Play all audios:
CD95 (APO-1, Fas) is a member of the tumor necrosis factor (TNF) receptor superfamily expressed in various tissues; however, expression of its ligand CD95L, a type II transmembrane protein
of the TNF family, is restricted to a few cell types, such as T cells, macrophages, and cells of immune-privileged tissues like testis and eye. In lymphoid tissues CD95 is constitutively
expressed; by contrast, CD95L is not present in resting T cells, but is highly expressed upon T-cell activation. The binding of CD95L to CD95 results in rapid apoptosis. Many features of
apoptosis in T cells induced by the CD95/CD95L system have been worked out: CD95 mediated apoptosis is now known to be a mechanism for maintenance of peripheral tolerance and for termination
of an ongoing immune response.1,2 CD95/CD95L interaction also initiates cell death in many other systems, and its deregulation leads to severe diseases.1,2 Thus, to explain the
pathophysiology of these diseases and to identify potential therapeutic targets it is important to understand the molecular mechanisms of CD95L activation. ACTIVATION OF CD95L VIA THE
CA2+/CALCINEURIN SIGNALING PATHWAY The earliest signaling events upon T-cell receptor (TCR) engagement are the sequential activation of tyrosine kinases (TPKs) including Lck and ZAP-70.
ZAP-70 and Lck are shown to be involved in up-regulation of CD95L expression in activation-induced apoptosis.3,4 Both Lck and ZAP-70 are required for calcium mobilization in T cells.
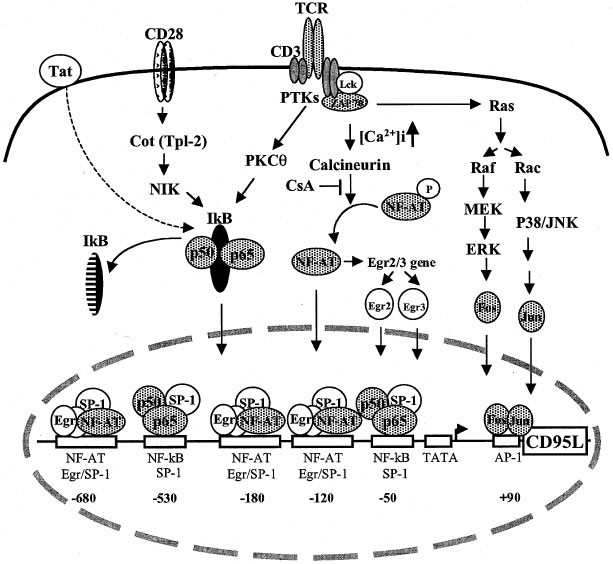
Increase in [Ca2+]i leads to activation of the Ca2+- and calmodulin-dependent phosphatase calcineurin and, consequently, activates calcineurin-regulated transcription factors (Figure 1).
CD95L mRNA expression and activation-induced-cell-death (AICD) in T cells can be blocked by the immunosuppressive drug Cyclosporin A (CsA), an inhibitor of calcineurin. This finding has
directed the search for CsA-sensitive transcription factors involved in CD95L activation. NF-AT proteins are major targets of CsA. In resting T cells, NF-AT proteins are phosphorylated, have
low affinity for DNA and reside in the cytoplasm. Activation of T cells through the TCR leads to calcineurin activation and results in rapid dephosphorylation of NF-AT proteins and their
translocation to the nucleus. The dephosphorylated NF-AT proteins exert increased affinity for DNA. CsA blocks calcineurin-dependent dephosphorylation and prevents nuclear translocation of
NF-AT. The first functional NF-AT binding site was identified at the −180 region of the human CD95L promoter and was shown to confer CsA-sensitive transcription to CD95L5,6 (Figure 1).
Following the identification of the NF-AT site, a cis-acting motif in the −120 region was found to bind members of the Early Growth Response Gene (Egr1, 2, 3 and 4) family of transcription
factors (Figure 1). Among these factors, Egr2 and Egr3 were proposed to confer most of the activation-induced expression to CD95L.7,8 Although Egr factors activate transcription in a
CsA-insensitive manner, Egr2 and Egr3 themselves are NF-AT target genes and their expression is sensitive to CsA7,8 (Figure 1). A recent study proposed that activation of CD95L is
sequentially regulated by NF-AT and Egr3 and the effect of NF-AT on CD95L expression is largely indirect acting through its induction of Egr2 and Egr3 transcription.9 However, the bulk of
studies favors a direct role of NF-AT in CD95L activation.5,6,10,11,12 In addition, it was found that both the −180 NF-AT and the −120 Egr regions are Egr/NF-AT composite sites for NF-AT and
Egr transcription factors.11 Three such Egr/NF-AT composite sites have been identified in the CD95L promoter (−680, −180 and −120) (Figure 1). NF-AT and Egr were shown to display a
cooperative and synergistic activation of the CD95L promoter at these three sites.11 ACTIVATION OF CD95L VIA THE PKCΘ SIGNALING PATHWAY The protein kinase C (PKC) iso-form PKCθ was found to
play an important role in induction of CD95L expression following TCR/CD3 stimulation.13 Of the various PKC isoforms present in T cells, PKCθ is essential for TCR-mediated T-cell activation
and is required for TCR-induced NF-κB activation in mature T lymphocytes.14 Normally, NF-κB remains sequestered in an inactive state by the cytoplasmic inhibitor-of-κB (IκB) proteins. T-cell
activation leads to phosphorylation, ubiquitinylation, and the subsequent degradation of IκB proteins. The DNA-binding subunits of NF-κB (p65, p50) immigrate into the nucleus and activate
expression of target genes (Figure 1). Several studies have indicated that high expression of CD95L in activated T cells requires NF-κB. Two NF-κB binding sites are localized at the −530 and
−50 region of the human CD95L promoter15 (Figure 1). Mutation at each of these sites resulted in 30–70% reduction of promoter activity indicating that NF-κB is required for optimal
activation of the CD95L promoter.15 Another NF-κB binding site has been reported at the −980 region of the CD95L promoter.16 The −980 NF-κB binding sequence described by this study, however,
differs from the DNA sequence described by other groups. Conflicting results have been obtained by mutation or deletion studies of the −980 site that argue against the role of this site in
expression of CD95L.15 AICD is also a major cause of depletion of CD4+ T cells in HIV-1 infected individuals. Increased expression of both CD95 and CD95L has been observed in HIV-1 infected
individuals. HIV-1 Tat protein is essential for efficient HIV-1 gene expression and has been implicated in sensitization of CD95-mediated apoptosis and in upregulation of CD95L expression in
T cells.17 Ectopic expression of Tat protein in T cells results in enhanced binding of NF-κB to its target DNA.15 The NF-κB sites of the CD95L promoter may contribute the Tat-enhanced CD95L
expression, at least in part.15 ACTIVATION OF CD95L VIA THE RAS SIGNALING PATHWAY Activation of T cells via TCR engagement also leads to a Ras-activated cascade of kinase activity including
Raf, MEK, ERK and p38 mitogen-activated protein kinase (MAPK) (Figure 1). It has been shown that the Ras signaling pathway is required for optimal TCR-mediated expression of CD95L.18,19 The
Ras pathway is involved in induction of AP-1 (Fos/Jun) transcription factors. An AP-1 binding site was recently localized in the +90 region of the CD95L promoter20 (Figure 1). This AP-1
site was originally identified to be essential for anti-cancer-drug-induced CD95L expression, and we found that it is also involved in upregulation of CD95L expression in T cells
(unpublished data). A distal AP-1 binding site at the −950 region which was shown to respond to DNA-damaging-agent-induced CD95L expression by another group, however, is not required for
T-cell activation-induced CD95L promoter activity.16 THE ROLE OF CONSTITUTIVE TRANSCRIPTION FACTOR SP-1 SP-1 is constitutively present in T cells and preferentially interacts with GC rich
DNA sequences. All NF-κB and Egr/NF-AT composite sites contain GC rich DNA sequences and are found to interact with SP-110,12,15 (Figure 1). SP-1 has been implicated in maintaining the basal
level of CD95L in T cells and in constitutive expression of CD95L in other tissues. DIFFERENTIAL REGULATION OF CD95L EXPRESSION IN TH1 AND TH2 CELLS During T-cell development, CD4+ T-helper
(Th) cell differentiate in response to antigen stimulation into different types of Th cells that can be grouped based on their cytokine expression patterns. Th1 cells produce IFN-γ, TNF and
IL-2 are important for the cell-mediated immune response against intracellular pathogens. Th2 cells produce IL-4, IL-5, IL-6 and IL-10, and are involved in humoral immunity. Activated Th1
cells are shown to express CD95L and are sensitive to CD95-mediated apoptosis, whereas Th2 cells express less or no CD95L and are resistant to AICD. A recent report by Rengarajan _et al_
proposed that enriched Egr3 expression in Th1 cells and deficient expression of Egr3 in Th2 cells may account for differential regulation of CD95L expression in Th1 and Th2 cells.9 However,
conflicting results concerning this proposal are reported by another group showing that Egr3 is not expressed in Th1 cells and, instead, is expressed in Th2 cells.12 Thus, the lack of CD95L
expression by Th2 cells is apparently not due to a deficiency of Egr3. Absence of Egr3 in Th1 cells shown in this study further argues against the role of Egr3 in preferential expression of
CD95L in Th1 _vs_ Th2 cells. Thus, the molecular basis for the differential expression of CD95L in Th1 and Th2 cells remains unknown. Taken together, activation-induced CD95L expression in T
cells involves multiple transcription factors. NF-AT and Egr proteins seem to play a major role for CD95L expression upon TCR-stimulation. However, other factors may be also important. For
example, in HIV-1 infected individuals, NF-κB activity may be enhanced through virus infection and, consequently, may lead to increased CD95L expression and accelerated apoptosis. T-cell
apoptosis can also be elicited by other stimuli, which change the oxidation/reduction (redox) status of the cell such as UV irradiation, chemotherapeutic agents, and stress. Activities of
NF-kB and AP-1 are known to be largely affected by the cellular redox status. In these cases, NF-κB and AP-1 may exert a major role in induction of the CD95L promoter. Thus, different sets
of transcription factors may be used for CD95L expression in T cells in different situations. REFERENCES * Krammer PH . 2000 _Nature_ 407: 789–795 Article CAS Google Scholar * Krammer PH
. 1999 _Adv. Immunol._ 71: 163–210 * Eischen CM _et al_. 1997 _J. Immunol._ 159: 1135–1139 * Gonzalez-Garcia A _et al_. 1997 _J. Immunol._ 158: 4104–4112 * Latinis MK _et al_. 1997 _J. Biol.
Chem._ 272: 31427–31434 Article CAS Google Scholar * Holtz-Heppelmann CJ _et al_. 1998 _J. Biol. Chem._ 273: 4416–4423 Article CAS Google Scholar * Mittelstadt PR, Ashwell JD . 1998
_Mol. Cell. Biol._ 18: 3744–3751 Article CAS Google Scholar * Mittelstadt PR, Ashwell JD . 1999 _J. Biol. Chem._ 274: 3222–3227 Article CAS Google Scholar * Rengarajan J _et al_. 2000
_Immunity_ 12: 293–300 Article CAS Google Scholar * Li-Weber M _et al_. 1998 _Eur. J. Immunol._ 28: 2373–2383 Article CAS Google Scholar * Li-Weber M _et al_. 1999 _Eur. J. Immunol._
29: 3017–3027 Article CAS Google Scholar * Dzialo-Hatton R _et al_. 2001 _J. Immunol._ 166: 4534–4542 * Villalba M _et al_. 1999 _J. Immunol._ 163: 5813–5819 * Sun Z _et al_. 2000
_Nature_ 404: 402–407 Article CAS Google Scholar * Li-Weber M _et al_. 2000 _Eur. J. Immunol._ 30: 661–670 Article CAS Google Scholar * Kasibhatla S _et al_. 1999 _J. Biol. Chem._ 274:
987–992 Article CAS Google Scholar * Westendorp MO _et al_. 1995 _Nature_ 375: 497–500 Article CAS Google Scholar * Latinis KM _et al_. 1997 _J. Immunol._ 158: 4602–4611 * Hsu SC _et
al_. 1999 _J. Biol. Chem._ 274: 25769–25776 Article CAS Google Scholar * Eichhorst ST _et al_. 2000 _Mol. Cell. Biol._ 20: 7826–7837 Download references AUTHOR INFORMATION AUTHORS AND
AFFILIATIONS * Tumor Immunology Program, German Cancer Research Center, Heidelberg, Germany M Li-Weber & P H Krammer Authors * M Li-Weber View author publications You can also search for
this author inPubMed Google Scholar * P H Krammer View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to P H Krammer.
RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Li-Weber, M., Krammer, P. The death of a T-cell: expression of the CD95 ligand. _Cell Death Differ_ 9,
101–103 (2002). https://doi.org/10.1038/sj.cdd.4400984 Download citation * Issue Date: 01 February 2002 * DOI: https://doi.org/10.1038/sj.cdd.4400984 SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative