Play all audios:
ABSTRACT Endothelial dysfunction is a characteristic of many vascular related diseases such as hypertension. Peroxisome proliferator activated receptor gamma, coactivator 1α (PGC-1α) is a
unique stress sensor that largely acts to promote adaptive responses. Therefore, we sought to define the role of endothelial PGC-1α in vascular function using mice with endothelial specific
loss of function (PGC-1α EC KO) and endothelial specific gain of function (PGC-1α EC TG). Here we report that endothelial PGC-1α is suppressed in angiotensin-II (ATII)-induced hypertension.
Deletion of endothelial PGC-1α sensitized mice to endothelial dysfunction and hypertension in response to ATII, whereas PGC-1α EC TG mice were protected. Mechanistically, PGC-1α promotes
eNOS expression and activity, which is necessary for protection from ATII-induced dysfunction as mice either treated with an eNOS inhibitor (LNAME) or lacking eNOS were no longer responsive
to transgenic endothelial PGC-1α expression. Finally, we determined that the orphan nuclear receptor, estrogen related receptor α (ERRα) is required to coordinate the PGC-1α -induced eNOS
expression. In conclusion, endothelial PGC-1α expression protects from vascular dysfunction by promoting NO• bioactivity through ERRα induced expression of eNOS. SIMILAR CONTENT BEING VIEWED
BY OTHERS ENDOTHELIAL SP1/SP3 ARE ESSENTIAL TO THE EFFECT OF CAPTOPRIL ON BLOOD PRESSURE IN MALE MICE Article Open access 21 September 2023 ENDOTHELIAL EPIDERMAL GROWTH FACTOR RECEPTOR IS
OF MINOR IMPORTANCE FOR VASCULAR AND RENAL FUNCTION AND OBESITY-INDUCED DYSFUNCTION IN MICE Article Open access 31 March 2021 NPRC DELETION MITIGATED ATHEROSCLEROSIS BY INHIBITING OXIDATIVE
STRESS, INFLAMMATION AND APOPTOSIS IN APOE KNOCKOUT MICE Article Open access 09 August 2023 INTRODUCTION Hypertension is the most prevalent risk factor for vascular disease worldwide, with
expectations that this condition will impact up to 1.56 billion people by the year 20251. Patients with hypertension are at increased risk of heart attack and stroke, two major sources of
cardiovascular morbidity and mortality. Among the sequelae of hypertension, endothelial dysfunction, characterized by compromised nitric oxide (NO•) bioavailability2,3, is particularly
prominent. Indeed, endothelial dysfunction has proven a powerful predictor of cardiovascular events4. Thus, there is considerable interest in developing strategies to improve endothelial
function and NO• bioactivity as a means to ameliorate the consequences of hypertension, including cardiovascular disease. Metabolic pathways have garnered increasing attention as being
relevant to vascular disease. In this regard, caloric restriction and exercise – two conditions that alter the balance between energy storage and utilization, have emerged as strategies to
mitigate chronic disease states, such as hypertension and its impact on the vasculature. Both caloric restriction and exercise activate the energy-sensitive enzyme, AMP-activated protein
kinase (AMPK), that can limit the development of endothelial dysfunction through decreased vascular reactive oxygen species (ROS) and improved NO• bioavailability5. One important downstream
target of AMPK is peroxisome proliferator activated receptor gamma, coactivator 1α (PGC-1α)6, a transcriptional coactivator important for the regulation of both mitochondrial and cellular
genes involved in metabolism and stress adaptation7,8. PGC-1α expression is decreased in aging9 and may be important for the aging associated decline of vascular function10. _In vitro_,
endothelial PGC-1α demonstrates improved ROS detoxification and enhanced endothelial resistance to cellular injury11,12. However, the role of PGC-1α on the endothelium _in vivo_ is not yet
clear. Thus, we probed the impact of endothelial specific PGC-1α manipulation on mouse models of endothelial dysfunction and hypertension. RESULTS LOSS OF ENDOTHELIAL PGC-1Α IMPAIRS
ENDOTHELIAL NO• BIOACTIVITY Previously we have demonstrated that the endothelium is modulated by mediators of mitochondrial function such as AMP kinase5 and uncoupling protein-213. As PGC-1α
is well-established to mediate mitochondrial biogenesis and metabolic stress adaptation in other cell types6, we examined PGC-1α expression in endothelial cells from wild-type (WT) mice
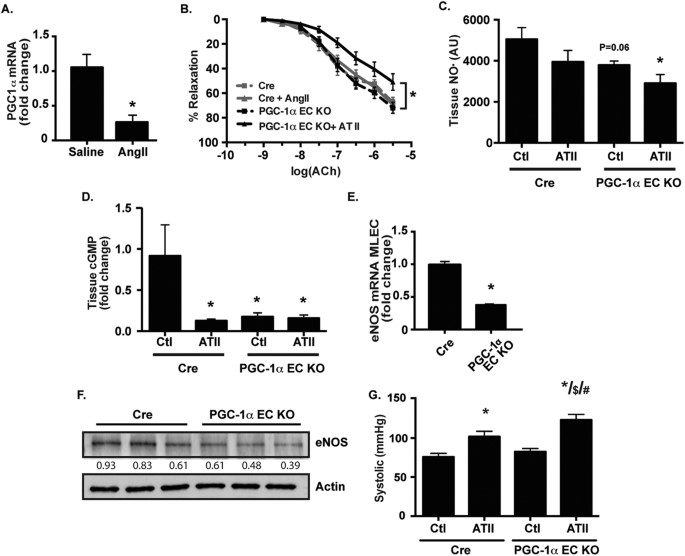
treated with a pressor dose (i.e, a dose known to induce endothelial dysfunction in WT mice) of angiotensin II (ATII). We observed a significant decrease in endothelial PGC-1α expression
(Fig. 1A). These data suggest that suppression of endothelial PGC-1α expression may be important in promoting endothelial dysfunction in response to ATII. To examine if loss of endothelial
PGC-1α primes the endothelium for dysfunction, we created endothelial-specific loss of function (PGC-1α EC KO) mice which demonstrated a significant reduction in PGC-1α expression
(Supplementary Figure 1A and B). We then examined endothelial function in response to ATII at a subpressor dose (i.e, a dose that does not significantly compromise endothelial function in WT
mice). We found that loss of endothelial PGC-1α significantly sensitized the endothelium to dysfunction induced by ATII (Fig. 1B). Similarly, tissue NO• abundance (Fig. 1C) and eNOS
expression (Supplementary Figure 1B and 1C) was decreased in the PGC-1α EC KO mice treated with ATII. Tissue cGMP, a downstream read out of bioactive NO•, was attenuated in the PGC-1α EC KO
mice (Fig. 1D). In addition, endothelial cells from PGC-1α EC KO mice exhibited decreased eNOS mRNA (Fig. 1E) and protein (Fig. 1F) expression. These data suggest that loss of PGC-1α leads
to compromised endothelial function, through decreases in eNOS expression and NO• bioactivity. Finally, we tested a pressor ATII dose (Fig. 1G) and found that the PGC-1α EC KO mice had
significantly increased blood pressure compared to the Cre control mice. ENDOTHELIAL PGC-1Α PROTECTS FROM ANGIOTENSIN II-INDUCED HYPERTENSION AND DYSFUNCTION _IN VIVO_ To determine if
upregulation of endothelial PGC-1α impacts NO• bioactivity, we created transgenic mice with constitutive human PGC-1α expression under the control of the vascular endothelial cadherin
promoter (PGC-1α EC TG; Fig. 2A). These mice demonstrated increased endothelial PGC-1α expression (Fig. 2B and C). After a pressor dose of ATII, relaxation in response to acetylcholine (Ach)
was attenuated in WT aorta, but largely preserved in vessels from PGC-1α EC TG mice (Fig. 2D). Similarly, NO• bioactivity after ATII treatment was preserved in PGC-1α EC TG mice as
demonstrated by aortic tissue cGMP levels (Fig. 2E). We then tested the impact of preserved endothelial NO• bioactivity on ATII-induced hypertension. In WT mice, ATII infusion caused a
significant increase in blood pressure as previously reported (Fig. 2F)14. This increase was prevented in PGC-1α EC TG mice (Fig. 2F). Together, these data confirm that augmentation of
endothelial PGC-1α expression _in vivo_ results in protection from endothelial dysfunction. PGC-1Α EXPRESSION PROTECTS THE ENDOTHELIUM THROUGH ENHANCED ENOS EXPRESSION AND NO• BIOACTIVITY It
is known _in vitro_ that PGC-1α can protect the endothelium from ROS-induced damage11,12. However, the impact of PGC1α on endothelial NO• generation is not known _in vivo_. Thus, we sought
to determine if PGC-1α overexpression influences NO• production and eNOS expression. In human aortic endothelial cells (HAECs), forced adenoviral overexpression of PGC-1α (Ad- PGC-1α) was
associated with enhanced NO• bioactivity assessed through cGMP production (Fig. 3A) coincident with a significant increase in eNOS mRNA (Fig. 3B) and protein expression (Fig. 3C and D). We
observed a modest increase in phosphorylated eNOS (Ser1177), likely due to the total eNOS increase (Fig. 3D). Consistent with these findings, there was increased eNOS expression in
endothelial cells from the PGC-1α EC TG mice (Fig. 3E). Aortic segments from PGC-1α EC TG mice exhibited reduced constriction in response to phenylephrine (Fig. 3F), consistent with
increased basal NO• bioactivity. This response was specific to eNOS as it was abrogated by the eNOS inhibitor, L-Nitroarginine Methyl Ester (LNAME; 300 μM; Fig. 3G). We examined the ability
of endothelial PGC-1α expression to prevent ATII - induced hypertension in a prospective study where the WT and PGC-1α EC TG cohorts were administered LNAME (0.5 g/L) in the drinking water
for 7 days followed by 7 days of LNAME + ATII. Results from this experiment demonstrated that eNOS was necessary for PGC-1α protection from hypertension (Fig. 3H). The requirement for eNOS
was further tested by breeding the PGC-1α EC TG mice onto the eNOS-null background (eNOS−/−). Endothelial nitric oxide synthase null mice are spontaneously hypertensive and exhibit
endothelial dysfunction15. In these mice, the endothelial specific expression of PGC-1α was not protective from spontaneous hypertension (Fig. 3I). These data confirm that eNOS is a key
contributor in mediating PGC-1α protection of the endothelium. PGC-1Α PROMOTES ENOS EXPRESSION THROUGH ERRΑ One common mechanism of eNOS activation and upregulation is via the
serine/threonine kinase, Akt16,17, therefore, we examined Akt expression and activation. In our studies, PGC-1α did not increase Akt activation or expression (Supplementary Figure 2A).
Another mechanism of eNOS activation is through reactive oxygen species (ROS) as we and others have previously demonstrated that ROS can increase eNOS activity and expression18,19. However,
we found decreased ROS production in endothelial cells from PGC-1α EC TG mice compared to WT mice (Supplementary Figure 2B). Thus, we turned our attention to well-known PGC-1α targets such
as Estrogen Related Receptor α (ERRα and Peroxisome Proliferator-Activated Receptor γ (PPARγ), that can modify eNOS expression or activity20. Forced expression of PGC-1α in HAECs produced no
change in PPARγ expression, but a significant increase in ERRα (Fig. 4A). Endothelial cells harvested from WT mice treated with ATII demonstrated suppressed ERRα expression (Fig. 4B),
reminiscent of the effect on PGC-1α (Fig. 1A). Furthermore, PGC-1α EC KO endothelial cells exhibit decreased ERRα expression (Fig. 4C), demonstrating that in endothelial cells, ERRα
expression closely mimics the expression pattern of PGC-1α. We then investigated whether ERRα was necessary for the increased expression of eNOS. As ERRα has been shown to induce eNOS
expression20, we used siRNA to knock down ERRα in HUVECs (Fig. 4D) and endothelial cells from our PGC-1α EC TG mice (Fig. 4E). In both of these cell types, PGC-1α mediated increases in eNOS
expression were attenuated as demonstrated in summary (Fig. 4F). DISCUSSION The data presented here indicate that PGC-1α expression plays an important role in the pathophysiology of
angiotensin II-induced hypertension. Downregulation of PGC-1α was observed during ATII-infusion and loss of PGC-1α facilitated the development of endothelial cell dysfunction, whereas
persistent endothelial PGC-1α expression attenuated the response to ATII. Of particular note, we identified a new role for PGC-1α in maintaining eNOS expression: PGC-1α loss of function was
associated with a reduction in eNOS expression and, conversely, PGC-1α gain of function increased basal eNOS expression. Furthermore, PGC-1α required ERRα to enhance eNOS expression.
Collectively, these data indicate that PGC-1α is a key novel determinant of endothelial cell eNOS expression and, as a consequence, NO• bioactivity. Although previous data has linked PGC-1α
to nitric oxide dependent responses, generally the literature has been focused on the effects of NO• on PGC-1α, rather than vice versa as we describe here. For example, NO• has been shown to
upregulate PGC-1α and stimulate mitochondrial biogenesis21. Likewise, caloric restriction, a state associated with increased longevity, causes mitochondrial biogenesis and PGC-1α
upregulation in an eNOS-dependent manner22. The data presented here suggest a bidirectional relationship between PGC-1α and NO•, with the former being required to maintain normal eNOS
expression in the endothelium. Furthermore, we demonstrate that PGC-1α expression dictates endothelial function in the context of angiotensin II-induced hypertension, a disease model
characterized by impaired NO• bioactivity and increased vascular ROS production23. With regard to the latter, there are several PGC-1α-dependent genes known to coordinate antioxidant gene
regulation, including SOD2, Prx3, Prx5, Trx 2, and catalase12. These data, combined with observations that PGC-1α protects the endothelium from ROS-mediated damage11,12,24, suggest that one
potential mechanism for our observations may also be the antioxidant program promoted by PGC-1α. This idea is supported by observations that manipulation of vascular ROS scavenging are
associated with improved NO• bioactivity and preservation of endothelial function25. However, the relative importance to the endothelium of PGC-1α action on ROS scavenging versus its effect
on eNOS expression remains to be determined. Nevertheless, our data indicate that PGC-1α cannot significantly impact the consequences of ATII in the absence of eNOS. In addition to ROS
scavenging, PGC-1α expression has been implicated in protection from inflammation. For example, PGC-1α overexpression results in decreased endothelial inflammation in response to tumor
necrosis factor-α26 PGC-1α deficient mice bred onto the ApoE−/− background displayed increased inflammatory markers in plaques27. As ATII-induced hypertension is associated with a vascular
inflammatory response with increased endothelial inflammatory gene expression28, one might speculate that endothelial PGC-1α manipulation may impact vascular inflammatory responses. While,
multiple studies in endothelial cells have demonstrated protection from ROS12,22, cell death29, and inflammation26, in conditions of high glucose, PGC-1α may have a detrimental effect and
lead to impaired endothelial function. For instance, under diabetic conditions, PGC-1α overexpression led to decreased angiogenesis, whereas PGC-1α loss of function improved endothelial
angiogenesis30. Therefore, endothelial PGC-1α could potentially serve multiple contextual roles wherein conditions of hypertension, lead to improved eNOS bioavailability, but in conditions
of hyperglycemia, endothelial angiogenesis is impeded. Further investigation will be required for a complete picture as to how PGC-1α contributes to endothelial phenotype in health and
disease. The data presented here demonstrate a novel paradigm wherein PGC-1α induces expression of ERRα, which is then necessary to enhance eNOS expression (Fig. 4D,E). In our study,
endothelial ERRα expression mirrored PGC-1α expression. Although it was known that the eNOS promoter contains an ERRα binding site20, we have now elucidated a functional connection to eNOS
_in vivo_. The link between ERRα and PGC-1α is consistent with studies in mice lacking ERRα, that phenocopy the heart failure seen in PGC-1α knockout mice31. However, studies with
endothelial-specific ERRα manipulation _in vivo_ will be needed to further clarify its role in vascular function. In summary, the work presented here links endothelial PGC-1α to NO•
bioactivity via eNOS regulation. This indicates that endothelial PGC-1α expression may be an important determinant of vascular health (Fig. 4F) as it is protective from endothelial
dysfunction in response to ATII. It will be of particular interest to determine if the molecular pathway described here impacts other critical processes involved in endothelial dysfunction,
such as mitochondrial function, metabolism, and inflammation. METHODS MATERIALS Antibodies: PGC-1α (Abcam); phospho-Ser1177 eNOS (Upstate Biotechnology); eNOS (BD Transduction); ERRα
(Abcam); β-actin (Sigma). Micro-osmotic pumps were purchased from DURECT corporation (Cupertino, CA). Other chemicals were obtained from Sigma. GENERATION OF ENDOTHELIAL SPECIFIC PGC-1Α
MANIPULATION IN MICE Human PGC-1α cDNA (Origene, Rockville, MD) was linearized and inserted into pBSmVELacZ (Obtained from Kenneth Walsh, Ph.D. Boston University, Boston, MA) to replace the
LacZ open reading frame through the NotI site to produce human PGC-1α expression under the control of the mouse vascular endothelial cadherin promoter (VE-Cad; Fig. 2A). The resulting
construct was microinjected into fertilized embryos harvested from C57BL/6 mice in the UMMS Transgenic Core to create the endothelial specific PGC-1α transgenic mouse line (PGC-1α EC TG).
Genotyping was performed with mouse tail DNA and primers of (ggctggtaccttggaactga) and (aatccgtcttcatccacagg). Two separate PGC-1α EC TG mouse lines were used for experiments. The PGC-1α EC
TG mice were bred onto the background of eNOS−/− mice (Jackson Laboratory) to generate PGC1α-EC TG/eNOS−/− mice. For the endothelial specific knockout line, the PGC-1α allele containing
_LoxP_ sites flanking exons 3–5 of the PGC-1α gene (_PGC1α__flox/flox_)32 was obtained from Bruce Spiegelman (Harvard University) and bred with the Tie2-Cre mouse line on the C57 background.
These endothelial-specific PGC-1α knockout mice (PGC-1α EC KO) were compared to Tie2-Cre mice (Cre). Approval for animal care and use for these experiments was granted by the Institutional
Animal Care and Use Committee (IACUC) of the University of Massachusetts Medical School and the Ethics Committee of the University Hospital Mainz and all experiments were carried out in
accordance with the guidelines from these institutions. CELL CULTURE Primary cultures of human aortic endothelial cells (HAECs) or human umbilical vein endothelial cells (HUVECs) were
obtained from Lonza Group Ltd. (Switzerland) and used in experiments during passages 2–8. Cultures were maintained in EBM-2 media with supplements (Lonza). Primary cultures of rat aortic
smooth muscle cells (RASMCs) were obtained from VEC Technologies (Rensselaer, NY, USA) and used in experiments during passages 2–8. RASMC cultures were maintained in DMEM (Cellgro) that
contained 10% fetal bovine serum, 500 IU/ml penicillin, and 500 IU/ml streptomycin. Mouse lung endothelial cells were prepared by immunoselection with anti-ICAM-2 antibody as previously
described33. Adenovirus was used for both overexpression (24 h) and knockdown (48 h) of PGC-1α (kind gift from the laboratory of Bruce Spiegelman) and ERRα (kind gift from laboratory of
Anastasia Kralli, Scripps Research Institute34). Control viruses (siCtl, LacZ, and GFP) from Vector BioLabs (Malvern, PA). MRNA EXTRACTION AND RT-PCR RNA was extracted from endothelial cells
or mice aorta using RNeasy mini kit (Qiagen) or Trizol tissue extraction. For mRNA expression analysis both TaqMan (Life Technologies) and SYBR Green (Bio-Rad) methods were used
(Supplementary Table 1). The ΔΔcycle threshold method was used for relative mRNA quantification and the gene expression was normalized to a housekeeping gene (TBP or HPRT). OSMOTIC PUMP
IMPLANTATION Mice were anesthetized with an intraperitoneal ketamine-xylazine mixture. Osmotic pumps (Alzet model 1007D) were implanted subcutaneously to allow infusion of ATII or vehicle
(NaCl 0.9%) at one of two rates for 7d: i) the suppressor rate of 0.5 mg · kg–1 · d–1 that does not significantly compromise endothelial function in wild type (WT) mice35 or a pressor rate
of 1.0 mg · kg–1 · d–1 for 7 days which induces endothelial dysfunction and hypertension in WT mice5. BLOOD PRESSURE Non-invasive determinations were performed in conscious mice by tail cuff
sphygmomanometer using BP-2000 pressure analysis system (Visitech Systems, Apex, NC) which we previously validated against telemetry monitoring35. The mice were trained daily for one week
prior to recorded measurements. A minimum of 10 preliminary measurements with 20 actual measurements were performed in each session. Measurements were made at the same time each day to
account for diurnal variation. Five consecutive daily measurements were averaged for each of the groups. Where indicated, NG-nitro-L-arginine methyl ester (LNAME) was administered (0.5 g/L)
in the drinking water for 7–14 days. WESTERN BLOT The cell and tissue lysates were denatured with Laemmli sample buffer (Cell Signaling) and then resolved by SDS-PAGE followed by western
blotting with antibodies as indicated. The protein bands were visualized using the AlphaImager© imaging system and bands quantified with Image J (NIH). Numbers below immunoblots represent
densitometry relative to actin. TISSUE NO• MEASUREMENT Electron paramagnetic resonance spectrometry (EPR) was used to assess vascular NO• synthesis using colloid
Fe(II)-diethyldithiocarbamate (Fe(DETC)2) as spin trap with an X-band table-top spectrometer MS200 (Magnettech, Berlin, Germany). The instrument settings were: 10 mW microwave power, 0.8 mT
amplitude modulation, 100 kHz modulation frequency, 327 mT center field, 10 mT sweep width, 60 s sweep time and 3 scans. Total NO• production was assessed by measurement amplitude of the
characteristic triplet EPR signal and expressed in arbitrary units, AU/(mg dry weight × h)35. TISSUE CGMP Tissue cGMP from aortas was measured as described33. _IN VITRO_ CGMP Endothelial NO•
bioactivity in culture was estimated as cGMP production in a RASMC reporter assay as previously described33. ISOMETRIC MEASUREMENTS OF AORTIC FUNCTION Thoracic aortic rings (2 mm in length)
were mounted on 200 μm pins in a 6-mL chamber vessel myograph (Danish Myo Technology; 610 M) with 1 g basal tension as previously described33. Aortic rings were subjected to
concentration-response curves to increasing concentrations of phenylephrine (PE) and acetylcholine (Ach), the latter in vessels precontracted with a submaximal (70–80%) concentration of PE
(10–7 M). STATISTICS Numerical data are presented as mean ± SE. Overall differences were analyzed using one-way or two-way ANOVA and tested with Tukey-Kramer Multiple-Comparison Test for
determining differences between the means when more than two groups were compared. An independent _t_-test was used when only two groups were compared. In all tests, significance was
accepted at P < 0.05. ADDITIONAL INFORMATION HOW TO CITE THIS ARTICLE: Craige, S. M. _et al_. PGC-1α dictates endothelial function through regulation of eNOS expression. _Sci. Rep._ 6,
38210; doi: 10.1038/srep38210 (2016). PUBLISHER'S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. REFERENCES
* Kearney, P. M. et al. Global burden of hypertension: analysis of worldwide data. Lancet 365, 217–223 (2005). Google Scholar * Vita, J. A. & Keaney, J. F. Endothelial function: a
barometer for cardiovascular risk? Circulation 106, 640–642 (2002). Article Google Scholar * Vita, J. A. et al. Coronary vasomotor response to acetylcholine relates to risk factors for
coronary artery disease. Circulation 81, 491–497 (1990). Article CAS Google Scholar * Gokce, N. et al. Risk stratification for postoperative cardiovascular events via noninvasive
assessment of endothelial function: a prospective study. Circulation 105, 1567–1572 (2002). Article Google Scholar * Schulz, E. et al. Suppression of the JNK pathway by induction of a
metabolic stress response prevents vascular injury and dysfunction. Circulation 118, 1347–1357 (2008). Article CAS Google Scholar * Patten, I. S. & Arany, Z. PGC-1 coactivators in the
cardiovascular system. Trends Endocrinol. Metab. 23, 90–97 (2012). Article CAS Google Scholar * Puigserver, P. et al. A cold-inducible coactivator of nuclear receptors linked to adaptive
thermogenesis. Cell 92, 829–839 (1998). Article CAS Google Scholar * Finck, B. N. & Kelly, D. P. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease.
J. Clin. Invest. 116, 615–622 (2006). Article CAS Google Scholar * Anderson, R. & Prolla, T. PGC-1alpha in aging and anti-aging interventions. Biochim. Biophys. Acta 1790, 1059–1066
(2009). Article CAS Google Scholar * Xiong, S. et al. Peroxisome proliferator-activated receptor γ coactivator-1α is a central negative regulator of vascular senescence. Arteriosclerosis,
Thrombosis, and Vascular Biology 33, 988–998 (2013). Article CAS Google Scholar * Olmos, Y. et al. Mutual dependence of Foxo3a and PGC-1alpha in the induction of oxidative stress genes.
J. Biol. Chem. 284, 14476–14484 (2009). Article CAS Google Scholar * Valle, I. I., Alvarez-Barrientos, A. A., Arza, E. E., Lamas, S. S. & Monsalve, M. M. PGC-1@a regulates the
mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res. 66, 12–12 (2005). Article Google Scholar * Shimasaki, Y. et al. Uncoupling protein 2 impacts
endothelial phenotype via p53-mediated control of mitochondrial dynamics. Circ. Res. 113, 891–901 (2013). Article CAS Google Scholar * Kawada, N., Imai, E., Karber, A., Welch, W. J. &
Wilcox, C. S. A mouse model of angiotensin II slow pressor response: role of oxidative stress. J. Am. Soc. Nephrol. 13, 2860–2868 (2002). Article CAS Google Scholar * Shesely, E. G. et
al. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA. 93, 13176–13181 (1996). Article ADS CAS Google Scholar * Dimmeler, S. et al.
Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399, 601–605 (1999). Article ADS CAS Google Scholar * Fulton, D. et al. Regulation of
endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399, 597–601 (1999). Article ADS CAS Google Scholar * Drummond, G. R., Cai, H., Davis, M. E., Ramasamy, S.
& Harrison, D. G. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circ. Res. 86, 347–354 (2000). Article CAS
Google Scholar * Thomas, S. R., Chen, K. & Keaney, J. F. Hydrogen peroxide activates endothelial nitric-oxide synthase through coordinated phosphorylation and dephosphorylation via a
phosphoinositide 3-kinase-dependent signaling pathway. J. Biol. Chem. 277, 6017–6024 (2002). Article CAS Google Scholar * Sumi, D. & Ignarro, L. J. Estrogen-related receptor alpha 1
up-regulates endothelial nitric oxide synthase expression. Proc. Natl. Acad. Sci. USA. 100, 14451–14456 (2003). Article ADS CAS Google Scholar * Nisoli, E. et al. Mitochondrial
biogenesis in mammals: the role of endogenous nitric oxide. Science 299, 896–899 (2003). Article ADS CAS Google Scholar * Borniquel, S., Valle, I., Cadenas, S., Lamas, S. & Monsalve,
M. Nitric oxide regulates mitochondrial oxidative stress protection via the transcriptional coactivator PGC-1alpha. FASEB J. 20, 1889–1891 (2006). Article CAS Google Scholar * Mollnau,
H. et al. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ. Res. 90, E58–65 (2002). Article Google
Scholar * Ali, F. et al. PPARdelta and PGC1alpha act cooperatively to induce haem oxygenase-1 and enhance vascular endothelial cell resistance to stress. Cardiovasc. Res. 85, 701–710
(2010). Article CAS Google Scholar * Daugherty, A. A., Manning, M. W. M. & Cassis, L. A. L. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient
mice. J. Clin. Invest. 105, 1605–1612 (2000). Article CAS Google Scholar * Kim, H.-J. et al. Effects of PGC-1α on TNF-α–Induced MCP-1 and VCAM-1 Expression and NF-κB Activation in Human
Aortic Smooth Muscle and Endothelial Cells. Antioxid. Redox Signal. 9, 301–307 (2007). Article CAS Google Scholar * Stein, S. et al. ApoE−/− PGC-1α−/− mice display reduced IL-18 levels
and do not develop enhanced atherosclerosis. PLoS ONE 5, e13539 (2010). Article ADS Google Scholar * Pacurari, M., Kafoury, R., Tchounwou, P. B. & Ndebele, K. The
Renin-Angiotensin-aldosterone system in vascular inflammation and remodeling. Int J Inflam 2014, 689360 (2014). * Won, J. C. et al. Peroxisome proliferator-activated receptor-gamma
coactivator 1-alpha overexpression prevents endothelial apoptosis by increasing ATP/ADP translocase activity. Arteriosclerosis, Thrombosis, and Vascular Biology 30, 290–297 (2010). Article
CAS Google Scholar * Sawada, N. et al. Endothelial PGC-1a Mediates Vascular Dysfunction in Diabetes. Cell Metab. 19, 246–258 (2014). Article CAS Google Scholar * Huss, J. M. et al. The
nuclear receptor ERRalpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. 6, 25–37 (2007). Article CAS Google Scholar * Lin, J. et al.
Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 119, 121–135 (2004). Article CAS Google Scholar * Craige, S. M. et al. NADPH oxidase 4
promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation 124, 731–740 (2011). Article CAS Google Scholar * Schreiber, S. N., Knutti, D., Brogli,
K., Uhlmann, T. & Kralli, A. The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor alpha (ERRalpha). J.
Biol. Chem. 278, 9013–9018 (2003). Article CAS Google Scholar * Kröller-Schön, S. et al. Peroxisome proliferator-activated receptor γ, coactivator 1α deletion induces angiotensin
II-associated vascular dysfunction by increasing mitochondrial oxidative stress and vascular inflammation. Arteriosclerosis, Thrombosis, and Vascular Biology 33, 1928–1935 (2013). Article
Google Scholar Download references ACKNOWLEDGEMENTS We would like to thank Heather Learnard, Michaella Reif, Xiaoyun Huang, Yukio Shimasaki, and Bettina Mros for their technical assistance.
This work was supported by F32HL099282 (to SMC), R01HL092122 and R01HL098407 (to JFK) from the NHLBI, 16SDG29660007 from American Heart Association (to SK) and KR 4011/2-1 from Deutsche
Forschungsgemeinschaft (DFG; to ES). AUTHOR INFORMATION Author notes * Craige Siobhan M., Kröller-Schön Swenja, Schulz Eberhard and Keaney John F. contributed equally to this work. AUTHORS
AND AFFILIATIONS * Division of Cardiovascular Medicine, Department of Medicine, University of Massachusetts Medical School, Worcester, MA, USA Siobhan M. Craige, Chunying Li, Shashi Kant,
Shenghe Cai, Mayur M. Contractor, Yongmei Pei & John F. Keaney Jr * Department of Cardiology Medizinische Klinik und Poliklinik Universitätsmedizin Mainz, Mainz, Germany Swenja
Kröller-Schön, Kai Chen & Eberhard Schulz * University of Connecticut Health Center, Farmington, CT, USA Kai Chen Authors * Siobhan M. Craige View author publications You can also search
for this author inPubMed Google Scholar * Swenja Kröller-Schön View author publications You can also search for this author inPubMed Google Scholar * Chunying Li View author publications
You can also search for this author inPubMed Google Scholar * Shashi Kant View author publications You can also search for this author inPubMed Google Scholar * Shenghe Cai View author
publications You can also search for this author inPubMed Google Scholar * Kai Chen View author publications You can also search for this author inPubMed Google Scholar * Mayur M. Contractor
View author publications You can also search for this author inPubMed Google Scholar * Yongmei Pei View author publications You can also search for this author inPubMed Google Scholar *
Eberhard Schulz View author publications You can also search for this author inPubMed Google Scholar * John F. Keaney Jr View author publications You can also search for this author inPubMed
Google Scholar CONTRIBUTIONS This study was conceived and designed by S.M.C., S.K.S., C.L., E.S., and J.F.K. The data was collected and analyzed by S.M.C., S.K.S., C.L., S.K., S.C., K.C.,
M.M.C., and Y.P. The manuscript was primarily written by S.M.C., S.K., and J.F.K. with contributions by all the listed authors. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no
competing financial interests. ELECTRONIC SUPPLEMENTARY MATERIAL SUPPLEMENTARY INFORMATION RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution 4.0
International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the
material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Craige, S., Kröller-Schön, S., Li, C. _et al._ PGC-1α dictates endothelial function
through regulation of eNOS expression. _Sci Rep_ 6, 38210 (2016). https://doi.org/10.1038/srep38210 Download citation * Received: 02 June 2016 * Accepted: 07 November 2016 * Published: 02
December 2016 * DOI: https://doi.org/10.1038/srep38210 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable
link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative