Play all audios:
ABSTRACT The study was designed to investigate the role of endogenous sulfur dioxide (SO2) in collagen remodeling and its mechanisms in vascular smooth muscle cells (VSMCs). Overexpression
of endogenous SO2 synthase aspartate aminotransferase (AAT) 1 or 2 increased SO2 levels and inhibited collagen I and III expressions induced by transforming growth factor (TGF)-β1 in VSMCs.
In contrast, AAT1 or AAT2 knockdown induced a severe collagen deposition in TGF-β1-treated VSMCs. Furthermore, AAT1 or AAT2 overexpression suppressed procollagen I and III mRNA, upregulated
matrix metalloproteinase (MMP)-13 expression, downregulated tissue inhibitors of MMP-1 level and vice versa. Mechanistically, AAT1 or AAT2 overexpression inhibited phosphorylation of type I
TGF-β receptor (TβRI) and Smad2/3 in TGF-β1-stimulated VSMCs. Whereas SB431542, an inhibitor of TGF-β1/Smad signaling pathway, attenuated excessive collagen deposition induced by AAT
knockdown. Most importantly, ectopically expressing AAT or exogenous addition of 100 μM SO2 blocked AAT deficiency-aggravated collagen accumulation in TGF-β1-stimulatd VSMCs, while no
inhibition was observed at 100 μM ethyl pyruvate. These findings indicated that endogenous SO2 alleviated collagen remodeling by controlling TGF-β1/TβRI/Smad2/3-mediated modulation of
collagen synthesis and degradation. SIMILAR CONTENT BEING VIEWED BY OTHERS DEHYDROCORYDALINE MAINTAINS THE VASCULAR SMOOTH MUSCLE CELL CONTRACTILE PHENOTYPE BY UPREGULATING SPTA1 Article
Open access 20 January 2025 HIF-1Α KNOCKDOWN ATTENUATES PHENOTYPIC TRANSFORMATION AND OXIDATIVE STRESS INDUCED BY HIGH SALT IN HUMAN AORTIC VASCULAR SMOOTH MUSCLE CELLS Article Open access
15 November 2024 MICRORNA‑331 INHIBITS ISOPROTERENOL‑INDUCED EXPRESSION OF PROFIBROTIC GENES IN CARDIAC MYOFIBROBLASTS VIA THE TGFΒ/SMAD3 SIGNALING PATHWAY Article Open access 28 January
2021 INTRODUCTION Severe structural changes in vascular walls characterized by vascular collagen remodeling are central to the pathophysiology of vascular diseases such as hypertension,
atherosclerosis and restenosis after coronary angioplasty1,2. Vascular collagen mainly consists of collagen I and III. Collagen I is associated with the tenacity and tensile strength of
vascular wall, while collagen III is associated with the elasticity of vascular wall, both play important role in maintaining the integrity of vascular structure. Previous study indicated
that vascular collagen remodeling was the consequence of an imbalance between collagen synthesis and degradation, characterized by excessive deposition of collagen, disequilibrium of
collagen types (increased ratio of collagen I/III) and disorganized collagen arrangement3. Among them, the excessive deposition of collagen I and III in vascular wall is one of the most
important factors of vascular remodeling and is also a common consequence of many cardiovascular diseases. The synthesis and degradation of collagen in VSMC are critical to vascular
remodeling3,4, but much less is known about the pathogenetic mechanism of collagen remodeling in VSMC under pathological conditions, especially the regulatory mechanism of abnormal synthesis
and (or) degradation of collagen has not been fully elucidated. More and more research has suggested that endogenous gaseous signaling molecules play important function in cardiovascular
system. It was reported that the gasotransmitter hydrogen sulfide inhibited the abnormal accumulation of vascular collagen and alleviated vascular remodeling in spontaneously hypertensive
rats5,6. Sulfur dioxide (SO2), another gasotransmitter, shares the same substrate with hydrogen sulfide, which also can be endogenously generated from a sulfur-containing amino acid7.
L-cysteine can be oxidized by cysteine dioxygenase to L-cysteine sulfinate, which can be transformed into β-sulfinylpyruvate by aspartate aminotransferase (AAT) and then spontaneously
decomposes to pyruvate and SO28. Study has shown that SO2 could inhibit hypoxic pulmonary vascular remodeling9. However, it’s still unclear about the regulation of endogenous SO2 on the
collagen remodeling in VSMCs and its possible mechanism. Transforming growth factor-β1 (TGF-β1) is widely known as a key factor in vascular collagen remodeling, participating in the
development of vascular injury in a variety of cardiovascular diseases. There are three isoforms of TGF-β, TGF-β1, TGF-β2 and TGF-β3. TGF-β1 is the major isoform of the TGF-β superfamily,
can be produced by VSMCs and regulates growth, differentiation, migration and proliferation of VSMCs as well as extracellular matrix deposition10,11. Although TGF-β1 action involve many
downstream signaling pathways and cross-talk, the intracellular Smad signaling pathway is considered to play a crucial role in mediating the intracellular response to TGF-β112. Activated
TGF-β1 binds tightly to transmembrane type II TGF-β receptor (TβRII), a serine/threonine kinase which phosphorylates type I TGF-β receptor (TβRI). Then the phosphorylated TβRI triggers Smad2
and Smad3 phosphorylation. The phosphorylated Smad2/3 forms a complex with Smad4 and translocate from cytoplasm into the nucleus and acts as a transcription factor to enhance the
transcription of collagen, matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs (TIMPs). TGF-β1 directly promotes collagen synthesis and inhibits collagen degradation, thus
resulting in the abnormal deposition of collagen13,14. Based on these discoveries, we designed experiments to explore the role of endogenous SO2 in the TGF-β1-induced collagen remodeling in
VSMCs and its possible mechanisms. RESULTS ENDOGENOUS SULFUR DIOXIDE WAS ASSOCIATED WITH THE INHIBITION OF TGF-Β1-INDUCED COLLAGEN REMODELING IN VSMCS Sulfur dioxide could be endogenously
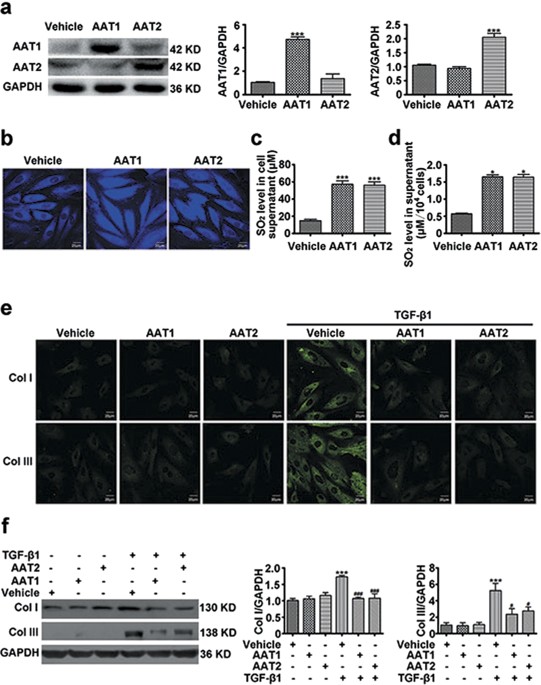
produced from L-cysteine in mammals through transamination by AAT. To investigate the effect of endogenous SO2 on collagen remodeling in VSMCs, we overexpressed two isozymes of AAT, AAT1 and
AAT2 in VSMCs respectively. Transfection of VSMCs with AAT1 or AAT2 plasmid could significantly increase AAT1 or AAT2 protein expression as compared with vehicle (Fig. 1a). In accordance,
the endogenous SO2 level was obviously elevated in VSMCs transfected with AAT1 or AAT2 plasmid (Fig. 1b). HPLC-FD assay also showed that much higher SO2 content in the supernatant from VSMCs
transfected with AAT1 or AAT2 plasmid than vehicle (Fig. 1c). And the concentration of SO2 corrected by the number of cells was also increased significantly by AAT overexpression (Fig. 1d).
Both immunofluorescence method and Western blot analysis showed that TGF-β1 could upregulated protein expression of collagen I and III in VSMCs, while AAT1 or AAT2 overexpression could
significantly inhibit the TGF-β1-induced collagen I and III expression (Fig. 1e,f). Therefore, these results indicated that endogenous SO2 might suppress TGF-β1-induced excessive collagen
deposition in VSMCs. ENDOGENOUS SULFUR DIOXIDE DEFICIENCY AGGRAVATED THE TGF-Β1-INDUCED COLLAGEN REMODELING IN VSMCS To further investigate the potential causal role of endogenous SO2 in
vascular collagen remodeling, we used shRNA to knock down AAT1 or AAT2. Specific knockdown of AAT1 or AAT2 was verified at protein level by Western blot analysis (Fig. 2a). In agreement,
transfection with AAT1 shRNA or AAT2 shRNA significantly decreased endogenous SO2 in VSMCs, as well as the SO2 level in the supernatant (Fig. 2b,c). As compared with control shRNA,
endogenous SO2 silencing greatly exacerbated TGF-β1-induced collagen I and III expression in VSMCs (Fig. 2d,e). These data probably supported a significant role of endogenous SO2 in the
regulation of collagen remodeling in VSMCs. ENDOGENOUS SULFUR DIOXIDE LIKELY INHIBITED THE TGF-Β1-INDUCED COLLAGEN REMODELING IN VSMCS VIA SUPPRESSING COLLAGEN SYNTHESIS Collagen synthesis
is one major link in collagen expression regulation, mainly reflected in the transcription and translation of procollagen gene. Real-time quantitative PCR (RT-PCR) analysis showed that
TGF-β1 stimulation increased the mRNA levels of procollagen I and III, while AAT1 or AAT2 overexpression could significantly inhibit the TGF-β1-induced mRNA expression of procollagen I and
III (Fig. 3a,b). In contrast, AAT1 or AAT2 knockdown further exacerbated it (Fig. 3c,d). These results indicated that endogenous SO2 likely inhibited the collagen remodeling in VSMCs via
inhibiting collagen synthesis. ENDOGENOUS SULFUR DIOXIDE POSSIBLY INHIBITED THE TGF-Β1-INDUCED COLLAGEN REMODELING IN VSMCS VIA PROMOTING COLLAGEN DEGRADATION Collagen degradation is the
other major link in collagen expression regulation. The key molecules regulating the collagen degradation in VSMCs are matrix metalloproteinase-13 (MMP-13) promoting collagen degradation and
tissue inhibitor of metalloproteinase-1 (TIMP-1) suppressing collagen degradation. RT-PCR and Western blot analysis showed that TGF-β1 downregulated the MMP-13 mRNA and protein levels in
VSMCs, whereas increased the TIMP-1 mRNA and protein levels (Fig. 4a–c). AAT1 or AAT2 overexpression in VSMCs could obviously elevate the TGF-β1-downregulated MMP-13 mRNA and protein levels
and suppress the TGF-β1-upregulated TIMP-1 mRNA and protein levels (Fig. 4a–c). In contrast, AAT1 or AAT2 knockdown could further inhibit the TGF-β1-downregulated mRNA and protein
expressions of MMP-13 and further promote the TGF-β1-upregulated mRNA and protein expressions of TIMP-1 (Fig. 4d–f). These results suggested that endogenous SO2 possibly inhibited the
collagen remodeling in VSMCs via promoting collagen degradation. ENDOGENOUS SULFUR DIOXIDE MIGHT INHIBIT THE SMAD2/3 SIGNALING PATHWAY DURING TGF-Β1-INDUCED COLLAGEN REMODELING IN VSMCS
Considering TGF-β/Smad2/3 signal is the key pathway in regulating collagen remodeling, next we observed whether this signaling pathway was involved in the regulation of endogenous SO2 on
collagen remodeling in VSMCs. Western blot analysis showed that TGF-β1 promoted the phosphorylation of Smad2 and Smad3 as compared with control, while AAT1 or AAT2 overexpression could
markedly inhibit the TGF-β1-induced phosphorylation of Smad2/3 (Fig. 5a). To examine if the expression of gene downstream of Smad2/3 was influenced by AAT overexpression, we detected the
expression of plasminogen activator inhibitor-1 (PAI-1) using RT-PCR. The data showed that AAT1 or AAT2 overexpression could significantly reduce the TGF-β1-upregulated mRNA expression of
PAI-1 (Fig. 5b). On the contrary, AAT1 or AAT2 deficiency dramatically enhanced the TGF-β1-induced phosphorylation of Smad2/3 (Fig. 5c). These data indicated that Smad2/3 signaling pathway
might be involved in the modulation of endogenous SO2 on collagen remodeling in VSMCs. ENDOGENOUS SULFUR DIOXIDE PROBABLY INHIBITED TΒRI PHOSPHORYLATION DURING TGF-Β1-INDUCED COLLAGEN
REMODELING IN VSMCS TGF-β1 binds to TβRII, which phosphorylates TβRI. Then the phosphorylated TβRI triggers Smad2 and Smad3 activation. Therefore, we tested whether endogenous SO2 could
interfere with phosphorylation of TβRI. Interestingly, TGF-β1 promoted TβRI phosphorylation in VSMCs, while AAT1 or AAT2 overexpression indeed downregulated the TGF-β1-induced TβRI
phosphorylation (Fig. 6a). In contrast, AAT1 or AAT2 silencing aggravated the TGF-β1-induced TβRI phosphorylation (Fig. 6b). These results suggested that endogenous SO2 might inactivate the
TGF-β/Smad2/3 signaling pathway during collagen remodeling of VSMCs through inhibiting TβRI phosphorylation. ENDOGENOUS SULFUR DIOXIDE DEFICIENCY PROMOTED COLLAGEN REMODELING MEDIATED BY
TGF-Β1/SMAD SIGNALING PATHWAY IN VSMCS To further explore whether TGF-β1/Smad pathway mediated the regulatory effect of endogenous SO2 on collagen remodeling in VSMCs, we applied SB431542,
an inhibitor of TGF-β1/Smad signaling pathway, to VSMCs transfected with AAT1 or AAT2 shRNA. RT-PCR showed SB431542 almost completely abolished the AAT1- or AAT2-silencing induction in mRNA
expression of procollagen I and III (Fig. 7a,b). Moreover, enhanced collagen I and III protein levels evoked by AAT1 or AAT2 knockdown were also greatly repressed by SB431542 (Fig. 7c,d),
suggesting endogenous SO2 likely inhibited collagen remodeling at least in part by blocking the activation of TGF-β1/Smad signaling pathway. ECTOPICALLY EXPRESSING AAT ABOLISHED AAT
DEFICIENCY-EXACERBATED COLLAGEN DEPOSITION IN TGF-Β1-STIMULATD VSMCS In order to exclude the collateral effects that were often produced by shRNA constructs, we ectopically expressed AAT1 or
AAT2 in AAT1 or AAT2 deficiency VSMCs, respectively. Western blot analysis showed that transfecting with AAT1 plasmid increased the inhibited protein expression of AAT1 in AAT1-scilencing
VSMCs and transfecting with AAT2 plasmid normalized AAT2 expression in AAT2-knocked down cells (Fig. 8a). Moreover, ectopic AAT1 or AAT2 expression significantly upregulated the inhibited
SO2 level in AAT deficient VSMCs (Fig. 8b). Of note, AAT1 or AAT2 overexpression substantially abolished the excessive deposition of collagen I and III induced by AAT1 or AAT2 knocked-down
in the TGF-β1-treated VSMCs (Fig. 8a). And the enhanced mRNA levels of procollagen I and III by AAT1 or AAT2 deficiency were also retarded by ectopic AAT1 or AAT2 expression (Fig. 8c,d).
These data verified that the effects produced by AAT shRNA constructs on collagen remodeling resulted from AAT deficiency. SULFUR DIOXIDE BUT NOT PYRUVATE PROTECTED AGAINST AAT
DEFICIENCY-INDUCED COLLAGEN REMODELING IN TGF-Β1-TREATED VSMCS Since SO2 and pyruvate were generated equimolar from L-cysteine catalyzed by AAT, we next observed the effects of 100 μM SO2
derivatives (Na2SO3/NaHSO3, 3:1 mole ratios) and 100 μM ethyl pyruvate on the collagen remodeling induced by AAT deficiency. HPLC-FD analysis showed that SO2 derivatives at 100 μM
significantly upregulated the inhibited SO2 level by AAT1 or AAT2 deficiency, while ethyl pyruvate did not impact endogenous SO2 content (Fig. 9b). Western blot analysis demonstrated that
100 μM SO2 derivatives could markedly inhibit AAT silencing-aggravated collagen I and III deposition in TGF-β1-treatd VSMCs, while no inhibition was observed at 100 μM ethyl pyruvate (Fig.
9a). Furthermore, 100 μM SO2 derivatives but not 100 μM ethyl pyruvate reduced excessive collagen synthesis in AAT1- or AAT2-knocked down plus TGF-β1-treated VSMCs (Fig. 9c,d). These data
suggested that SO2 but not pyruvate exerted a crucially protective role in AAT deficiency promoted-collagen remodeling in TGF-β1-treated VSMCs. DISCUSSION With the increasing understanding
about the function of hydrogen sulfide as gaseous signaling molecule in cardiovascular system, more and more attention has been paid to the physiological and pathophysiological functions of
SO2, another important gasotransmitter in cardiovascular regulation. Recent studies have found that SO2 can be endogenously generated from sulfur-containing amino acid metabolism in human
body7. L-cysteine can be oxidized into L-cysteine sulfinate by cysteine dioxygenase, the latter can be transformed into β-sulfinylpyruvate by AAT, then spontaneously decomposes to pyruvate
and SO28. A part of SO2 _in vivo_ can quickly combine with water to form sulphite, while other exists in gaseous form. Previous Studies found that exogenous SO2 donor inhibited the hypoxic
pulmonary vascular remodeling9. In addition, Liu _et al._ reported that SO2 suppressed aortic VSMCs proliferation15. However, it’s still unclear about whether endogenous SO2 regulates
collagen remodeling in VSMCs and its underlying mechanisms. Therefore, we overexpressed SO2 generating enzyme, AAT in VSMCs or knocked down AAT with shRNA, demonstrating endogenous SO2
inhibited the TGF-β1-induced excessive collagen deposition in VSMCs and its possible mechanisms. Maintenance of extracellular matrix (ECM) is one of the major functions of VSMCs. Disturbance
of ECM homeostasis leads to excessive ECM deposition, much of which is produced by VSMCs, is a common consequence of cardiovascular diseases such as atherosclerosis, hypertension and
restenosis after coronary angioplasty16,17,18. Most of ECM proteins within vascular walls are collagen I and III18. Therefore, searching for endogenous molecules regulating collagen
remodeling in VSMCs, can help to protect from the occurrence and development of cardiovascular disease. SO2, a gaseous signaling molecule, can be endogenously generated in many cells,
including VSMCs19. To explore the effect of endogenous SO2 on collagen remodeling in VSMCs, we used immunofluorescence staining and Western blot to detect the protein expression of collagen
I and III in VSMCs. The data showed that endogenous SO2 significantly inhibited the TGF-β1-induced collagen I and III protein levels. Collagen remodeling contains two major links, namely
excessively increased collagen synthesis and/or reduced collagen degradation. Collagen synthesis reflects in the transcription and translation of procollagen gene. So next we detected the
effect of endogenous SO2 on collagen synthesis. We found that endogenous SO2 markedly suppressed the TGF-β1-induced procollagen I and III mRNA levels, suggesting that endogenous SO2 could
inhibit collagen synthesis in VSMCs. The balance between degradation of ECM is guaranteed by MMPs and TIMPs20,21. MMPs consist of a family of Zn-dependent endopeptidases that degrade ECM and
basement membrane. They take part in tissue remodeling, cell infiltration and tumor invasion. For example, MMP-2, MMP-9 and MMP-13 are reported to have pivotal roles in vascular remodeling
in several disease states20,21,22. MMP-2 mainly degrades type-IV, V, VII, X and XI collagen, gelatin, fibronectin, laminin and elastin. MMP-9 mainly degrades type-IV, V, VII, X collagen,
gelatin, elastin, laminin, proteoglycan, fibronectin and entacin. Of note, MMP-13 degrades type-I, II, III, IV, IX, X and XIV collagen, fibronectin, tenascin-C, proteoglycan and gelatin22.
MMPs activation can be suppressed by their endogenous inhibitors, the TIMPs. Four types of TIMPs have been found, of which TIMP-1 is the important inhibitor of MMP-1, -3, -9 and -1323.
Therefore, in this study we detected the effect of endogenous SO2 on MMP-13 and TIMP-1 mRNA and protein expressions in VSMCs. We found that TGF-β1 downregulated the mRNA and protein
expressions of MMP-13 and upregulated the mRNA and protein levels of TIMP-1. Endogenous SO2 could dramatically increase the TGF-β1-downregulated MMP-13 level and decrease the
TGF-β1-upregulated TIMP-1 level, suggesting endogenous SO2 could promote collagen degradation. TGF-β1 is a potent profibrotic factor that is involved in vascular fibrosis24,25,26. Previous
study reported that TGF-β1 could stimulate collagen synthesis4,27,28 and inhibit collagen degradation3,29,30. Classic TGF signaling pathway is realized by Smad family members3,4,12.
Activated TGF-β1 combines to TβRII, which phosphorylates TβRI. Then the phosphorylated TβRI triggers Smad2 and Smad3 phosphorylation. The phosphorylated Smad2/3 forms a complex with Smad4,
translocate from cytoplasm into the nucleus and activate the transcription of procollagen13,14. Previous studies have showed that TGF-β1 activated collagen remodeling in VSMCs mainly through
TGF-β/Smad signaling pathway3,4,31. TGF-β1 could significantly increase the collagen protein expression in VSMCs through Smad2/3 signaling pathway4,16. Our present study showed that
endogenous SO2 markedly inhibited the TGF-β1-induced phosphorylation of Smad2 and Smad3 in VSMCs. In order to further explore how endogenous SO2 blocked the Smad signaling pathway, we
detected the phosphorylation level of TβRI. The data indicated that endogenous SO2 suppressed the phosphorylation of TβRI. And SB431542, an inhibitor of TGF-β1/Smad signaling pathway, could
significantly abolish the promoting effect of endogenous SO2 deficiency on TGF-β1-induced collagen remodeling in VSMCs. These results suggested that endogenous SO2 alleviated the collagen
remodeling in VSMCs at least in part by inhibiting TGF-β/Smad signaling pathway. The limitation of the present study involved the employment of exclusively one shRNA construct targeting
either AAT1 or AAT2 and compared to only one control for all experiments. Considering the collateral effects that were often produced by such constructs, we used ectopically expressing AAT
in knocked-down cells. The data showed that ectopic AAT1 or AAT2 expression upregulated the repressed SO2 level in AAT knocked down-VSMCs. As a consequence, AAT1 or AAT2 overexpression
abolished AAT silencing-induced collagen I and III deposition in TGF-β1-treated VSMCs possibly via reducing collagen synthesis. These results supported the assumption that AAT1 and AAT2
shRNA used in the present study targeted AAT1 and AAT2 genes, respectively. AAT shRNA-induced exacerbation of collagen remodeling might result from AAT deficiency, other than the off-target
effects. Endogenous SO2 was produced from L-cysteine in a two-step reaction, in which the later step was catalyzed by AAT and resulted in equimolar pyruvate as well7,8,32. Previous studies
showed that pyruvate could modulate TGF-β signaling33,34. However, whether the effects of AAT manipulation on collagen remodeling were caused by SO2 or pyruvate had not been elucidated.
Here, we added exogenous SO2 derivatives or pyruvate in AAT-knocked down VSMCs. Considering the instability of pyruvate, ethyl pyruvate, a simple aliphatic ester derived from pyruvic acid,
was widely implicated in scientific research instead of pyruvate34,35. Since SO2 and pyruvate were generated equimolar from L-cysteine catalyzed by AAT7,8,32, VSMCs were pretreated with 100
μM SO2 derivatives or 100 μM ethyl pyruvate in the present study. The results showed that SO2 derivatives at 100 μM significantly inhibited AAT silencing-exacerbated collagen I and III
deposition in TGF-β1-treated VSMCs, while no inhibition was observed at 100 μM ethyl pyruvate. These data demonstrated that SO2 generated by AAT but not pyruvate played a crucially
protective role against collagen remodeling in AAT knocked-down VSMCs with TGF-β1 stimulation. In conclusion, we discovered an inhibitory effect of endogenous SO2 on TGF-β1-induced collagen
remodeling in VSMCs via suppressing collagen synthesis and promoting collagen degradation. The underlying mechanism might involve inhibiting the TβRI phosphorylation by endogenous SO2 to
block the Smad2/3 signaling pathway activation. These findings suggest that endogenous SO2 may be a promising therapeutic target for vascular collagen remodeling related cardiovascular
diseases such as hypertension. METHODS CELL CULTURE Rat A7r5 VSMCs were obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in Dulbecco’s modified
Eagle’s medium (DMEM) containing 10% FBS, 2 mmol/l glutamine and 20 mmol/l HEPES (pH 7.4) in an atmosphere of 5% CO2 at 37 °C. OVEREXPRESSION OF AAT1 OR AAT2 IN VSMCS The cDNA fragment
encoding the full-length rat AAT1 (NM_012571.2) or AAT2 (NM_013177.2) was amplified by PCR and inserted into the pIRES2 vector and the resultant plasmid AAT1 or AAT2 was verified by DNA
sequencing. A7r5 VSMCs were then transfected with AAT1, AAT2 or vehicle plasmid using JetPEI reagent according to the manufacturer’s instructions (Polyplus Transfection, Illkirch, France).
KNOCKDOWN OF AAT1 OR AAT2 IN VSMCS Rat AAT1 shRNA and AAT2 shRNA were both from OriGene Technologies (Rockville, MD, USA). A7r5 VSMCs were seeded in 6-well plates to 60–80% confluence. AAT1
shRNA, AAT2 shRNA and control shRNA were respectively transfected into A7r5 VSMCs using the JetPEI reagent according to the manufacturer’s instructions (Polyplus Transfection, Illkirch,
France). DETERMINATION OF MRNA EXPRESSIONS OF PROCOLLAGEN I AND III, MMP-13 AND TIMP-1 BY QUANTITATIVE REAL-TIME POLYMERASE CHAIN REACTION Total RNA was extracted using the Trizol reagent
and transcribed into cDNA using oligo (dT) primer and M-MLV reverse transcriptase. Quantitative real-time polymerase chain reaction (RT-PCR) was performed on an ABI PRISM 7300 instrument
(Applied Biosystems, Foster, CA, USA). The amplification conditions for the cDNA were: denaturing at 94 °C for 30 s, annealing and polymerizing at 60 °C for 1 min for 40 cycles. Samples and
standards were determined in triplicate. TaqMan probes were modified by 5′-FAM and 3′-TAMRA. Sequences of the primers and probes were: for procollagen I, forward, 5′-CTTGTTGCTGAGGGCAACAG-3′,
reverse, 5′-GCAGGCGAGATGGCTTATTC-3′, Taqman probe, 5′-ATTCACCTACACTGTCCTTGTCGATGGCTG-3′; for procollagen III, forward, 5′-GAAAAAACCCTGCTCGGAATT-3′, reverse, 5′-ATCCATCTTGCAGCCTTGGT-3′,
Taqman probe, 5′-AGAGACCTGAAATTCTGCCACCCTGAACTC-3′; for MMP-13, forward, 5′- CTTCTGGCACACGCTTTTCC-3′, reverse, 5′-GCTCATGGGCAGCAACAATA -3′, Taqman probe, 5′-CCTGGACCAAACCTTGGCGG -3′; for
TIMP-1, forward, 5′-AGCCCTGCTCAGCAAAAGG-3′, reverse, 5′- CTGTCCACAAGCAATGACTGTCA-3′, Taqman probe, 5′- CTTCGTAAAGACCTATAGTGCTGGCTG-3′; for PAI-1, forward, 5′-GGTCAAGATCGAGGTGAACGA-3′,
reverse, 5′-GCGGGCTGAGACTAGAATGG, Taqman probe, 5′-CGGCACAGTGGCGTCTTCCTCC-3′; and for β-actin, forward, 5′-ACCCGCGAGTACAACCTTCTT-3′, reverse, 5′-TATCGTCATCCATGGCGAACT-3′, Taqman probe,
5′-CCTCCGTCGCCGGTCCACAC-3′. WESTERN BLOT ANALYSIS A7r5 cells ( 5 × 104) were incubated in 6-well plates. When cells were grown to 60–70% confluences, they were treated as follows. In the
first series, cells were transfected with 2 μg of vehicle, AAT1 or AAT2 plasmid. After 24 h, they were starved in DMEM with 0.5% FBS before TGF-β1 (10 ng/ml) treatment for 24 h or 1 h. In
the second series, cells were transfected with 2 μg of control shRNA, AAT1 shRNA or AAT2 shRNA for 24 h. Then they were starved for 24 h followed by TGF-β1 (10 ng/ml) treatment for 24 h or 1
h. In the third series, cells were transfected with control shRNA, AAT1 shRNA or AAT2 shRNA for 24 h, staved for 24 h, treated with SB431542 (5 μmol/L) for 1 h and then stimulated with
TGF-β1 (10 ng/ml) for 24 h. These samples were harvested and lysed in lysis buffer (0.5 mmol/L EDTA, 10 mmol/L Tris–HCl, pH 7.4, 0.3 mol/L sucrose and protease inhibitor cocktail) as
previously described15. Equal amounts of protein were resolved on SDS-PAGE gels and transferred onto nitrocellulose membranes. Non-specific bindings were blocked by incubation in 5% milk
blocking buffer. The primary antibodies anti-AAT1 and anti-AAT2 were from Sigma-Aldrich Corporation (St Louis, MO, USA), anti-collagen I and anti-collagen III were from Abcam (Cambridge, MA,
USA), anti-MMP-13 and anti-TIMP-1 were from Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti-phospho-Smad2, anti-phospho-Smad3 and anti-Smad2/3 were from Cell Signaling Technology
(Danvers, MA, USA) and anti-GAPDH were form Kangcheng (Shanghai, China). After incubation, each primary antibody bound with their respective specific horseradish peroxidase-conjugated
secondary antibodies (Santa Cruz Biotechnology) and the bands were visualized using enhanced chemiluminescence detection kit (Thermo Scientific, Rockford, IL, USA). The densitometric
analysis of the positive bands was performed using AlphaEaseFC (Alpha Innotech Corporation, San Leandro, CA, USA). COLLAGEN I AND III EXPRESSION IN VSMCS BY IMMUNOFLUORESCENCE AND CONFOCAL
MICROSCOPY VSMCs were plated on glass coverslips. When cells were grown to 60–70% confluences, they were treated as follows. In the first series, cells were transfected with vehicle, AAT1 or
AAT2 plasmid. After 24 h, they were starved in DMEM with 0.5% FBS before TGF-β1 (10 ng/ml) treatment for 24 h. In the second series, cells were transfected with control shRNA, AAT1 shRNA or
AAT2 shRNA for 24 h. Then they were starved for 24 h followed by TGF-β1 treatment for 24 h. In the third series, cells were transfected with control shRNA, AAT1 shRNA or AAT2 shRNA for 24
h, staved for 24 h, treated with SB431542 (5 μmol/L) for 1 h and then stimulated with TGF-β1 for 24 h. The cells were fixed with 4% paraformaldehyde (0.01 mol/L PBS, pH 6.8), washed with
PBS, incubated with rabbit anti-collagen I primary antibody (1:25; Sigma-Aldrich) and rabbit anti-collagen III primary antibody (1:25; Sigma-Aldrich) respectively at 4 °C overnight. Next
day, after washing, the coverslips were incubated with secondary antibody (1:100) (Invitrogen, Carlsbad, CA) in the dark for 90 min at room temperature. The slides were viewed using a
Fluoview laser scanning confocal microscope (Olympus, Tokyo, Japan). In the negative control group, IgG was used instead of the first antibody. MEASUREMENT OF SO2 CONCENTRATION IN VSMCS
SUPERNATANT BY HIGH-PERFORMANCE LIQUID CHROMATOGRAPHY WITH FLUORESCENCE DETECTION VSMCs supernatants were collected for SO2 content determination. SO2 concentrations were measured using
high-performance liquid chromatography with fluorescence detection (HPLC-FD, Agilent 1200 series, Agilent Technologies, Palo Alto, CA, USA)9. Briefly, 100 μL of VSMCs supernatant was mixed
with 70 μL of 0.212 M sodium borohydride in 0.05 M Tris-HCl (pH 8.5) and incubated at 28 °C for 30 min. The sample was then mixed with 10 μL of 70 mM mBrB in acetonitrile, incubated for 10
min at 42 °C and then mixed with 40 μL of 1.5 M perchloric acid. Protein precipitate in the mixture was removed by centrifugation at 12400×g for 10 min at 25 °C. The supernatant was
immediately neutralized by adding 10 μL of 2 M Tris-HCl (pH 3.0) and centrifuged at 12400×g for 10 min. The neutralized supernatant was used for HPLC-FD. Sulfitebimane was measured by
excitation at 392 nm and emission at 479 nm. Quantification was carried out by the standardization of sodium sulfite. MEASUREMENT OF ENDOGENOUS SO2 CONTENT IN VSMCS BY A FLUORESCENT PROBE
Endogenous SO2 in VSMCs was measured using a fluorescent probe (kindly provided by Professor Kun Li, College of Chemistry, Sichuan University, Sichuan, China). The culture supernatant of
VSMCs was discarded and washed with PBS for three times. Then, the cells were stained in the working liquid of fluorescent probe (10 μM) for 1 h at 37 °C, washed with PBS and then fixed with
4% paraformaldehyde for 15 min at room temperature. After washed with PBS, the cells were detected as blue fluorescent by confocal microscopy. PHOSPHORYLATION OF TΒRI IN VSMCS EVALUATED BY
IMMUNOPRECIPITATION VSMCs were seeded in 100-mm dishes for immunoprecipitation. When cells were grown to 60–70% confluences, they were treated as follows. In the first series, cells were
transfected with vehicle, AAT1 or AAT2 plasmid. After 24 h, they were starved in DMEM with 0.5% FBS before TGF-β1 (10 ng/ml) treatment for 1 h. In the second series, cells were transfected
with control shRNA, AAT1 shRNA or AAT2 shRNA for 24 h. Then they were starved for 24 h followed by TGF-β1 treatment for 1 h. The cells were then harvested with the antibody against TβRI
(Santa Cruz Biotechnology, catalog sc-398) before immunoprecipitation with protein A/G agarose beads (Thermo Fisher Scientific, Waltham, MA, USA)20,36. The precipitated proteins were
resolved by 10% SDS-PAGE and then immunoblotted with antibody against phosphoserine (Abcam, Cambridge, MA, USA)20. STATISTICAL ANALYSIS Results are presented as mean ± SD. Statistical
comparisons were performed with PRISM5 software (GraphPad). Comparisons among more than 2 groups involved one-way ANOVA followed by the Student-Newman-Keuls test for post-hoc comparison as
appropriate. _P_ < 0.05 was considered significant. ADDITIONAL INFORMATION HOW TO CITE THIS ARTICLE: Huang, Y. _et al._ Endogenous sulfur dioxide alleviates collagen remodeling via
inhibiting TGF-β/Smad pathway in vascular smooth muscle cells. _Sci. Rep._ 6, 19503; doi: 10.1038/srep19503 (2016). REFERENCES * Rocnik, E. F., Chan, B. M. & Pickering, J. G. Evidence
for a role of collagen synthesis in arterial smooth muscle cell migration. J. Clin. Invest. 101, 1889–98 (1998). Article CAS Google Scholar * Intengan, H. D. & Schiffrin, E. L.
Vascular remodeling in hypertension: roles of apoptosis, inflammation and fibrosis. Hypertension. 38, 581–7 (2001). Article CAS Google Scholar * Kim, H. J. et al. Peroxisome
proliferator-activated receptor {delta} regulates extracellular matrix and apoptosis of vascular smooth muscle cells through the activation of transforming growth factor-{beta}1/Smad3. Circ.
Res. 105, 16–24 (2009). Article CAS Google Scholar * Zhao, J. et al. Urotensin II-induced collagen synthesis in cultured smooth muscle cells from rat aortic media and a possible
involvement of transforming growth factor-β1/Smad2/3 signaling pathway. Regul. Pept. 182, 53–8 (2013). Article CAS Google Scholar * Zhao, X. et al. Regulatory effect of hydrogen sulfide
on vascular collagen content in spontaneously hypertensive rats. Hypertens. Res. 31, 1619–30 (2008). Article CAS Google Scholar * Yan, H., Du, J. & Tang, C. The possible role of
hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochem. Biophys. Res. Commun. 313, 22–7 (2004). Article CAS Google Scholar * Stipanuk, M. H. Metabolism of
sulfur-containing amino acids. Annu. Rev. Nutr. 6, 179–209 (1986). Article CAS Google Scholar * Shapiro, R. Genetic effects of bisulfite (sulfur dioxide). Mutat. Res. 39, 149–75 (1977).
Article CAS Google Scholar * Sun, Y. et al. Effects of sulfur dioxide on hypoxic pulmonary vascular structural remodeling. Lab. Invest. 90, 68–82 (2010). Article CAS Google Scholar *
Pardali, E., Goumans, M. J. & ten Dijke, P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends. Cell. Biol. 20, 556–67 (2010). Article CAS Google
Scholar * Border, W. A. & Ruoslahti, E. Transforming growth factor-beta in disease: the dark side of tissue repair. J. Clin. Invest. 90, 1–7 (1992). Article CAS Google Scholar *
Massagué, J. Wounding Smad. Nat. Cell. Biol. 1, E117–9 (1999). Article Google Scholar * Li, W. et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J. Clin.
Invest. 124, 755–67 (2014). Article CAS Google Scholar * Mullen, A. C. et al. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell. 147, 565–76
(2011). Article CAS Google Scholar * Liu, D. et al. Sulfur dioxide inhibits vascular smooth muscle cell proliferation via suppressing the Erk/MAP kinase pathway mediated by cAMP/PKA
signaling. Cell. Death. Dis. 5, e1251 (2014). Article CAS Google Scholar * Sun, H. J. et al. Salusin-β contributes to vascular remodeling associated with hypertension via promoting
vascular smooth muscle cell proliferation and vascular fibrosis. Biochim. Biophys. Acta. 1852, 1709–1718 (2015). Article CAS Google Scholar * Xu, J. & Shi, G. P. Vascular wall
extracellular matrix proteins and vascular diseases. Biochim. Biophys. Acta. 1842, 2106–2119 (2014). Article CAS Google Scholar * Jacob, M. P. et al. Extracellular matrix remodeling in
the vascular wall. Pathol. Biol (Paris). 49, 326–32 (2001). Article CAS Google Scholar * Du, S. et al. Endogenously generated sulfur dioxide and its vasorelaxant effect in rats. Acta.
Pharmacol. Sin. 29, 923–930 (2008). Article CAS Google Scholar * Sun, L. et al. Hydrogen sulfide alleviates myocardial collagen remodeling in association with inhibition of TGF-β/Smad
signaling pathway in spontaneously hypertensive rats. Mol. Med. 20, 503–15 (2015). Article Google Scholar * Giannandrea, M. & Parks, W. C. Diverse functions of matrix
metalloproteinases during fibrosis. Dis. Model. Mech. 7, 193–203 (2014). Article CAS Google Scholar * Mori, S. et al. Expression and Roles of MMP-2, MMP-9, MMP-13, TIMP-1 and TIMP-2 in
Allergic Nasal Mucosa. Allergy. Asthma. Immunol. Res. 4, 231–9 (2012). Article CAS Google Scholar * Cevik, C. et al. Rosuvastatin therapy does not affect serum MMP-13 or TIMP-1 levels in
hypercholesterolemic patients. Tex. Heart. Inst. J. 38, 229–33 (2011). PubMed PubMed Central Google Scholar * Verrecchia, F. & Mauviel, A. Transforming growth factor-beta signaling
through the Smad pathway: role in extracellular matrix gene expression and regulation. J. Invest. Dermatol. 118, 211–5 (2002). Article CAS Google Scholar * Ghosh, J. et al. The role of
transforming growth factor beta1 in the vascular system. Cardiovasc. Pathol. 14, 28–36 (2005). Article CAS Google Scholar * Lambers, C. et al. The interaction of endothelin-1 and TGF-β1
mediates vascular cell remodeling. PLoS One. 8, e73399 (2013). Article ADS CAS Google Scholar * González-Ramos, M. et al. HSP70 increases extracellular matrix production by human
vascular smooth muscle through TGF-β1 up-regulation. Int. J. Biochem. Cell. Biol. 45, 232–242 (2013). Article Google Scholar * Stow, R. C., Mallawaarachchi, C. M. & Weissberg, P. L.
Migration of adventitia myofibroblasts following vascular balloon injury: insights from _in vivo_ gene transfer to rat carotid arteries. Cardiovasc. Res. 59, 212–21 (2003). Article Google
Scholar * Schiller, M., Javelaud, D. & Mauviel, A. TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J. Dermatol.
Sci. 35, 83–92 (2004). Article CAS Google Scholar * Mauviel, A. Transforming growth factor-beta: a key mediator of fibrosis. Methods. Mol. Med. 117, 69–80 (2005). CAS PubMed Google
Scholar * Dai, X. et al. SMAD3 deficiency promotes vessel wall remodeling, collagen fiber reorganization and leukocyte infiltration in an inflammatory abdominal aortic aneurysm mouse model.
Sci. Rep. 5, 10180 (2015). Article ADS CAS Google Scholar * Singer, T. P. & Kearney, E. B. Intermediary metabolism of L-cysteinesulfinic acid in animal tissues. Arch Biochem
Biophys. 61, 397–409 (1956). Article CAS Google Scholar * Harvey, S. A. et al. Responses of cultured human keratocytes and myofibroblasts to ethyl pyruvate: a microarray analysis of gene
expression. Invest Ophthalmol Vis Sci. 51, 2917–27 (2010). Article Google Scholar * Ju, K. D. et al. Ethyl pyruvate ameliorates albuminuria and glomerular injury in the animal model of
diabetic nephropathy. Am J Physiol Renal Physiol. 302, F606–13 (2012). Article CAS Google Scholar * Varma, S., Hegde, K. & Kovtun, S. Oxidative damage to lens in culture:
reversibility by pyruvate and ethyl pyruvate. Ophthalmologica. 220, 52–57 (2000). Article Google Scholar * Kang, S. H. et al. Transcriptional repression of the transforming growth
factor-beta type I receptor gene by DNA methylation results in the development of TGF-beta resistance in human gastric cancer. Oncogene. 18, 7280–6 (1999). Article CAS Google Scholar
Download references ACKNOWLEDGEMENTS We thank Dr Jia Liu for her assistances in cell culture and SO2 measurement. This work was supported by the National Natural Science Foundation of China
(Grant Nos. 81400311, 31440052 and 91439110) and by the Major Basic Research Development Program of China (Nos. 2012CB517806 and 2013CB933801). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS *
Department of Pediatrics, Peking University First Hospital, Beijing, 100034, P.R. China Yaqian Huang, Zhizhou Shen, Qinghua Chen, Pan Huang, Chunyu Zhang, Junbao Du & Hongfang Jin *
Department of Endocrinology, Beijing Chao-Yang Hospital, Capital Medical University, Beijing, 100020, P.R. China Heng Zhang * Department of Pediatrics, Capital Medical University Shijitan
Hospital, Beijing, 100038, P.R. China Shuxu Du * Department of Physiology and Pathophysiology, Peking University Health Science Centre, Beijing, 100191, P.R. China Bin Geng & Chaoshu
Tang * Key Laboratory of Green Chemistry and Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu, 610064, P.R. China Kun Li * Key Laboratory of Molecular
Cardiology, Ministry of Education, Beijing, 100191, P.R. China Chaoshu Tang & Junbao Du Authors * Yaqian Huang View author publications You can also search for this author inPubMed
Google Scholar * Zhizhou Shen View author publications You can also search for this author inPubMed Google Scholar * Qinghua Chen View author publications You can also search for this author
inPubMed Google Scholar * Pan Huang View author publications You can also search for this author inPubMed Google Scholar * Heng Zhang View author publications You can also search for this
author inPubMed Google Scholar * Shuxu Du View author publications You can also search for this author inPubMed Google Scholar * Bin Geng View author publications You can also search for
this author inPubMed Google Scholar * Chunyu Zhang View author publications You can also search for this author inPubMed Google Scholar * Kun Li View author publications You can also search
for this author inPubMed Google Scholar * Chaoshu Tang View author publications You can also search for this author inPubMed Google Scholar * Junbao Du View author publications You can also
search for this author inPubMed Google Scholar * Hongfang Jin View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Y.H. designed and executed
most of the experiments and analyzed the results. Z.S. and Q.C. detected the SO2 concentration by HPLC-FD and analyzed the results. P.H. and H.Z. operated the confocal microscope, took
pictures and analyzed the results. S.D., B.G. and C.Z. analyzed the data and interpretated the results. K.L. prepared the fluorescent probe of SO2 and executed the experiments. C.T., J.D.
and H.J. designed the project, analyzed the data, interpretated the results and revised the manuscript. Y.H., J.D. and H.J. prepared the manuscript. ETHICS DECLARATIONS COMPETING INTERESTS
The authors declare no competing financial interests. RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third
party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative
Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Huang, Y., Shen, Z., Chen, Q. _et al._ Endogenous sulfur dioxide alleviates collagen remodeling via inhibiting TGF-β/Smad
pathway in vascular smooth muscle cells. _Sci Rep_ 6, 19503 (2016). https://doi.org/10.1038/srep19503 Download citation * Received: 09 September 2015 * Accepted: 09 December 2015 *
Published: 14 January 2016 * DOI: https://doi.org/10.1038/srep19503 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative