Play all audios:
ABSTRACT Glioblastoma is a fatal and incurable cancer with the hyper-activated mTOR pathway. HSP60, a major chaperone for maintenance of mitochondrial proteostasis, is highly expressed in
glioblastoma patients. To understand the effects of HSP60 on glioblastoma tumorigenesis and progression, we characterized the HSP60-knockdowned glioblastoma cells and revealed that HSP60
silencing markedly suppressed cell proliferation and promoted cell to undergo the epithelial-mesenchymal transition (EMT). Proteomic analysis showed that ribosomal proteins were
significantly downregulated whereas EMT-associated proteins were up-regulated in HSP60-knockdowned U87 cells as confirmed by a distinct enrichment pattern in newly synthesized proteins with
azido-homoalanine labeling. Biochemical analysis revealed that HSP60 knockdown increased reactive oxygen species (ROS) production that led to AMPK activation, similarly to the complex I
inhibitor rotenone-induced AMPK activation. Activated AMPK suppressed mTORC1 mediated S6K and 4EBP1 phosphorylation to decrease protein translation, which slowed down cell growth and
proliferation. On the other hand, high levels of ROS in HSP60 knockdowned or rotenone-treated U87 cells contributed to EMT. These results indicate that HSP60 silencing deactivates the mTOR
pathway to suppress glioblastoma progression, suggesting that HSP60 is a potential therapeutic target for glioblastoma treatment. SIMILAR CONTENT BEING VIEWED BY OTHERS QUANTITATIVE
PROTEOMICS ANALYSIS OF GLIOBLASTOMA CELL LINES AFTER LNCRNA HULC SILENCING Article Open access 15 June 2021 MIR-204-5P MEDIATES PERK INHIBITION TO SUPPRESS GROWTH AND INDUCE APOPTOSIS IN
OVARIAN CANCER THROUGH THE EIF2Α/ATF-4/CHOP PATHWAY Article Open access 11 April 2025 HYPOXIA-INDUCED CIRCADAMTS6 IN A TDP43-DEPENDENT MANNER ACCELERATES GLIOBLASTOMA PROGRESSION VIA ANXA2/
NF-ΚB PATHWAY Article 17 November 2022 INTRODUCTION Glioblastoma (GBM) is the most common and lethal brain tumor in adults1. GBM is classified by the World Health Organization as the grade
IV gliomas. Complete surgical removal of GBM tumors is difficult due to its invasion to the surrounding brain tissues2. GBM patients are not well responded to radiotherapy and
chemotherapy3,4, which leads to an extremely poor prognosis in GBM patients5. The median survival of GBM patients is less than one year after diagnosis6 and the five year survival rates of
GBM patients are less than 10%7. Three dysregulated pathways were identified in GBM cells including receptor tyrosine kinase (RTK) signaling, retinoblastoma (RB) signaling and TP53
signaling8. As a result of the enhanced RTK signaling, mTORC1 signaling is hyperactivated in GBM that promotes growth and proliferation and can be a compelling therapeutic target in GBM
treatment2. However, rapamycin, the mTORC1 inhibitor, was failed in clinical trials due to insufficiently suppression of mTORC1 signaling, rapamycin resistance or activation of the
downstream effectors9. Finding a new target to suppress mTORC1 signaling is, therefore, important for development of new therapeutic strategies for GBM. It is known that activated AMPK
phosphorylates raptor to decrease mTORC1 activity10. However, AMPK mediated growth reduction in GBM has not yet been reported. Heat shock protein 60 (HSP60) is one of the major chaperones in
mitochondria for assisting protein folding, transportation and degradation to maintain mitochondrial proteostasis11,12,13,14. HSP60 plays tumor-type dependent roles with pro-apoptotic or
pro-survival functions in tumorigenesis and progression15. Samali _et al._16 reported that HSP60 interacted with pro-caspase3 that accelerated its activation _in vitro_ and _in vivo_.
However, Kirchhoff _et al._17 showed that cytosolic HSP60 played an anti-apoptotic role in cardiac myocytes through interactions with pro-apoptotic Bax and Bak proteins that were also
considered as an important factor in preventing apoptosis in tumor cells. As the result, HSP60 was down-regulated in bronchial adenocarcinoma, vesical transitional cell carcinoma and
carcinosarcoma18,19,20,21 whereas the high HSP60 expression was presented in cervical cancer, prostate cancer, breast cancer and GBM22,23,24,25. Effects of HSP60 on GBM progression has not
been well examined. In the present work, we established multiple stable cell lines, in which HSP60 expression was knocked down. We carried out both quantitative proteomics and AHA labeling
experiments to understand the mechanisms underlying HSP60 mediated cellular processes. AHA, an analog of methionine with azide group, is recognized by methionine tRNA and incorporated into
newly synthesized proteins. These newly synthesized proteins can be enriched by click chemistry and subsequently identified by LC-MS/MS analysis. AHA labeling was a method of choice for
studying regulation of protein synthesis. Our results revealed that HSP60 silencing in U87 cells enhanced cellular ROS production through disruption of respiratory complex I and markedly
suppressed cell growth and proliferation through activating the ROS-AMPK-mTOR pathway, suggesting that HSP60 is a potential target for GBM treatment. We also demonstrated that bioorthogonal
noncanonical amino acid tagging is a useful tool for examining cellular proteostasis network. RESULTS HSP60 KNOCKDOWN SUPPRESSES CELL GROWTH AND PROLIFERATION AND INCREASES SUSCEPTIBILITY TO
OXIDATIVE STRESS To explore the effects of HSP60 expression on GBM cell proliferation, we constructed stable cell lines in which HSP60 expression was knocked down by small hairpin RNA
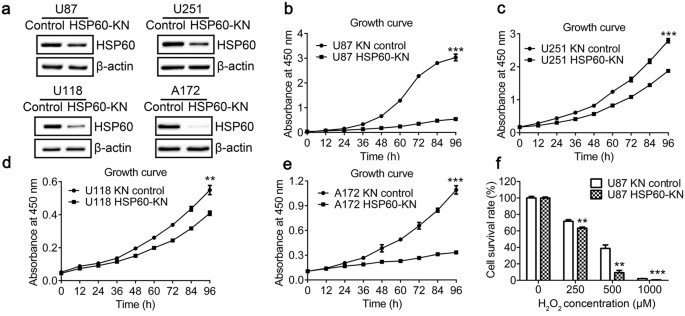
interference. The cells transfected with a scrambled shRNA with no homology in human genome was used as the control. HSP60 silencing efficacy in U87, U251, U118 and A172 cells was examined
by qPCR and western blotting (Fig. 1(a) and Supplementary Fig. S1), showing that the expression levels of HSP60 were decreased by approximately 50% in HSP60-knockdown cells. HSP60 knockdown
significantly decreased the cell growth rate in all four cell lines (Fig. 1(b–e)), but did not decrease the growth in other cell lines such as 293T, A549, A2780 and 786-O cells (data not
shown). The results indicate that HSP60 knockdown only suppresses cell proliferation in GBM cells. Next, we examined whether HSP60 knockdown cells were more sensitive to oxidative stress.
The cell viability of H2O2-treated cells was determined by CCK-8 assay (Fig. 1(f)). When cells were treated with 500 μM H2O2 for 12 h, cell survival rates were 40% and 10% for the control
and HSP60-KN-U87 cells, respectively. A significant decrease in cell survival confirmed that HSP60-KN-U87 cells are more susceptible to oxidative stress. IDENTIFICATION OF THE DIFFERENTIALLY
EXPRESSED PROTEINS BETWEEN THE CONTROL AND HSP60-KN CELLS Quantitative proteomics analysis of the control and HSP60-KN-U87 cells identified 6877 proteins in biological triplicates. Based on
the average reporter ion ratios from TMT analysis, 100 proteins were significantly up-regulated (ratio > 1.5, p value < 0.05) and 170 proteins were significantly down-regulated (ratio
< 0.67, p value < 0.05) in HSP60-KN-U87 cells compared to control cells (Supplementary Tables S1 and S2). The Gene Ontology (GO) was used to analyze the biological relevance of the
differentially expressed proteins with Panther (www.pantherdb.org/). As shown in Fig. 2, the majority of the differentially expressed proteins participated in the cellular process and the
metabolic process. Among the down-regulated proteins, 116 proteins are associated with the primary metabolic process, which are the dominant difference between HSP60-KN-U87 and the control
cells (Fig. 2(a)). We also found that ten ribosomal proteins were down-regulated in HSP60-KN-U87 cells (Fig. 2(b) and Table 1), suggesting that the protein synthesis rate in HSP60-KN cells
was decreased. Among the up-regulated proteins, 26 proteins are associated with cell communication (Fig. 2(c)), in which seven proteins associated with the WNT/Catenin pathway were
up-regulated in HSP60-KN cells (Fig. 2(d) and Table 1). HSP60 knockdown induced changes in the expression levels of RPS9, RPL18A, CDH2 (N-cadherin), CTNNB1 (β-catenin), SRC and LEF1 were
further confirmed by western blot analysis (Supplementary Fig. S2). _HSP60-silencing or rotenone treatment induces ROS overproduction in U87 cells._ Proteomics showed that seven subunits of
respiratory complex I were downregulated in three biological replicate analysis of the control and HSP60-KN-U87 cells (Fig. 3(a) and Table 2) as confirmed by western blotting of NADH
dehydrogenase ubiquinone 1 alpha subcomplex subunit 5 (NDUFA5), NADH dehydrogenase ubiquinone flavoprotein 1 (NDUFV1), NADH dehydrogenase ubiquinone flavoprotein 2 (NDUFV2) and NADH
dehydrogenase ubiquinone iron-sulfur protein 3 (NDUFS3) in HSP60-KN-U87 cells (Fig. 4(a) and Supplementary Fig. S2), indicating that the integrity of the respiratory complexes in
HSP60-KN-U87 cells was compromised. To clarify the underlying mechanism responsible for the downregulation of respiratory complex I, we carried out AHA experiment to enrich newly synthesized
proteins. By quantitative proteomics, we identified that the synthesis rate of all 10 subunits of respiratory complex I was greatly decreased in HSP60-KN-U87 cells (Fig. 3(b) and
Supplementary Table S3). This demonstrates that HSP60 silencing disrupts the mitochondrial proteostasis via slowing down protein synthesis rate. It has been known that the cellular ROS was
mainly produced from the respiratory complex I and respiratory complex III26. We, therefore, tested whether HSP60 knockdown caused ROS overproduction. The cellular ROS level was measured
with the CellROX® Deep Red kit and showed that HSP60 knockdown increased ROS production by about 3 fold, indicating that both mitochondrial proteostasis and ROS homeostasis were disrupted in
HSP60-KN-U87 cells (Fig. 3(c)). To further confirm that HSP60-silencing induced downregulation of respiratory complex I was responsible for ROS overproduction, we treated U87 cells with 100
nM rotenone for 1 h, an inhibitor of respiratory complex I, which also resulted in ROS overproduction (Fig. 3(d)). Furthermore, we demonstrated that rotenone treatment also slowed down the
growth and proliferation of U87 cells (Fig. 3(e,f)), similarly as those observed for HSP60-KN-U87 cells. HSP60-SILENCING SUPPRESSED CELL GROWTH THROUGH THE ROS/AMPK/MTOR PATHWAY Western
blotting was employed to investigate the ROS-downstream signaling pathways underlying HSP60 silencing mediated suppression of cell proliferation. Phosphorylation of AMPKα at T172 were
significantly increased in HSP60-KN-U87 cells (Fig. 4(a) and Supplementary Fig. S3), indicating that ROS activated AMPK pathway. Targets of activated AMPK pathway include the mTOR pathway
and fatty acid synthesis, in which AMPK directly phosphorylates raptor to prevent the formation of mTORC127. Indeed, western blotting showed that the phosphorylation of raptor was
significantly increased (Fig. 4(a) and Supplementary Fig. S3), which consequently resulted in significantly decreased S6K and 4EBP1 phosphorylation (Fig. 4(a) and Supplementary Fig. S3),
suggesting that protein translation was inhibited in HSP60-KN cells through the ROS/AMPK/mTOR pathway. These results were further validated by the AHA labeling. Using this approach, we
identified 3704 newly synthesized proteins, in which the levels of 706 proteins were significantly decreased (ratio < 0.67, p value < 0.05) and 143 proteins were significantly
increased (ratio > 1.5, p value < 0.05) in HSP60-KN-U87 cells compared to the control cells (Supplementary Table S3 and S4). The difference in numbers of proteins with altered
synthesis rates between these two cell lines indicates that HSP60 knockdown slows down protein translation. This was further validated by the western blot analysis of AHA labeled proteins,
showing that HSP60 knockdown inhibited the protein translation (Supplementary Fig. S4). GO analysis showed that majority of the decreased synthetic proteins were associated to the metabolic
process and cellular process (Fig. 4(b,c)). Synthesis rates for proteins associated with protein metabolism were lower in HSP60-KN-U87 cells (Fig. 4(d)). Especially, levels of 91 newly
synthesized proteins related to ribosomes were lower in HSP60-KN-U87 cells than those in the control cells (Supplementary Table S3), in consistent with the proteomic results. On the other
hand, levels of newly synthesized proteins associated with cell communication were higher in HSP60-KN-U87 cells than those in the control cells (Supplementary Table S4). Thus, HSP60
knockdown induced a distinct enrichment pattern in newly synthesized proteins, demonstrating that proteostasis state was regulated by protein synthesis and HSP60 functions as a switch for
tuning expressions of proteins of anabolic process and those associated with cell communication. Furthermore, levels of 12 newly synthesized proteins related to lipid metabolism were
significantly lower in HSP60-KN-U87 cells than those in the control cells (Fig. 5(a) and Supplementary Fig. S5), suggesting that HSP60 knockdown mediated activation of AMPK pathway affected
lipid metabolism. We conducted the lipidomic analysis and showed that five classes of phospholipids were decreased in HSP60-KN-U87 cells (Fig. 5(b)). Nile red staining also indicated that
phospholipids in HSP60-KN-U87 cells were reduced (Fig. 5(c–f)), in consistent with the proteomics and western blot data showing that fatty acid synthase and acetyl-CoA carboxylase were
down-regulated in HSP60-KN-U87 cells (Fig. 4(a)). All these results declared that HSP60-silencing suppressed cell proliferation and growth through ROS/AMPK axis. HSP60-SILENCING PROMOTED
CELL TO UNDERGO EMT AHA labeling results determined that levels of newly synthesized proteins in cell communication were higher in HSP60-KN-U87 cells than those in the control cells. For
examples, HSP60 knockdown induced upregulation of the newly synthesized proteins associated with integrin pathway including Laminin subunit gamma-1, Focal adhesion kinase 1, Proto-oncogene
tyrosine-protein kinase Src (SRC), Dedicator of cytokinesis protein 1, Integrin alpha-V, Paxillin, Engulfment and cell motility protein 2, Filamin-B, Integrin alpha-6, Alpha-actinin-1 and
Collagen alpha-1(I) chain. Similarly, the WNT pathway associated proteins N-cadherin (CDH2), Catenin alpha-1(CTNNA1), β-catenin (CTNNB1), Catenin delta-1 (CTNND1), Proto-cadherin gamma-C3
(PCDHGC3), Protocadherin-7 (PCDH7), Inositol 1,4,5-trisphosphate receptor type 3 (ITPR3), UDP-glucose 6-dehydrogenase (UGDH) and Inositol 1,4,5-trisphosphate receptor type 1 (ITPR1) were
increasingly synthesized in HSP60-KN-U87 cells compared to the control cells (Fig. 6(a) and Supplementary Table S4). HSP60 knockdown mediated activation of integrin and WNT pathway proposes
that cells undergo EMT process as confirmed by the western blotting, showing that EMT markers N-cadherin and vimentin were significantly increased (Fig. 6(b) and Supplementary Fig. S6).
Western blot analysis also revealed that both the cytoplasmic and nuclear levels of β-catenin were higher in HSP60-KN-U87 cells (Fig. 6(b) and Supplementary Fig. S7). It has been reported
that nuclear accumulation of β-catenin initiated EMT in pancreatic cancer28. Consistent with these results, our data suggested that β-catenin played an important role in HSP60 silencing
mediated EMT process. Moreover, cell invasion assay showed that HSP60-KN-U87 cells displayed a higher invasiveness than control cells did (Fig. 6(c)). To clarify whether HSP60 knockdown
mediated ROS/AMPK activation causes EMT, U87 cells were treated with rotenone and expression changes in selected proteins were probed by western blotting. As we expected, rotenone also
induced activation of AMPK pathway and upregulation of EMT markers (Fig. 6(d) and Supplementary Fig. S6), which confirmed that ROS-induced AMPK activation was the underlying mechanism for
EMT process in GBM cells. DISCUSSION GBM is the most common brain tumor with high relapse and mortality rate29. The current therapy is based on surgical excision followed by temozolomide
chemotherapy and/or radiation30,31. New therapeutic approaches are urgently needed. Consistent with previous results that the high HSP60 expression was detected in GBM patients, we confirmed
that HSP60-silencing suppressed the growth and proliferation in multiple GBM cell lines32. The high cellular ROS level and mesenchymal phenotype were also presented in HSP60-knockdown
cells. Proteomic analysis and AHA labeling shed light on the mechanisms underlying HSP60-silencing enhanced ROS production, in which multiple subunits of the respiratory complex I were
down-regulated in HSP60-KN-U87 cells. The respiratory complex I contains 45 subunits33, which must assemble together to form a mature holoenzyme for dehydrogenating NADH and transferring
electrons to coenzyme Q. As a major site for ROS production34, early studies have shown that the down-regulation of complex I subunits causes dysfunction of the respiratory chain to generate
excessive ROS35,36, which supports our finding that the high level of ROS is present in HSP60-KN-U87 cells. We further showed that AMPK was activated in HSP60-KN cells. Similar results were
observed when cells were treated with rotenone. These results demonstrate that both HSP60 knockdown and rotenone-treatment activates ROS/AMPK pathway. It has been known that AMPK regulates
several cellular processes, including protein synthesis, fatty acid metabolism and glucose uptake37,38,39,40. Two different mechanisms were proposed for AMPK mediated inhibition of mTORC127.
AMPK phosphorylates TSC2 to inactivate Rheb41,42, an essential activator of mTORC143; or AMPK directly phosphorylates raptor to prevent the formation of mTORC1 complex44. Our data showed
that raptor phosphorylation was increased in HSP60-KN cells, which caused deactivation of the mTOR signaling. Consequently, targets of mTORC1 complex, S6K and 4EBP1 phosphorylation were
decreased45,46, which ultimately led to 4EBP1 binding to eIF4E and prevented protein synthesis47,48. It was confirmed by the fact that the quantity of the newly synthesized proteins in
HSP60-KN-U87 cells was significantly less than that synthesized in the control cells, especially for the ribosomal proteins and the complex I subunits. Activated AMPK also regulated lipid
metabolism including fatty acid synthesis and β-oxidation37. It has been known that AMPK phosphorylates acetyl CoA carboxylase 1, a rate limiting enzymes in fatty acid synthesis, to
down-regulate fatty acid synthesis49 while AMPK also inhibits the activity of SREBP1, the transcription factor for ACC1 and FASN, to suppress the expression of ACC1 and FASN50. Consistent
with those findings, we found that ACC and FASN were down-regulated in HSP60-KN cells, resulting in a markedly decrease in lipid synthesis, especially for phospholipids. We further
demonstrated that HSP60-knockdowned or rotenone treated cells underwent EMT with enhanced cell motilities. CONCLUSIONS In summary, our results provide mechanistic information for
understanding functions of HSP60 in tumorigenesis and progression of GBM cells. We demonstrate that HSP60 played a key role in maintenance of mitochondrial proteostasis and ROS homeostasis.
HSP60 silencing or rotenone treatment disrupted the integrity of the respiratory chain, leading to ROS overproduction and activation of AMPK pathway, which inhibited protein synthesis and
drove cell to undergo EMT. Our data propose that HSP60 silencing attenuates the mTOR pathway to suppress cell proliferation and HSP60 is a potential therapeutic target for GBM treatment.
METHODS CELL LINES Human glioblastoma cell line U87, U251, U118, A172 and human embryonic kidney 293T were obtained from cell bank of Chinese Academy of Sciences (Shanghai, China). U87
cells, U251 cells, U118 cells, A172 cells and 293T cells were grown in DMEM media (Wisent, Montreal, QC) supplemented with 10% fetal bovine serum (Wisent, Montreal, QC) and 1%
penicillin/streptomycin (Wisent, Montreal, QC) at 37 °C in a humidified incubator with 5% CO2. HSP60 KNOCK DOWN STABLE TRANSFECTION CELL LINE The shRNA sequence used for targeting HSP60 was
based on previous report51 and further confirmed by NCBI BLAST. A scrambled shRNA with no homologous sequence been found in the human genome was used as negative control. The shRNA sequences
were shown in Supplementary Table S5. The shRNAs were cloned into plasmid pLL3.7 and co-transfected with pMD2.G, pMDLg/pRRE and pREV-Rev into 293T cells using Lipofectamine 2000
(Invitrogen, NY) to package lentiviral particles. After transfection for 48 h, Supernatants containing lentiviral particles were harvested and used to infect U87, U251, U118 and A172 cells
in the presence of 6 μg/ml of polybrene for 5 h.Cells were cultured in fresh medium for 96 h after infections and sorted by flow cytometer with GFP positive to generate stable cell lines.
CELL PROLIFERATION ASSAY WITH CCK-8 Cells were seeded in 96-well plates (2,000 cells per well). Cell proliferation rate was measured with the Cell Counting Kit-8 (CCK-8) according to the
manufacturer’s instructions (Dojindo Laboratories, Japan). Briefly, CCK-8 reagents were added into wells after cells grew for 0, 12, 24, 36, 48, 72, 84, 96 h respectively or treated with
rotenone for 0, 12, 24, 36, 48, 60 and 72 h. The plates were incubated at 37 °C for 2 h. Absorbance at 450 nm was measured after incubation. Cell viability assay of HSP60 knockdown cells
treated with hydrogen peroxide. Effects of hydrogen peroxide on control and HSP60 knockdown cells were analyzed with the CCK-8 kit. Briefly, Cells were treated with hydrogen peroxide (0,
250, 500 and 1,000 μM) for 12 h. The CCK-8 reagent was added to treated cells and incubated at 37 °C for 2 h. Optical density (OD) was measured at 450 nm with a microplate reader. PROTEOMIC
ANALYSIS OF NEWLY SYNTHESIZED PROTEINS 5 × 106 cells were seeded onto 100 mm dishes supplemented with 10 ml DMEM medium and incubated in CO2 incubator overnight. Cells were washed twice with
PBS and supplemented with DMEM medium with 40 μM L-methionine or L-Azidohomoalanine. Cells were then incubated in CO2 incubator for 4 h. Cells were harvested and washed twice with PBS and
lysed in PBS buffer containing 0.1% SDS, 1% NP-40 and protease inhibitor cocktail. After sonication, cell lysates were centrifuged at 14,000 × g for 20 min at 4 °C. Proteins were quantified
with BCA kit. 500 μg proteins were reacted with biotin alkyne (Thermo, Waltham, MA) supplemented with 1 mM CuSO4 and 5 mM vitamin C for 1 h at room temperature. After methanol precipitation,
the precipitates were redissolved in RIPA buffer and incubated with 40 μl streptavidin sepharose overnight at 4 °C. The beads were washed three times with PBS buffer containing 1% NP-40,
then washed three times with PBS buffer. Finally, the newly synthesized proteins were eluted with 2× loading buffer and loaded onto SDS-PAGE gel. The gel bands were excised and digested with
the standard in-gel digestion protocol. The digested peptides were extracted twice with 50% acetonitrile aqueous solution containing 0.1% formic acid for 60 min. Extracts were then dried in
a Speed Vac and labelled with the TMT reagent (Thermo, Waltham, MA) according to the manufacturer’s protocol. Labelled peptides were mixed together and analyzed by LC-MS/MS. The peak lists
from LC-MS/MS analysis were generated with the SEQUESTTM searching algorithm in the Proteome Discoverer software (version 1.4.1.14). The generated MS/MS spectra were searched against the
Uniprot Human database (release date of November 20, 2015; 20193 entries) using the SEQUEST HT searching engine. The search criteria were as follows: full tryptic specificity was required;
two missed cleavage was allowed; carbamidomethylation (C) and TMT sixplex (K and N-terminal) were set as the fixed modifications; the oxidation (M) was set as the variable modification;
precursor ion mass tolerances were set at 10 ppm for all MS acquired in an orbitrap mass analyzer; and the fragment ion mass tolerance was set at 0.02 Da for all MS2 spectra acquired. The
peptide false discovery rate was calculated using Percolator provided by Proteome Discoverer software. When the q value was smaller than 1%, the peptide spectrum match was considered to be
correct. False discovery was determined based on peptide spectrum match when searched against the reverse, decoy database. Peptides only assigned to a given protein group were considered as
unique. Relative protein quantification was performed using the Proteome Discoverer software (Version 1.4.1.14) according to manufacturer’s instructions on the six reporter ion intensities
per peptide. Protein ratios were calculated as the median of all peptide hits belonging to a protein. Proteins with ratios smaller than 0.67 and p-value < 0.05 were considered as
down-regulated proteins while proteins with ratios greater than 1.5 and p-value < 0.05 were considered as up-regulated proteins. Tryptic peptides from bovine serum albumin were used as
the quality control (QC) sample to evaluate the performance of mass spectrometry system. PROTEIN SEPARATION BY TWO DIMENSIONAL HPLC AND PROTEOMICS ANALYSIS Proteomic analysis was carried out
as previously described52. Briefly, Proteins of infected U87 cells were extracted with 8 M urea and 100 μg of protein was reduced by adding 5 mM dithiotheitol for 1 h at room temperature
and then alkylated by 12.5 mM iodoacetamide for 1 h at room temperature protected from light. The protein samples were finally digested with trypsin for 20 h at room temperature. The
digested peptides were purified using Sep-Pak C18 cartridges and labeled with TMT reagents (Thermo, Waltham, MA) according to the manufacturer’s protocol. Labelled peptides were mixed
together, desalted by Sep-Pak C18 cartridges and separated by reverse phase (RP) chromatography. The first dimension RP separation by microLC was performed on an Ultimate 3000 System
(Thermo, Waltham, MA) by using a Xbridge C18 RP column (5 μm, 150 Å, 250 mm × 4.6 mm i.d., Waters). Mobile phases A (2% acetonitrile, adjusted pH to 10.0 using NH3·H2O) and B (98%
acetonitrile, adjusted pH to 10.0 using NH3·H2O) were used to develop a gradient. The eluted peptides were monitored at 214 nm and collected every minute. The fractions were dried and
reconstituted in 20 μL of 0.1% (v/v) FA in water for the second dimension LC-MS/MS analysis at a low pH value. The peptides were separated by a C18 column (75 μm inner-diameter, 150 mm
length; Upchurch, Oak Harbor, USA) with a flow rate of 250 nL/min. Mobile phase A consisted of 0.1% formic acid and mobile phase B consisted of 100% acetonitrile and 0.1% formic acid. The
Orbitrap Q-Exactive mass spectrometer was operated in the data-dependent acquisition mode using Xcalibur 3.0 software. A full-scan followed by 20 data-dependent MS/MS scans were acquired
using higher energy collisional dissociation with normalized collision energy of 35%. The data analysis was the same as described above. Proteomic analysis was carried out in biological
triplicates. DETECTION OF CELLULAR REACTIVE OXYGEN SPECIES (ROS) The cellular ROS was measured using CellROX® Deep Red Reagents (Invitrogen, Grand Island, NY) following manufacturer’s
instructions. Briefly, cells were stained with 5 μM CellROX® Deep Red Reagent and incubated at 37 °C for 30 min. Then, cells were washed with PBS and analyzed on a BD FACS Aria II Flow
Cytometer (BD Biosciences, San Jose, CA). WESTERN BLOTTING Cells were lysed in RIPA buffer supplemented with protease inhibitor cocktail and phosphatase inhibitor cocktail (Thermo, Waltham,
MA). Equal amount of proteins were separated on the 12% SDS-PAGE gel and transferred onto a PVDF transfer membrane. Western blot analysis followed a standard procedure. P70 S6 Kinase,
phospho-p70 S6 Kinase (Thr389) β-catenin and FASN antibodies were obtained from Sigma (St Louis, MO). AMPKα, phospho-AMPKα (Thr172), Raptor, phosphor-Raptor (Ser792), 4EBP1, phospho-4E-BP1
(Thr37/46), ACC, N-cadherin, SRC, LEF1 and β-actin were obtained from Cell Signaling Technology (Danvers, MA). NDUFV1, NDUFV2, NDUFS3, RPS9 and RPL18A were obtained from Proteintech
(Chicago, IL). NDUFA5 antibody was obtained from Pierce (Rockford, IL). HSP60 antibody was obtained from Stressgen (Victoria, BC). Vimentin antibody was obtained from Proteintech (Chicago,
IL). Histone H3 antibody was obtained from Beyotime(shanghai, China). QUANTITATIVE REAL-TIME PCR (QPCR) Total RNA was isolated using the SV Total RNA Isolation System (Promega, Fitchburg,
WI) according to the manufacturer’s instructions. cDNA were synthesized using the GoScriptTM Reverse Transcription System (Promega, Fitchburg, WI).All qPCRs were performed using the Roche
LightCycler 96 System with SYBR green incorporation (Promega, Fitchburg, WI). The primers for all genes used in this study are from primerbank
(https://pga.mgh.harvard.edu/primerbank/index.html) and listed in Supplementary Table S6. 18SrRNA was used as internal control. The relative mRNA level was calculated using 2−ΔΔCt method.
LIPID ANALYSIS Lipids were extracted according to the method of Bligh and Dyer53. 5 × 106 cells were suspended in 500 μl PBS, followed by addition of 3 ml of mixture of chloroform and
methanol (V:V = 2:1). Samples were vortex for 30 s and centrifuged at 1,000 rpm for 5 min. The lower chloroform layer was collected and dried by nitrogen. Samples were reconstituted in
chloroform and methanol solution (V:V = 2:1), separated with Cortecs C18 column (2.1 × 100 mm, Waters) and analyzed by an Q Exactive orbitrap mass spectrometer that was operated in both
positive and negative mode using Xcalibur 3.0 software. A full-scan followed by 10 data-dependent MS/MS scans were acquired using higher energy collisional dissociation with stepped
normalized collision energy of 15%, 30% and 45%. The data analysis was performed on LipidSearch software (Thermo, Waltham, MA). The search criteria were as follows: product search type was
used, precursor ion mass tolerances were set at 8 ppm for all MS acquired in an orbitrap mass analyzer and the fragment ion mass tolerance was set at 15 ppm for all MS2 spectra acquired.
Sample mixture was used as quality control to evaluate the performance of mass spectrometry. NILE RED STAINING Cells were fixed with 4% paraformaldehyde for 20 min and washed with PBS for
three times. Then, cells were incubated with the Nile red solution (1 μg/ml fresh solution) for 20 min, washed with PBS for three times and imaged with co-focal microscopy (Zeiss, Jena,
Germany). INVASION ASSAY Invasion assay was performed using QCM 24-well cell invasion assay kit (Millipore, Boston, MA) according to the manufacturer’s instructions. Briefly, cells were
starved for 24 h prior to analysis. Cells were harvested and re-suspended in 1 ml Quenching Medium. 1.25 × 105 cells were seeded in the insert containing serum free media while the serum
containing media were added to the lower chamber. Cells then were incubated for 24 h in a CO2 incubator. Cells that invaded through the ECMatrix-coated membrane were lysed in Lysis
Buffer/Dye Solution and incubated for 15 min at room temperature. RFU values were read with PerkinElmer’s EnSpire Multimode plate reader (Waltham, MA) in fluorescence mode using 480/520 nm
filter set. STATISTICAL ANALYSIS Statistical analysis was carried out with GraphPad Prism 6.0 software by two sided unpaired _t_ tests. P values of < 0.05 were considered significant.
ADDITIONAL INFORMATION HOW TO CITE THIS ARTICLE: Tang, H. _et al._ Down-regulation of HSP60 Suppresses the Proliferation of Glioblastoma Cells via the ROS/AMPK/mTOR Pathway. _Sci. Rep._ 6,
28388; doi: 10.1038/srep28388 (2016). REFERENCES * Legler, J. M. et al. Cancer surveillance series: brain and other central nervous system cancers: recent trends in incidence and mortality.
J. Natl. Cancer Inst. 91, 1382–1390 (1999). Article CAS PubMed Google Scholar * Cloughesy, T. F., Cavenee, W. K. & Mischel, P. S. Glioblastoma: from molecular pathology to targeted
treatment. Annu. Rev. Pathol. 9, 1–25 (2014). Article CAS PubMed Google Scholar * Dunn, G. P. et al. Emerging insights into the molecular and cellular basis of glioblastoma. Gene Dev.
26, 756–784 (2012). Article CAS PubMed Google Scholar * Masui, K., Cloughesy, T. F. & Mischel, P. S. Molecular pathology in adult high-grade gliomas: from molecular diagnostics to
target therapies. Neuropath. Appl. Neuro. 38, 271–291 (2012). Article CAS Google Scholar * Mrugala, M. M. Advances and challenges in the treatment of glioblastoma: a clinician’s
perspective. Discov. Med. 15, 221–230 (2013). PubMed Google Scholar * Stewart, L. A. Chemotherapy in adult high-grade glioma: a systematic review and meta-analysis of individual patient
data from 12 randomised trials. Lancet 359, 1011–1018 (2002). Article CAS PubMed Google Scholar * Stupp, R. et al. Effects of radiotherapy with concomitant and adjuvant temozolomide
versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466 (2009). Article CAS PubMed Google
Scholar * Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008). Article ADS CAS
Google Scholar * Cloughesy, T. F. et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLos Med. 5, e8 (2008). Article CAS
PubMed PubMed Central Google Scholar * Gwinn, D. M. et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 (2008). Article CAS PubMed PubMed
Central Google Scholar * Lianos, G. D. et al. The role of heat shock proteins in cancer. Cancer Lett. 360, 114–118 (2015). Article CAS PubMed Google Scholar * Bukau, B. & Horwich,
A. L. The Hsp70 and Hsp60 chaperone machines. Cell 92, 351–366 (1998). Article CAS Google Scholar * Fersht, A. R. & Daggett, V. Protein folding and unfolding at atomic resolution.
Cell 108, 573–582 (2002). Article CAS PubMed Google Scholar * White, G. W. N. et al. Simulation and experiment conspire to reveal cryptic intermediates and a slide from the
nucleation-condensation to framework mechanism of folding. J. Mol. Biol. 350, 757–775 (2005). Article CAS PubMed Google Scholar * Arya, R., Mallik, M. & Lakhotia, S. C. Heat shock
genes - integrating cell survival and death. J. Biosci. 32, 595–610 (2007). Article CAS PubMed Google Scholar * Samali, A., Cai, J., Zhivotovsky, B., Jones, D. P. & Orrenius, S.
Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. EMBO J. 18, 2040–2048 (1999). Article CAS PubMed PubMed Central
Google Scholar * Kirchhoff, S. R., Gupta, S. & Knowlton, A. A. Cytosolic heat shock protein 60, apoptosis and myocardial injury. Circulation 105, 2899–2904 (2002). Article CAS PubMed
Google Scholar * Cappello, F., Di Stefano, A., D’Anna, S. E., Donner, C. F. & Zummo, G. Immunopositivity of heat shock protein 60 as a biomarker of bronchial carcinogenesis. Lancet
Oncol. 6, 816–816 (2005). Article CAS PubMed Google Scholar * Cappello, F. et al. HSP60 and HSP10 down-regulation predicts bronchial epithelial carcinogenesis in smokers with chronic
obstructive pulmonary disease. Cancer 107, 2417–2424 (2006). Article CAS PubMed Google Scholar * Lebret, T. et al. Heat shock proteins HSP27, HSP60, HSP70 and HSP90: expression in
bladder carcinoma. Cancer 98, 970–977 (2003). Article CAS PubMed Google Scholar * Kamishima, T. et al. Carcinosarcoma of the urinary bladder: expression of epithelial markers and
different expression of heat shock proteins between epithelial and sarcomatous elements. Pathol. Int. 47, 166–173 (1997). Article CAS PubMed Google Scholar * Hwang, Y. J. et al.
Expression of Heat Shock Protein 60 kDa Is Upregulated in Cervical Cancer. Yonsei Med. J. 50, 399–406 (2009). Article CAS PubMed PubMed Central Google Scholar * Castilla, C. et al.
Immunohistochemical Expression of Hsp60 Correlates With Tumor Progression and Hormone Resistance in Prostate Cancer. Urology 76, 1017.e1–1017.e6 (2010). Article Google Scholar * Hamrita,
B. et al. Identification of tumor antigens that elicit a humoral immune response in breast cancer patients’ sera by serological proteome analysis (SERPA). Clin. Chim. Acta 393, 95–102
(2008). Article CAS PubMed Google Scholar * Ghosh, J. C., Dohi, T., Kang, B. H. & Altieri, D. C. Hsp60 regulation of tumor cell apoptosis. J. Biol. Chem. 283, 5188–5194 (2008).
Article CAS PubMed Google Scholar * Sabharwal, S. S. & Schumacker, P. T. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 14, 709–721
(2014). Article CAS PubMed PubMed Central Google Scholar * Krishan, S., Richardson, D. R. & Sahni, S. Adenosine monophosphate-activated kinase and its key role in catabolism:
structure, regulation, biological activity and pharmacological activation. Mol. Pharmacol. 87, 363–377 (2015). Article CAS PubMed Google Scholar * Roy, L. D. et al. MUC1 enhances
invasiveness of pancreatic cancer cells by inducing epithelial to mesenchymal transition. Oncogene 30, 1449–1459 (2011). Article CAS PubMed Google Scholar * Xu, L., Chow, K. K. H., Lim,
M. & Li, G. Current Vaccine Trials in Glioblastoma: A Review. J. Immunol Res. (2014). * Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New
Engl. J. Med. 352, 987–996 (2005). Article CAS PubMed Google Scholar * Stupp, R. et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on
survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466 (2009). Article CAS PubMed Google Scholar * Ghosh, J. C.,
Siegelin, M. D., Dohi, T. & Altieri, D. C. Heat shock protein 60 regulation of the mitochondrial permeability transition pore in tumor cells. Cancer Res. 70, 8988–8993 (2010). Article
CAS PubMed PubMed Central Google Scholar * Carroll, J. et al. Bovine complex I is a complex of 45 different subunits. J. Biol. Chem. 281, 32724–32727 (2006). Article CAS PubMed Google
Scholar * Sabharwal, S. S. & Schumacker, P. T. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 14, 709–721 (2014). Article CAS PubMed
PubMed Central Google Scholar * Rigoulet, M., Yoboue, E. D. & Devin, A. Mitochondrial ROS Generation and Its Regulation: Mechanisms Involved in H2O2 Signaling. Antioxid. Redox Sign.
14, 459–468 (2011). Article CAS Google Scholar * Sharma, L. K. et al. Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation.
Hum. Mol. Genet. 20, 4605–4616 (2011). Article CAS PubMed PubMed Central Google Scholar * Hardie, D. G. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat.
Rev. Mol. Cell Biol. 8, 774–785 (2007). Article CAS PubMed Google Scholar * Steinberg, G. R. & Kemp, B. E. AMPK in Health and Disease. Physiol. Rev. 89, 1025–1078 (2009). Article
CAS PubMed Google Scholar * Srivastava, R. A. et al. AMP-activated protein kinase: an emerging drug target to regulate imbalances in lipid and carbohydrate metabolism to treat
cardio-metabolic diseases. J. Lipid Res. 53, 2490–2514 (2012). Article CAS PubMed PubMed Central Google Scholar * Ruderman, N. B., Carling, D., Prentki, M. & Cacicedo, J. M. AMPK,
insulin resistance and the metabolic syndrome. J. Clin. Invest. 123, 2764–2772 (2013). Article CAS PubMed PubMed Central Google Scholar * Shaw, R. J. LKB1 and AMP-activated protein
kinase control of mTOR signalling and growth. Acta Physiol. (Oxf) 196, 65–80 (2009). Article CAS ADS Google Scholar * Inoki, K., Li, Y., Xu, T. & Guan, K. L. Rheb GTPase is a direct
target of TSC2 GAP activity and regulates mTOR signaling. Gene Dev. 17, 1829–1834 (2003). Article CAS PubMed Google Scholar * Dibble, C. C. & Manning, B. D. Signal integration by
mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 15, 555–564 (2013). Article CAS PubMed PubMed Central Google Scholar * Gwinn, D. M. et al. AMPK
phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 (2008). Article CAS PubMed PubMed Central Google Scholar * Wullschleger, S., Loewith, R. & Hall, M.
N. TOR signaling in growth and metabolism. Cell 124, 471–484 (2006). Article CAS PubMed Google Scholar * Holz, M. K., Ballif, B. A., Gygi, S. P. & Blenis, J. mTOR and S6K1 mediate
assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123, 569–580 (2005). Article CAS PubMed Google Scholar *
Haghighat, A., Mader, S., Pause, A. & Sonenberg, N. Repression of Cap-Dependent Translation by 4E-Binding Protein 1:Competition with P220 for Binding to Eukaryotic Initiation Factor-4E.
EMBO J. 14, 5701–5709 (1995). Article CAS PubMed PubMed Central Google Scholar * Gingras, A. C. et al. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Gene Dev. 13,
1422–1437 (1999). Article CAS PubMed Google Scholar * Hardie, D. G. & Pan, D. A. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem. Soc.
Trans. 30, 1064–1070 (2002). Article CAS PubMed Google Scholar * Li, Y. et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in
diet-induced insulin-resistant mice. Cell Metab. 13, 376–388 (2011). Article CAS PubMed PubMed Central Google Scholar * Tiedemann, R. E. et al. Identification of molecular
vulnerabilities in human multiple myeloma cells by RNA interference lethality screening of the druggable genome. Cancer Res. 72, 757–768 (2012). Article CAS PubMed Google Scholar * Gu,
L. et al. Functional Characterization of Sirtuin-like Protein in Mycobacterium smegmatis. J. Proteome Res. 14, 4441–4449 (2015). Article CAS PubMed Google Scholar * Bligh, E. G. &
Dyer, W. J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 (1959). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We
thank the Protein Chemistry Facility at the Center for Biomedical Analysis of Tsinghua University for sample analysis. This work was supported in part by NSFC 31270871 (H.T.D) and MOEC
2012Z02293 (H.T.D) and the Global Science Alliance Program of Thermo-Fisher Scientific. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * MOE Key Laboratory of Bioinformatics, School of Life
Sciences, Tsinghua University, Beijing, 100084, China Haiping Tang, Jin Li, Xiaohui Liu & Haiteng Deng * Department of Neurosurgery, Changgung Hospital Affiliated to Tsinghua University,
Beijing, 100084, China Guihuai Wang * Molecular Pharmacology and Chemistry Program, Memorial Sloan-Kettering Cancer Center, New York, 10065, United States of America Minkui Luo Authors *
Haiping Tang View author publications You can also search for this author inPubMed Google Scholar * Jin Li View author publications You can also search for this author inPubMed Google
Scholar * Xiaohui Liu View author publications You can also search for this author inPubMed Google Scholar * Guihuai Wang View author publications You can also search for this author
inPubMed Google Scholar * Minkui Luo View author publications You can also search for this author inPubMed Google Scholar * Haiteng Deng View author publications You can also search for this
author inPubMed Google Scholar CONTRIBUTIONS H.T. conducted the proteomics experiments. H.T. and X.L. conducted the lipidomic analysis. H.T. and J.L. analyzed data. G.W., M.L. and H.D.
designed the experiments and wrote the paper. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing financial interests. ELECTRONIC SUPPLEMENTARY MATERIAL SUPPLEMENTARY
TABLES SUPPLEMENTARY INFORMATION RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this
article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will
need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ Reprints and permissions ABOUT
THIS ARTICLE CITE THIS ARTICLE Tang, H., Li, J., Liu, X. _et al._ Down-regulation of HSP60 Suppresses the Proliferation of Glioblastoma Cells via the ROS/AMPK/mTOR Pathway. _Sci Rep_ 6,
28388 (2016). https://doi.org/10.1038/srep28388 Download citation * Received: 08 February 2016 * Accepted: 03 June 2016 * Published: 21 June 2016 * DOI: https://doi.org/10.1038/srep28388
SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to
clipboard Provided by the Springer Nature SharedIt content-sharing initiative