Play all audios:
ABSTRACT Many fundamental biological processes depend on intricate networks of interactions between proteins and nucleic acids and a quantitative description of these interactions is
important for understanding cellular mechanisms governing DNA replication, transcription, or translation. Here we present a versatile method for rapid and quantitative assessment of
protein/nucleic acid (NA) interactions. This method is based on protein induced fluorescence enhancement (PIFE), a phenomenon whereby protein binding increases the fluorescence of Cy3-like
dyes. PIFE has mainly been used in single molecule studies to detect protein association with DNA or RNA. Here we applied PIFE for steady state quantification of protein/NA interactions by
using microwell plate fluorescence readers (mwPIFE). We demonstrate the general applicability of mwPIFE for examining various aspects of protein/DNA interactions with examples from the
restriction enzyme _BamH_I, and the DNA repair complexes Ku and XPF/ERCC1. These include determination of sequence and structure binding specificities, dissociation constants, detection of
weak interactions, and the ability of a protein to translocate along DNA. mwPIFE represents an easy and high throughput method that does not require protein labeling and can be applied to a
wide range of applications involving protein/NA interactions. SIMILAR CONTENT BEING VIEWED BY OTHERS LARGE STOKES SHIFT FLUORESCENT RNAS FOR DUAL-EMISSION FLUORESCENCE AND BIOLUMINESCENCE
IMAGING IN LIVE CELLS Article 18 September 2023 SINGLE-MOLECULE FLUORESCENCE MULTIPLEXING BY MULTI-PARAMETER SPECTROSCOPIC DETECTION OF NANOSTRUCTURED FRET LABELS Article Open access 15 May
2024 SINGLE-MOLECULE KINETIC LOCKING ALLOWS FLUORESCENCE-FREE QUANTIFICATION OF PROTEIN/NUCLEIC-ACID BINDING Article Open access 15 September 2021 INTRODUCTION Interactions of proteins with
nucleic acids are crucial for a number of fundamental biological processes including DNA replication and repair, transcription, RNA processing, and translation. For example, binding
affinities of transcription factors to promoter and enhancer elements are important determinants of gene expression and a quantitative description of these parameters is crucial for
understanding the regulatory networks controlling developmental and physiological processes1,2. The strength and quality of protein/NA interactions are determined by atomic contacts between
amino acids and nucleotides underpinned by physical forces including electrostatic and hydrophobic interactions, hydrogen bonds, and van de Waals forces3. Protein/NA interactions are further
affected by neighboring proteins, small molecules, and the physical properties of the environment such as temperature or pH. These factors can modulate protein/NA affinities within cells,
providing a means for the regulation of biological mechanisms that rely on protein/NA interactions. Therefore, the quantitative description of these interactions under a number of
experimental conditions is essential for understanding both physiological processes and human pathologies, and are instrumental for smart drug design4. A wide variety of methods have been
developed to describe interactions between proteins and nucleic acids both _in vivo_ and _in vitro_. These include chromatin immunoprecipitation, yeast one hybrid assay, microplate capture
and ELISA based detection assays, filter binding assay, electrophoretic mobility shift assay (EMSA), fluorescence anisotropy, surface plasmon resonance, kinetic assays based on stopped-flow
systems, and single molecule techniques5. However, each method has its limitations and a combination of several techniques are often applied for the analysis of a particular interaction. For
example, EMSA is one of the most commonly used methods for determining protein/NA affinity and sequence specificity, but it is a rather laborious and time consuming technique with
relatively limited throughput that may be insensitive to low-affinity interactions. For screening even a moderate number of experimental parameters or chemical libraries for drug design,
high throughput methods are required. These methods usually use fluorescence that allows rapid detection by fluorescence scanners. Fluorescence anisotropy methods are based on the change in
fluorescence polarization upon protein binding to a labeled nucleic acid. In combination with special plate readers such methods can be used in a high throughput format6,7. Fluorescence
anisotropy can provide information about binding affinity and the molecular weight of the resulting complex, but it does not provide positional information. Methods exploiting Förster
resonance energy transfer (FRET) that occurs between donor and acceptor fluorophores placed on the protein and nucleic acid are also widely used8. These assays allow for a range of
experimental settings and can provide spatial information about the interaction. Nevertheless, one disadvantage of FRET-based assays is the requirement for protein labeling, which may be
laborious and interfere with the biochemical properties of the proteins being studied. Here we describe a quantitative, high throughput approach for studying protein/NA interactions without
the need for protein labeling. This method exploits protein induced fluorescence enhancement (PIFE), a phenomenon whereby an increase in fluorescence is observed when a protein binds to a
fluorescently labeled nucleic acid in the vicinity of a fluorophore9. Physical interaction of a protein with cyanine dyes sterically constrains their isomerization, blocking the formation of
a fluorescently inactive _cis_ isomer9. Similar effects have been shown in higher viscosity or lower temperature environments10,11. PIFE is very specific and has a higher spatial resolution
than FRET, showing a sharp response within a 0–3 nm range12. The short range and high sensitivity of PIFE proved to be excellent for studying protein/NA interactions in single molecule
(smPIFE) experiments12,13, as well as in kinetic studies using stopped-flow systems14,15,16. We modified PIFE for the quantification of steady-state protein/NA interactions using fluorescent
plate readers (microwell PIFE, mwPIFE). We demonstrate the general applicability and versatility of this method in a variety of experiments that probe distinct aspects of protein/NA
interactions. These include the association of a protein at different positions along DNA, defining minimal DNA-length binding requirements, determining sequence- or structure-specific DNA
binding affinities, and studying the effect of ions on protein binding and nuclease activity. mwPIFE is very simple yet highly quantitative, and can be applied to a broad range of questions
involving different aspects of protein/NA interactions. RESULTS DETECTION OF PROTEIN – DNA INTERACTION BY MWPIFE In single molecule experiments, the signal emitted from an individual
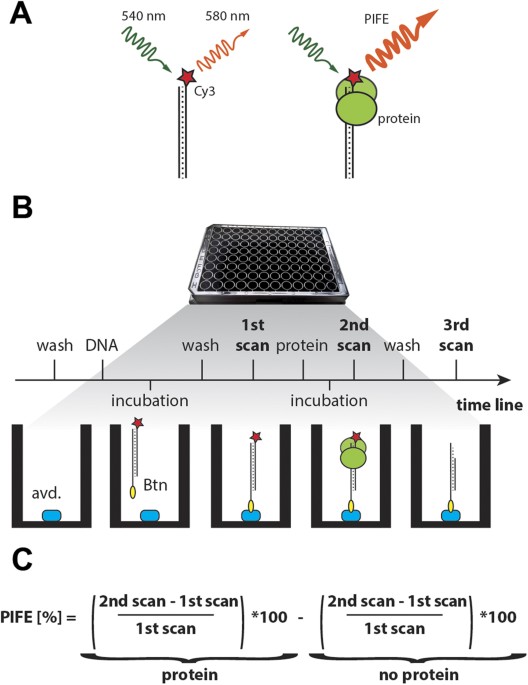
fluorophore can yield a 2- to 2.5-fold PIFE upon protein binding to a Cy3 labeled nucleic acid (Fig. 1A)12. We reasoned that the PIFE effect should also be detectable in a population of
molecules by standard fluorescence plate readers and would therefore be suitable for more routine protein/NA analyses. We designed an experimental protocol to detect the steady-state
interaction of an unlabeled protein with immobilized Cy3-labeled DNA probe by PIFE in a microwell (Fig. 1B). A Cy3-labelled oligonucleotide probe is immobilized to a black microwell coated
with neutravidin through biotin attached to either the 5′ or 3′ end of the oligonucleotide. Based on fluorescence measurement of a concentration range of immobilized oligonucleotides we
decided to use 2.5 pmol of Cy3- labelled probe (Supplementary Fig. 1). Fluorescence is measured using a plate reader and the signal from the first scan serves as a reference value for
determining PIFE. A second scan is performed after incubation of a protein with the DNA probe in the well (Fig. 1B). PIFE is calculated for each well as the relative difference between the
signals in the first and second scans. To correct for the effect of buffer and photobleaching on PIFE, a second scan is also performed in a control well where buffer with no protein is
added. The final PIFE value represents PIFE in the protein well minus PIFE in the control well (Fig. 1C). A third optional scan can be performed after protein washing in applications that
assess protein/DNA dissociation, effect of additional co-factors, or enzymatic activity. PIFE is then calculated relative to the first scan. To test whether the mwPIFE assay used in this
format is able to reliably detect protein-DNA association, we examined the interaction of the restriction endonuclease _Bam_HI with its target sequence. A similar experiment was previously
performed in a single molecule setup to analyze the distance dependence of PIFE between _Bam_HI and the fluorophore17. We prepared 60 bp duplex DNA probes containing _Bam_HI restriction
sites either 1, 16, 31, or 46 bp away from Cy3 placed at the 5′ end of one DNA strand, while the opposite strand was attached to microwells through its biotinylated 5′end (Fig. 2A,
Supplementary Table 1). A DNA probe of the same length without a _Bam_HI site was used as a control. In the presence of _Bam_HI and Ca2+ ions, we detected a 30% PIFE when the binding site
was located 1 nucleotide away from the Cy3. Importantly, the PIFE values with _Bam_HI showed a highly significant difference from control reactions with either bovine serum albumin (BSA) or
DNA probe with no BamHI site (P values range from 1.2 × 10−6 to 1.3 × 10−6; two tailed t-test) demonstrating that mwPIFE can reliably detect a protein/NA interaction in a quantitative
manner. No PIFE above background was detected with DNA probes where the restriction site was placed 16, 31, or 46 bp away from Cy3 (Fig. 2B), confirming data from the smPIFE study17. Thus,
similar to the single molecule setup, PIFE detection in a population of molecules is specific for short distance interactions. DETECTION OF DNA CLEAVAGE AND PROTEIN DISSOCIATION The mwPIFE
setup allows for the testing of a number of experimental conditions and their effect on protein/NA interaction. We demonstrate this by examining the effect of divalent cations on _Bam_HI/DNA
binding and cleavage. In the absence of cations, only a weak _Bam_HI induced PIFE was detected. Addition of Mg2+ or Ca2+ allows efficient binding of _Bam_HI to the restriction site and
induced 40–60% PIFE (Fig. 2C). To determine _Bam_HI cleavage activity, a third scan was performed after a 30 min incubation followed by buffer exchange. In the presence of Ca2+, mwPIFE was
reduced to background level, suggesting that only a small fraction of _Bam_HI remained associated with DNA after washing. The addition of Mg2+ ions resulted in a drastic decrease in
fluorescence and negative PIFE, which was presumably caused by cleavage and removal of the labeled DNA fragment by washing. These data are consistent with the known properties of _Bam_HI,
which has a relaxed requirement of divalent cations for binding, but requires Mg2+ for efficient cleavage18. This experiment shows that mwPIFE can also be adapted to study protein
dissociation and DNA cleavage. ASSESSMENT OF DNA BINDING SPECIFICITY Important aspects of protein/NA interaction are the sequence and structural specificities that can be assessed in
competition assays. The high throughout format of mwPIFE is well suited for these experiments as it allows parallel measurements of a number of different competitors in a broad range of
concentrations. As a proof of concept, we designed an mwPIFE experiment to test the sequence specificity of _Bam_HI in a competition assay. Binding of _Bam_HI to a Cy3 labeled
oligonucleotide containing the restriction site in the presence of Ca2+ and four different competitor DNAs was assessed by mwPIFE. When competitor DNA contained substitutions outside of the
recognition site (Fig. 3A), it competed with the labeled probe as efficiently as unlabeled probe. However, when a single base was changed within the _Bam_HI site, competition was no longer
observed and PIFE was comparable to the reactions where oligonucleotide lacking the BamHI site or no competitor control were added (Fig. 3B). This experiments show that mwPIFE can be used
for determining the sequence specificity of a protein/NA interaction. Many DNA processing proteins recognize their substrates via specific structures in a sequence independent manner. One of
them is the structure-selective endonuclease XPF/ERCC1 that is involved in a number of DNA repair mechanisms including nucleotide excision repair, DNA double strand break repair, and
interstrand crosslink repair19. XPF and ERCC1 form an obligate heterodimer20 that binds and processes branched DNA structures with double-to-single stranded transitions, such as bubbles,
stem loops, splayed arms, or overhangs21,22. To test whether mwPIFE can be used to determine the specificity of structure-dependent interactions, we assessed the association of the XPF/ERCC1
heterodimer with a labeled DNA substrate in the presence of different DNA structures as competitors. Because reconstitution of the full length XPF/ERCC1 in a heterologous expression system
is inefficient23, we used a truncated heterodimer lacking the N-terminal helicase-like domain of XPF which retains DNA binding and residual cleavage activity22,24. The mwPIFE binding
substrate consisted of a 25 bp duplex DNA with a 10 nt long 3′ overhang (Supplementary Table 1). The DNA probe was immobilized through the end of the single stranded overhang, while the Cy3
label was at the end of the duplex region (Fig. 4A). We observed 80% PIFE in the presence of XPF/ERCC1, indicating efficient binding of the complex to the DNA substrate. Next, we performed
the mwPIFE assay whereby the XPF/ERCC1 dimer was added in the presence of increasing concentrations of DNA competitors that included stem-loop, 3′overhang (Y/10), splayed DNA (Y), ssDNA
(10ss), and dsDNA (10ds) structures (Fig. 4B). These different competitors exhibited various effect on PIFE. While the stem-loop and splayed DNA exhibited the strongest inhibition of PIFE at
higher concentrations, the 3′ overhang showed an intermediate effect and no inhibition was detected for the short ss and dsDNAs (Fig. 4A). We validated this data by determining the
dissociation constants (Kd) for each competitor by fluorescence anisotropy (Fig. 4C). Kd values of the competitors are in agreement with their ability to compete for XPF/ERCC1 binding in the
mwPIFE assay, demonstrating the utility of mwPIFE in analyzing DNA-structure specific interactions. ANALYSIS OF KU-DNA INTERACTION BY MWPIFE We next analyzed DNA interactions of the human
Ku complex, a DNA repair factor with high affinity to DNA ends. Ku is a heterodimer consisting of Ku70 and Ku80 subunits, forming a ring-like structure that loads onto DNA through a free
end25. Ku-DNA interaction is sequence independent and EMSA has shown it requires at least 14 bp of duplex DNA26. We linked a linear 15 bp long dsDNA probe at one end to a microtiter well,
while the opposite Cy3 labeled end was available for Ku binding. mwPIFE increased with protein concentration, resulting in a binding curve that reached a plateau around 80% at the highest Ku
concentrations (Fig. 5A). The calculated Kd for Ku/DNA interaction using mwPIFE was 10 ± 5 nM, which is within the range previously determined by fluorescence anisotropy experiments27. Next
we used mwPIFE to determine the minimum DNA length requirements for Ku/DNA interaction. We measured mwPIFE with terminally labeled dsDNA probes ranging from 10 to 15 bps. An ~50% PIFE was
detected with 13, 14, and 15 bp probes, indicating that 13 bp of duplex DNA is sufficient for stable Ku binding. Nonetheless, we also detected a small (7%), but statistically significant
PIFE (P = 8.76E-3, two tailed t-test) with an 11 bp probe and a 20% PIFE with the 12 bp probe (Fig. 5B). These signals reflect less stably bound Ku with DNA termini, indicating that mwPIFE
can reliably quantify relatively weak and transient protein/NA interactions that may be difficult to detect by other methods. One of the advantages of PIFE is its dependence on the physical
proximity of the protein and fluorescent dye, allowing for the spatial mapping of protein-DNA interactions. Although Ku is known primarily as a DNA end binding protein, it can translocate
along naked DNA and reach internal positions. To assess Ku/DNA translocation, we used mwPIFE to measure Ku occupancy at different positions along DNA. We designed duplex DNA probes with Cy3
placed either terminally, or internally 15 or 60 bp away from the free end. We also used an internally labeled probe with both DNA termini blocked by neutravidin. While the strongest PIFE
was observed when Cy3 was placed terminally, a specific interaction was still detected at internal sites, albeit with PIFE decreasing with the distance from the free end (Fig. 5C). Only
negligible PIFE was detected on the biotin-blocked DNA probe, confirming that Ku requires a free end for efficient binding. This experiment demonstrates that mwPIFE can be used to study the
topology of protein/NA binding. DISCUSSION Since the first observations that increasing the viscosity of the local environment increases the fluorescence quantum yield of cyanine dyes, and
that this phenomenon can be used to study protein/NA interactions10,28,29, PIFE has gained in popularity particularly in single molecule studies. smPIFE has two major advantages over other
fluorescence techniques. First, it does not require protein labeling, which can be laborious and interfere with protein function. Second, it allows for the detection of weaker protein/NA
interactions, which would otherwise require high concentrations of fluorescently labeled proteins obscuring single molecule detection12. smPIFE has been used in a number of applications
including the determination of dissociation and association rate constants, polymerase conformation changes upon nucleotide binding, changes in DNA conformation upon protein binding,
helicase translocation along dsRNA, and transcription initiation13,29,30,31. These examples show the great versatility of the method in elucidating various biochemical processes involving
protein/NA interactions. Nevertheless, as with other single molecule approaches, smPIFE is a rather advanced technique relying on expensive and specifically tailored instrumentation operated
by skilled specialists. In this report, we demonstrate utility of the PIFE phenomenon in detecting steady-state protein/NA interactions in microwell plates using standard fluorescence plate
readers. This is a relatively simple assay that requires neither specialized equipment nor extensive training. Bulk measurement of PIFE in a microwell provides an immediate numerical
readout of the protein/NA interaction which, together with the possibility of the simultaneous analysis of tens to hundreds of samples, renders this technique highly quantitative. We
obtained 40–80% PIFE with three different protein-DNA binding systems, which permitted reliable detection of protein-DNA interactions. The lower mwPIFE values comparing to smPIFE, which can
reach up to 250%12, are expected as mwPIFE averages signal from large population of labeled oligonucleotides whereby not all of them are bound with a protein. We demonstrate that mwPIFE can
be used to determine dissociation constants and to rapidly screen for substrate specificities in competition experiments. The multiple-well plate format of the assay allows for testing a
broad range of competitors over a wide range of concentrations, which surpasses the sample processing efficiency of other conventional techniques currently used for this purpose, such as
EMSA or fluorescence anisotropy. Data on the affinity of XPF/ERCC1 to different DNA substrates showed that the results obtained by mwPIFE are comparable to those from fluorescence
anisotropy, but mwPIFE is more time and cost efficient. Hence, mwPIFE may be particularly useful in high-throughput applications such as searching for optimal binding sites of transcription
factors or screening chemical libraries for small molecules that inhibit a particular protein/NA interaction. smPIFE proved to be an excellent technique for detecting transient and weak
interactions, with potential to reveal reaction intermediates31,32. By analyzing Ku’s affinity to short end-labeled DNA fragments with mwPIFE, we were able to record weak but significant
signals even with 11 bp duplexes. The signal became more pronounced with 12 bp DNA, and reached a plateau with 13–15 bp DNA substrates. A similar experiment performed by EMSA determined that
the shortest DNA capable of forming a detectable complex with Ku is 14 bp26. We suggest that the signal detected by mwPIFE with the 11 and 12 bp probes represents less stable Ku-DNA
interactions where DNA does not span the entire Ku-DNA loading channel. This is consistent with photo-crosslinking and crystal structure studies showing that the lagging part of the Ku-DNA
loading channel is in direct contact with bases at positions 11 and 1325,26. Hence, mwPIFE seems to be superior to EMSA in detecting and quantifying weak protein/NA interactions. A unique
feature of PIFE is its dependence on protein binding in the immediate vicinity of the dye. An smPIFE study with three different proteins, including BamHI, established the high sensitivity of
the method within a 3 nm range from Cy317. Although we have not performed a fine-tuned distance calibration of mwPIFE, our observation of a marked decrease of PIFE when the binding site of
BamHI was moved 15 bp from the dye indicates a similar distance dependence as in the smPIFE setting. This property of PIFE was exploited to study the translocation of proteins along nucleic
acids. Typically in these experiments a dye is placed at the DNA termini and the directionality and velocity of the movement was extrapolated from changes in fluorescence over time measured
either in smPIFE or stopped-flow settings13,16,28. Here we used mwPIFE to assess the ability of Ku to translocate along duplex DNA by measuring the fluorescence intensity of Cy3 placed at
different distances from the DNA end. Since Ku can load onto duplex DNA exclusively from a free DNA end, PIFE at internal positions should reflect Ku’s ability to move along DNA. Similar
mwPIFE setups, where Cy3 is placed at different positions along a nucleic acid substrate, can be used to map the topology of protein/NA interactions or formation of higher order structures.
Although the PIFE phenomenon has so far only been used in a relatively limited number of studies, it offers a number of advantages over other assays for protein/NA interactions. Its
requirement for standard laboratory equipment, relative ease of the methodological procedure, high throughput format, direct quantitative readout, and wide range of experimental applications
are the major advantages that poise mwPIFE to become a wide-spread technique complementing, and perhaps in some cases substituting, other methods that are currently being used. MATERIALS
AND METHODS PROTEIN PREPARATION _BamH_I 10U/μl was purchased from Thermo Scientific (catalog number: ER0051). Human Ku heterodimer was reconstituted by co-expression of Ku70 and Ku80
subunits in HI5 insect cells. The Ku80 cDNA was PCR amplified from a HeLa cDNA library (kindly provided by Johannes Popov) using primers Hs80_HL/HR and cloned into BamHI/NotI sites of
pFastBac HTA (Invitrogen). The Ku80 cDNA was then reamplified from pFastBac HTA vector using primers Hs80_DL/DR and cloned into XhoI/NheI sites of pFastBac Dual. The Ku70 cDNA was amplified
from the HeLa cDNA library using primers Hs70_DL/DR and cloned into BamHI/SpeI sites of pFastBac Dual harboring Ku80. The resulting pFastBac Dual vector was transposed to EMBacY bacmid in
_E. coli_ and subsequently transfected to Sf9 cells for virus formation. Hi5 insect cells were infected with the virus and harvested 3 days past arrest. Cells were resuspended in lysis
buffer (50 mM Tris-Cl, 250 mM KCl, 10% v/v glycerol, 1 mM DTT, pH 8.0) supplemented with protease inhibitors (Roche) and frozen in liquid nitrogen. Thawed cells were spun down and the Ku
complex was bound to His Mag Sepharose Ni (GE health care). Beads were washed in lysis buffer containing 50 mM imidazole and Ku was eluted in lysis buffer with 250 mM imidazole. Proteins
were filtered using Nanosep centrifugal columns (Pall) and stored at 4 °C. The human XPF/ERCC1 complex was produced in _Escherichia coli_ strain BL21 (DE3) by co-expression of two plasmid
vectors. ERCC1 (residues 93–297) was expressed from a modified pET-24d vector and had a dual GST, His-6 tag at the N-terminus with a TEV cleavage site. XPF (residues 640–916) was expressed
from pCDF vector without a tag. The recombinant protein heterodimer was extracted from harvested _E. coli_ cells in 25 mM Tris-HCl buffer pH 8, 500 mM NaCl, 10 mM Imidazole, 0.01% (v/v)
NP40, 10% (v/v) glycerol, 2 mM beta-mercaptoethanol and protease inhibitors. The resulting lysate was applied to a HiTrap IMAC HP column charged with CoCl2 according to manufacturer’s
instructions (GE Healthcare) and step eluted in buffer supplemented with 500 mM imidazole. The protein containing fractions were incubated at 4 °C with 0.5 mg of TEV protease per 1 L culture
for 2 hours and then dialyzed overnight at 4 °C against 25 mM Tris-HCl buffer pH 8, 500 mM NaCl, 10 mM Imidazole, 10% (v/v) glycerol and 2 mM beta-mercaptoethanol. The dialyzed protein
sample was passed through the same HiTrap IMAC HP column and the non-bound protein was collected. The protein sample was again dialyzed overnight at 4 °C against 25 mM Tris-HCl pH 7.5, 15 mM
NaCl, 10% (v/v) glycerol and 2 mM beta-mercaptoethanol. The dialyzed protein was applied to a Mono Q 4.6/100 PE column (GE HealthCare) and gradient eluted with 10 column volumes of buffer
supplemented with 1 M NaCl. The protein containing fractions were combined and applied to a HiLoad 16/600 Superdex 75 pg gel filtration column (GE HealthCare) equilibrated with 25 mM
Tris-HCl buffer pH 7.5, 50 mM NaCl and 1 mM TCEP. MWPIFE A black 96 microwell plate coated with 100 μl of NeutrAvidin protein and blocked with 200 μl of SuperBlock blocking buffer (Thermo
Scientific, Prod. # 15117) was washed three times with a buffer that was specific for each protein. An oligonucleotide DNA probe of choice (Supplementary Table 1) was annealed beforehand and
2.5 pmol was applied in 100 μl of buffer and incubated for 2 hours in the well in dark. Wells were washed afterwards 3 times with 200 μl of buffer. The first scan was obtained after adding
100 μl of a buffer using the FLUOstar Omega scanner (BMG Labtech) in a well scanning mode with a scan matrix of 10 by 10 points within 6 mm diameter and 10 flashes per scan point. The
electrical power in the flash lamp is 0,065 W per flash. Ex540-10 and Em580-10 filters were used for fluorescence measurements of Cy3 labeled probes. After the scan, the buffer was replaced
with 100 ul of a buffer containing appropriate protein BamHI (50 U), BSA (15 pmol, Biorad), Ku (0.5–20 pmol) or XPF/ERCC1 (25 pmol). The following binding buffers were used: BamHI buffer (10
mM Tris-HCl pH 8, 100 mM KCl, 0.02% Triton X-100, 0.1 mg/ml BSA) with the addition of either 5 mM EDTA or MgCl2 or CaCl2; Ku binding buffer (35 mM Tris-Cl pH 7.9, 5.5% glycerol, 150 mM KCl,
1 mM EDTA, 0.1 mM DTT, 1.5 mM imidazol); and XPF/ERCC1 buffer (10 mM Tris-Cl pH 8, 10 mM NaCl). Optionally, DNA competitors were prepared by hybridization of forward and reverse strand
(Supplementary Table 1) and added to the mixture together with the protein. Incubations were carried out at room temperature for 30 minutes to reach binding equilibrium and the second scan
was performed. PIFE was calculated to correct for initial signal and no protein control (Fig. 1B and text in the result section). Optionally, wells were washed two times with 200 μl of the
appropriate buffer with no protein, and the third scan was performed in 100 μl of the buffer. FLUORESCENCE ANISOTROPY The binding analysis of XPF/ERCC1 was performed in 10 mM Tris-HCl buffer
pH 8, 10 mM NaCl, 10% (v/v) glycerol and 1 mM TCEP. Reactions containing 50 nM DNA substrate were incubated for 30 min at 25 °C and subsequently transferred to 384-well microplate and read
in a Tecan Microplate Reader Infinite F500 (Tecan group Ltd). The data for each protein concentration was averaged over 5 min intervals to remove instrumental noise and processed by
subtracting the anisotropy value obtained from a respective DNA substrate without protein. Equilibrium dissociation constants (Kd) were calculated by fitting the data in OriginPro (OriginLab
Corporation) to the following equation: FA = (([D] + [P] + Kd) − (([D] + [P] + Kd)2 − (4*[D]*[P]))1/2) * (A)/(2 * [D]), where [D] and [P] are concentrations of DNA and protein respectively,
and A is the maximum anisotropy value. ADDITIONAL INFORMATION HOW TO CITE THIS ARTICLE: Valuchova, S. _et al_. A rapid method for detecting protein-nucleic acid interactions by protein
induced fluorescence enhancement. _Sci. Rep._ 6, 39653; doi: 10.1038/srep39653 (2016). PUBLISHER'S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in
published maps and institutional affiliations. REFERENCES * Jolma, A. et al. DNA-binding specificities of human transcription factors. Cell 152, 327–339, doi: 10.1016/j.cell.2012.12.009
(2013). Article CAS PubMed Google Scholar * Hallikas, O. et al. Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell 124, 47–59,
doi: 10.1016/j.cell.2005.10.042 (2006). Article CAS PubMed Google Scholar * Luscombe, N. M., Laskowski, R. A. & Thornton, J. M. Amino acid-base interactions: a three-dimensional
analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res 29, 2860–2874 (2001). Article CAS PubMed PubMed Central Google Scholar * Yap, J. L. et al. Small-molecule
inhibitors of dimeric transcription factors: Antagonism of protein-protein and protein-DNA interactions. Medchemcomm 3, 541–551, doi: 10.1039/c2md00289b (2012). Article CAS Google Scholar
* Dey, B. et al. DNA-protein interactions: methods for detection and analysis. Molecular and cellular biochemistry 365, 279–299, doi: 10.1007/s11010-012-1269-z (2012). Article CAS PubMed
Google Scholar * Mahapatra, L., Mao, C., Andruska, N., Zhang, C. & Shapiro, D. J. High-throughput fluorescence anisotropy screen for inhibitors of the oncogenic mRNA binding protein,
IMP-1. Journal of biomolecular screening 19, 427–436, doi: 10.1177/1087057113499633 (2014). Article CAS PubMed Google Scholar * Owicki, J. C. Fluorescence polarization and anisotropy in
high throughput screening: perspectives and primer. Journal of biomolecular screening 5, 297–306 (2000). Article CAS PubMed Google Scholar * Heyduk, T. & Heyduk, E. Molecular beacons
for detecting DNA binding proteins. Nat Biotechnol 20, 171–176, doi: 10.1038/nbt0202-171 (2002). Article CAS PubMed Google Scholar * Stennett, E. M., Ciuba, M. A., Lin, S. &
Levitus, M. Demystifying PIFE: The Photophysics Behind the Protein-Induced Fluorescence Enhancement Phenomenon in Cy3. The journal of physical chemistry letters 6, 1819–1823, doi:
10.1021/acs.jpclett.5b00613 (2015). Article CAS PubMed Google Scholar * Aramendia, P. F., Negri, R. M. & Sanroman, E. Temperature-Dependence of Fluorescence and Photoisomerization in
Symmetrical Carbocyanines - Influence of Medium Viscosity and Molecular-Structure. J Phys Chem-Us 98, 3165–3173, doi: 10.1021/J100063a020 (1994). Article CAS Google Scholar * Sundstroem,
V. & Gillbro, T. Viscosity-dependent isomerization yields of some cyanine dyes. A picosecond laser spectroscopy study. J Phys Chem 1788–1794 (1982). * Hwang, H. & Myong, S. Protein
induced fluorescence enhancement (PIFE) for probing protein-nucleic acid interactions. Chemical Society reviews 43, 1221–1229, doi: 10.1039/c3cs60201j (2014). Article CAS PubMed PubMed
Central Google Scholar * Myong, S. et al. Cytosolic viral sensor RIG-I is a 5′-triphosphate-dependent translocase on double-stranded RNA. Science 323, 1070–1074, doi:
10.1126/science.1168352 (2009). Article CAS ADS PubMed PubMed Central Google Scholar * Tomko, E. J., Fischer, C. J. & Lohman, T. M. Ensemble methods for monitoring enzyme
translocation along single stranded nucleic acids. Methods 51, 269–276, doi: 10.1016/j.ymeth.2010.03.010 (2010). Article CAS PubMed PubMed Central Google Scholar * Fischer, C. J.,
Tomko, E. J., Wu, C. G. & Lohman, T. M. Fluorescence methods to study DNA translocation and unwinding kinetics by nucleic acid motors. Methods Mol Biol 875, 85–104, doi:
10.1007/978-1-61779-806-1_5 (2012). Article CAS PubMed PubMed Central Google Scholar * Antony, E. et al. Srs2 disassembles Rad51 filaments by a protein-protein interaction triggering
ATP turnover and dissociation of Rad51 from DNA. Mol Cell 35, 105–115, doi: 10.1016/j.molcel.2009.05.026 (2009). Article CAS PubMed PubMed Central Google Scholar * Hwang, H., Kim, H.
& Myong, S. Protein induced fluorescence enhancement as a single molecule assay with short distance sensitivity. Proc Natl Acad Sci USA 108, 7414–7418, doi: 10.1073/pnas.1017672108
(2011). Article ADS PubMed PubMed Central Google Scholar * Viadiu, H. & Aggarwal, A. K. The role of metals in catalysis by the restriction endonuclease BamHI. Nat Struct Biol 5,
910–916, doi: 10.1038/2352 (1998). Article CAS PubMed Google Scholar * Nowotny, M. & Gaur, V. Structure and mechanism of nucleases regulated by SLX4. Curr Opin Struct Biol 36,
97–105, doi: 10.1016/j.sbi.2016.01.003 (2016). Article CAS PubMed Google Scholar * Tripsianes, K. et al. The structure of the human ERCC1/XPF interaction domains reveals a complementary
role for the two proteins in nucleotide excision repair. Structure 13, 1849–1858, doi: 10.1016/j.str.2005.08.014 (2005). Article CAS PubMed Google Scholar * de Laat, W. L., Appeldoorn,
E., Jaspers, N. G. & Hoeijmakers, J. H. DNA structural elements required for ERCC1-XPF endonuclease activity. J Biol Chem 273, 7835–7842 (1998). Article CAS PubMed Google Scholar *
Bowles, M. et al. Fluorescence-based incision assay for human XPF–ERCC1 activity identifies important elements of DNA junction recognition. Nucleic Acids Research, doi: 10.1093/nar/gks284
(2012). * Enzlin, J. H. & Scharer, O. D. The active site of the DNA repair endonuclease XPF-ERCC1 forms a highly conserved nuclease motif. EMBO J 21, 2045–2053, doi:
10.1093/emboj/21.8.2045 (2002). Article CAS PubMed PubMed Central Google Scholar * Tsodikov, O. V., Enzlin, J. H., Scharer, O. D. & Ellenberger, T. Crystal structure and DNA binding
functions of ERCC1, a subunit of the DNA structure-specific endonuclease XPF-ERCC1. Proc Natl Acad Sci USA 102, 11236–11241, doi: 10.1073/pnas.0504341102 (2005). Article CAS ADS PubMed
PubMed Central Google Scholar * Walker, J. R., Corpina, R. A. & Goldberg, J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature
412, 607–614 (2001). Article CAS ADS PubMed Google Scholar * Yoo, S., Kimzey, A. & Dynan, W. S. Photocross-linking of an oriented DNA repair complex. Ku bound at a single DNA end. J
Biol Chem 274, 20034–20039 (1999). Article CAS PubMed Google Scholar * Arosio, D. et al. Studies on the mode of Ku interaction with DNA. J Biol Chem 277, 9741–9748 (2002). Article CAS
PubMed Google Scholar * Fischer, C. J., Maluf, N. K. & Lohman, T. M. Mechanism of ATP-dependent translocation of E.coli UvrD monomers along single-stranded DNA. J Mol Biol 344,
1287–1309, doi: 10.1016/j.jmb.2004.10.005 (2004). Article CAS PubMed Google Scholar * Luo, G., Wang, M., Konigsberg, W. H. & Xie, X. S. Single-molecule and ensemble fluorescence
assays for a functionally important conformational change in T7 DNA polymerase. Proc Natl Acad Sci USA 104, 12610–12615, doi: 10.1073/pnas.0700920104 (2007). Article CAS ADS PubMed
PubMed Central Google Scholar * Song, D., Graham, T. G. & Loparo, J. J. A general approach to visualize protein binding and DNA conformation without protein labelling. Nature
communications 7, 10976, doi: 10.1038/ncomms10976 (2016). Article CAS ADS PubMed PubMed Central Google Scholar * Sorokina, M., Koh, H. R., Patel, S. S. & Ha, T. Fluorescent
lifetime trajectories of a single fluorophore reveal reaction intermediates during transcription initiation. Journal of the American Chemical Society 131, 9630–9631, doi: 10.1021/ja902861f
(2009). Article CAS PubMed PubMed Central Google Scholar * Markiewicz, R. P., Vrtis, K. B., Rueda, D. & Romano, L. J. Single-molecule microscopy reveals new insights into nucleotide
selection by DNA polymerase I. Nucleic Acids Res 40, 7975–7984, doi: 10.1093/nar/gks523 (2012). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS Ku
expression was performed by the VBCF Protein Technologies Facility (www.vbcf.ac.at). This work was supported by the Grant Agency of the Czech Republic (14-22346S to K.R.; 15-22380Y to
K.T.), Marie Curie Career Integration Grant (618223) to K.T. and Marie Curie International Incoming Fellowship (624894) to A.P.P. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * CEITEC -
Central European Institute of Technology, Masaryk University, Brno, 625 00, Czech Republic Sona Valuchova, Jaroslav Fulnecek, Alexander P. Petrov, Konstantinos Tripsianes & Karel Riha
Authors * Sona Valuchova View author publications You can also search for this author inPubMed Google Scholar * Jaroslav Fulnecek View author publications You can also search for this author
inPubMed Google Scholar * Alexander P. Petrov View author publications You can also search for this author inPubMed Google Scholar * Konstantinos Tripsianes View author publications You can
also search for this author inPubMed Google Scholar * Karel Riha View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS S.V., J.F. and K.R.
designed mwPIFE experiments, S.V. and J.F. performed mwPIFE experiments and analyzed data, A.P.P. and K.T. designed and performed fluorescence anisotropy experiments, and S.V. and K.R. wrote
the manuscript. All authors reviewed the manuscript. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing financial interests. ELECTRONIC SUPPLEMENTARY MATERIAL
SUPPLEMENTARY INFORMATION RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this
article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will
need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ Reprints and permissions ABOUT
THIS ARTICLE CITE THIS ARTICLE Valuchova, S., Fulnecek, J., Petrov, A. _et al._ A rapid method for detecting protein-nucleic acid interactions by protein induced fluorescence enhancement.
_Sci Rep_ 6, 39653 (2016). https://doi.org/10.1038/srep39653 Download citation * Received: 21 September 2016 * Accepted: 24 November 2016 * Published: 23 December 2016 * DOI:
https://doi.org/10.1038/srep39653 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently
available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative