACKNOWLEDGEMENTS This work was supported by the National Natural Science Foundation of China (Grant No 31070954, 81271253,11172158), the Key Innovation Project of Shanghai Municipal
Education Commission (Grant No. 14ZZ090) and in part by the First-class Discipline of Universities in Shanghai. We are grateful for the support of Mr. Jie Tao in taking pictures of nude
mice. We also acknowledge the technical support of Mr.Pengfei Li for AFM observation. AUTHOR INFORMATION Author notes * Xie Yuqiong and Li Qiang contributed equally to this work. AUTHORS AND
AFFILIATIONS * Laboratory of Molecular Neural Biology, School of Life Sciences, Shanghai University, Shanghai, 200444, China Yuqiong Xie, Qiang Li, Qingbo Yang, Mei Yang, Zhifeng Zhang,
Huang Yan, Ruili Feng, Shiqing Zhang, Chen Huang & Tieqiao Wen * Institute of Systems Biology, Shanghai University, Shanghai, 200444, China Yuqiong Xie, Qiang Li, Mei Yang, Liucun Zhu,
Shiqing Zhang, Zengrong Liu & Tieqiao Wen Authors * Yuqiong Xie View author publications You can also search for this author inPubMed Google Scholar * Qiang Li View author publications
You can also search for this author inPubMed Google Scholar * Qingbo Yang View author publications You can also search for this author inPubMed Google Scholar * Mei Yang View author
publications You can also search for this author inPubMed Google Scholar * Zhifeng Zhang View author publications You can also search for this author inPubMed Google Scholar * Liucun Zhu
View author publications You can also search for this author inPubMed Google Scholar * Huang Yan View author publications You can also search for this author inPubMed Google Scholar * Ruili
Feng View author publications You can also search for this author inPubMed Google Scholar * Shiqing Zhang View author publications You can also search for this author inPubMed Google Scholar
* Chen Huang View author publications You can also search for this author inPubMed Google Scholar * Zengrong Liu View author publications You can also search for this author inPubMed Google
Scholar * Tieqiao Wen View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS T.W. designed and wrote the main manuscript text. Y.X., Q.L., Q.Y.,
M.Y., Z.Z., R.F., S.Z. and C.H. performed experiments. H.Y. prepared figures. L.Z. and Z.L. did the analysis of system biology. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no
competing financial interests. ELECTRONIC SUPPLEMENTARY MATERIAL SUPPLEMENTARY INFORMATION supplementary information RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons
Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/ Reprints and permissions ABOUT THIS ARTICLE
CITE THIS ARTICLE Xie, Y., Li, Q., Yang, Q. _et al._ Overexpression of DCF1 inhibits glioma through destruction of mitochondria and activation of apoptosis pathway. _Sci Rep_ 4, 3702
(2014). https://doi.org/10.1038/srep03702 Download citation * Received: 26 November 2013 * Accepted: 18 December 2013 * Published: 15 January 2014 * DOI: https://doi.org/10.1038/srep03702
SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to
clipboard Provided by the Springer Nature SharedIt content-sharing initiative