Play all audios:
_Dear Editor_, The release of the cytochrome _c_ (cyt. _c_) and several other protein factors from the mitochondrial intermembrane space results in the activation of caspases, proteases
responsible for cell destruction during programmed cell death (PCD) and is considered an initiation and commitment point in the apoptotic pathway. Release of these factors from the
mitochondria, in response to a broad range of signals or insults, is regulated by proteins in the Bcl-2 family.1, 2 This family consists of both proapoptotic proteins (e.g. Bax, Bak, Bid)
that promote the release of mitochondrial factors and antiapoptotic proteins (e.g. Bcl-XL and Bcl-2) that prevent the release of these factors in the absence of death signal. Proteins in
this family share up to four homologous BH1-4 (_B_cl-_h_omology) domains and many of them also have C-terminal hydrophobic domain that targets these proteins to membranes. A subset of
proapoptotic proteins (e.g. Bid) that contain only BH3 domain (BH3-only proteins) are located in various cellular compartments where they can be released in response to a variety of death
signals, allowing them to transfer the signal to the multidomain proapoptotic proteins Bax and Bak. When activated, in part, by interaction with BH3-only proteins, both Bax and Bak
oligomerize in the outer mitochondrial membrane. _In vitro_ studies support the idea that this oligomerization results in the formation of an aqueous pore of sufficient size to facilitate
translocation of cyt. _c_, and perhaps other components of the mitochondrial intermembrane space, into the cytosol where they then initiate and modulate the apoptotic cascade.3, 4, 5 The
precise molecular mechanisms by which multidomain proapoptotic proteins like Bax and Bak are activated by BH3-only proteins have not yet been completely resolved. Additionally, it is also
not clear whether pore formation forms the basis of the action of these proteins during PCD _in vivo_ and whether pore formation _in vivo_ requires additional components or is based on the
action of these proapoptotic proteins alone. Several studies have suggested that the process of pore formation requires, or is facilitated by, the interaction of Bax (or Bak) with a yet to
be identified cytosolic6 or mitochondrial protein.7 Cardiolipin (CL), a unique phospholipid of the inner mitochondrial membrane, has also been recently proposed to be required in the action
of proapoptotic Bcl-2 proteins.8, 9 These _in vitro_ studies have suggested that CL is required for the Bax-mediated permeabilization of phospholipid vesicles; Bax-mediated permeabilization
does not proceed in the absence of CL.9 In contrast, others10 have shown that recombinant Bax is able to induce release of cyt. _c_ from the mitochondria lacking CL following isolation of
these organelles from yeast strains in which the gene encoding for cardiolipin synthase (CL synthase; _CRD1_),11 required for CL synthesis, had been eliminated (_Δcrd1_ strains). However,
phosphatidylglycerol (PG), the intermediate in the synthesis of the CL, accumulates in the mitochondrial membranes of _Δcrd1_ strains and is known to be able to substitute functionally for
the CL.12, 13 Thus, it is not yet clear whether CL or, in the absence of CL, PG is required for the _in vivo_ mitochondrial action of proapoptotic molecules like Bax. To test directly the
requirement of either CL or PG in the _in vivo_ action of multidomain, proapoptotic members of the Bcl-2 family, we have assessed the response of yeast (_Saccharomyces cerevisiae_) lacking
genes required for the synthesis of both CL and PG to the expression of Bax and Bcl-XL. Although the yeast genome does not encode Bcl-2 family members or other proteins involved in PCD in
metazoan cells, expression of proapoptotic Bcl-2 family members like Bax results in yeast cell death.14, 15, 16 Once expressed, Bax is constitutively targeted to the outer mitochondrial
membrane where it mediates alterations in mitochondrial function similar to those observed in mammalian cells: release of cyt. _c_, mitochondrial swelling, alterations of mitochondrial
membrane potential, mitochondrial matrix alkalinization and cytosolic acidification.14, 17, 18, 19 In contrast, antiapoptotic Bcl-2 family members like Bcl-2 and Bcl-XL suppress Bax-induced
cell death when coexpressed in yeast and are also targeted to the outer mitochondrial membrane.16, 20, 21 All results generated in these studies employing yeast strongly indicate that Bcl-2
family members act directly upon highly conserved mitochondrial components that correspond directly to their apoptotic substrates in mammalian cells, and furthermore that all cellular
components necessary for the formation of pore by Bax are highly conserved and present in yeast mitochondria. Thus, the availability of yeast as an alternative system in which to study the
action of the proteins of the Bcl-2 family together with the powerful techniques of yeast genetics has made it possible to test the participation of various cellular components, such as the
voltage-dependent anion channel and ATP/ADP translocator, in the process of the permeabilization of the mitochondrial membrane by Bcl-2 family members.18, 22, 23, 24 To test whether
proapoptotic proteins like Bax require CL to kill cells _in vivo_, we examined Bax activity in yeast cells devoid of CL due to deletion of _CRD1_, the yeast gene encoding CL synthase that is
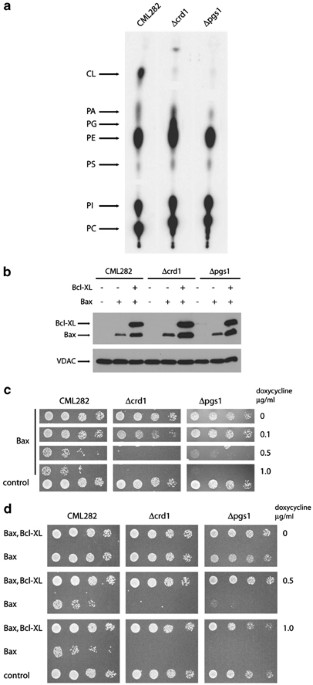
required for the biosynthesis of this unique mitochondrial phospholipid.11 _CRD1_ was eliminated by standard gene transplacement methods. _CRD1_-deficient strains (_Δcrd1_) contained no
detectable CL (Figure 1a). However, as had been noted in previous studies, _Δcrd1_ strains contained elevated level of PG, a biosynthetic precursor of CL (Figure 1a and Chang _et al_.11).
These earlier studies also demonstrated that PG is able to compensate partially for the absence of CL.12, 13 Consequently, we also generated CML282 derivatives in which _PGS1_, the gene
encoding PG-P synthase,12 has been inactivated. As expected, _PGS1_-deficient strains (_Δpgs1_) contained no detectable CL or PG (Figure 1a). Low amounts of radiolabel at the positions of PG
and CL do not migrate at positions of authentic PG or CL in two-dimensional systems (not shown). As expected, _Δpgs1_ strains were also respiratory incompetent (not shown).12 To test the
ability of Bax to kill cells lacking CL and PG, we took advantage of the fact that CML282 constitutively expresses a modified tetR protein that mediates the expression of target genes placed
downstream of _tetO_ sequences in response to tetracycline or its analogs (reverse or ‘tet-on’ system). Earlier studies had demonstrated that the level of Bax expression under these
conditions is proportional to the concentration of doxycycline present in the media; higher concentrations lead to higher levels of Bax expression.25 Accordingly, _Δcrd1_, _Δpgs1_ and
wild-type cells were transformed with plasmid-containing sequences encoding an N-terminally, HA-tagged Bax protein downstream of the Tet-inducible promoter in pCM252 in strain CML282,
allowing us to regulate expression of Bax following addition of doxycycline to the media.25 When grown on media containing 1 _μ_g/ml doxycycline, all three strains express similar levels of
Bax (Figure 1b). To evaluate the ability of Bax to inhibit growth in the absence of CL and PG, we assessed the viability of these strains following serial dilutions of cells onto plates
containing increasing concentrations of doxycycline (Figure 1c). The viability of the wild-type strain is dramatically compromised at concentrations of doxycycline higher than 0.1 _μ_g/ml,
as is each of the mutant strains; the apparent increase in sensitivity of the _Δcls1_ and _Δpgs1_ strains may reflect growth defects generated by these mutations. However, the levels of Bax
required to kill wild-type and mutant strains are identical, suggesting that the sensitivity to Bax _in vivo_ is not dependent on the presence of either CL or PG. We also assessed the
ability of an antiapoptotic protein such as Bcl-XL to abrogate the cell killing effects of a proapoptotic molecule like Bax in strains devoid of CL or PG. In wild-type cells, coexpression of
Bcl-XL is able to protect cells completely from the toxic effects of Bax expression.16, 18, 20, 25 Accordingly wild-type, _Δcrd1_ and _Δpgs1_ strains containing Bax expression plasmids were
cotransformed with plasmids that mediate the expression of N-terminally, HA-tagged Bcl-XL in response to the addition of doxycycline to the media; similar levels of Bcl-XL are expressed in
response to doxycycline addition in each strain (Figure 1b). Strain viability was again tested by the serial dilution of each strain onto the plates with varying concentrations of
doxycycline (Figure 1d). The viability of wild-type, _Δcls1_ and _Δpgs1_ strains were completely restored following coexpression of Bcl-XL at all concentration of doxycycline, suggesting
that the antiapoptotic Bcl-2 proteins do not require CL or PG for their action. Using similar systems, our earlier studies have defined the quantitative relations between antagonist actions
of Bax and Bcl-XL. These studies led to a model in which the action of Bax and Bcl-XL on the mitochondrial membranes involves the interaction of Bax and Bcl-XL with the hypothetical target
in the outer mitochondrial membrane.25 In this model, Bax and Bcl-XL compete for a common target; interaction of Bax with this target leads to outer membrane permeabilization and cell death,
while Bcl-XL, which interacts with the target preferentially, prevents the target from the interaction with Bax. Clearly, in this model the availability of the target would determine the
sensitivity of the cell to both Bax-mediated killing and Bcl-XL-mediated rescue. The fact that absence of CL or PG does not affect yeast cell sensitivity to the action of either Bax and
Bcl-XL indicates that the amount of target, the ability of target to interact with Bax and Bcl-XL and preference of the target for Bcl-XL remain unaffected by the absence of either CL or PG.
While the experiments described in this report demonstrate that the action of proapoptotic Bax and antiapoptotic Bcl-XL do not require CL or PG, the possibility that CL participates in the
mitochondrial response to apoptotic signals at a different point in the cascade remains open. It has been suggested that CL may participate in the release of cyt. _c_ by retaining a fraction
of cyt. _c_ bound to the outer surface of the inner mitochondrial membrane.10, 26 In addition, several reports suggest that CL, or CL metabolites, can partition to other intracellular
membranes during apoptosis.27 However, rather than active participation of CL, it may be that a decrease in CL levels due to downregulation of _de novo_ synthesis of CL as well as increased
turnover of existing CL during apoptosis, as it has been demonstrated in cardiomyocyte model,28 or the participation of CL breakdown products play a role during PCD. The precise mechanisms
by which CL may participate in the apoptotic response by such mechanisms remain to be firmly established and are not addressed by the studies reported here. REFERENCES * Gross A, McDonnell
JM and Korsmeyer SJ (1999) _Genes Dev._ 13: 1899–1911 * Cory S and Adams JM (2002) _Nat. Rev. Cancer._ 2: 647–656 * Schendel SL _et al_. (1997) _Proc. Natl. Acad. Sci. USA_ 94: 5113–5118 *
Saito M, Korsmeyer SJ and Schlesinger PH (2000) _Nat. Cell Biol._ 2: 553–555 * Pavlov EV _et al_. (2001) _J. Cell Biol._ 155: 725–731 * Kluck RM _et al_. (1999) _J. Cell Biol._ 147: 809–822
* Roucou X _et al_. (2002) _Biochem. J._ 368: 915–921 * Lutter M _et al_. (2000) _Nat. Cell Biol._ 2: 754–761 * Kuwana T _et al_. (2002) _Cell_ 111: 331–342 * Iverson SL _et al_. (2004) _J.
Biol. Chem._ 279: 1100–1107 * Chang SC _et al_. (1998) _J. Biol. Chem._ 273: 14933–14941 * Chang SC _et al_. (1998) _J. Biol. Chem._ 273: 9829–9836 * Zhang M, Mileykovskaya E and Dowhan W
(2002) _J. Biol. Chem._ 277: 43553–43556 * Ligr M _et al_. (1998) _FEBS Lett._ 438: 61–65 * Zha H _et al_. (1996) _Mol. Cell. Biol._ 16: 6494–6508 * Priault M _et al_. (1999) _FEBS Lett._
443: 225–228 * Manon S, Chaudhuri B and Guerin M (1997) _FEBS Lett._ 415: 29–32 * Gross A _et al_. (2000) _Mol. Cell. Biol._ 20: 3125–3136 * Matsuyama S _et al_. (2000) _Nat. Cell Biol._ 2:
318–325 * Greenhalf W, Stephan C and Chaudhuri B (1996) _FEBS Lett._ 380: 169–175 * Minn AJ _et al_. (1999) _EMBO J._ 18: 632–643 * Priault M _et al_. (1999) _Eur. J. Biochem._ 260: 684–691
* Priault M _et al_. (1999) _FEBS Lett._ 456: 232–238 * Kissova I _et al_. (2000) _FEBS Lett._ 471: 113–118 * Polcic P and Forte M (2003) _Biochem. J._ 374: 393–402 * Ott M _et al_. (2002)
_Proc. Natl. Acad. Sci. USA_ 99: 1259–1263 * Sorice M _et al_. (2004) _Cell Death Differ._ 11: 1133–1145 * Ostrander DB _et al_. (2001) _J. Biol. Chem._ 276: 38061–38067 * Ostrander DB _et
al_. (2001) _J. Biol. Chem._ 276: 25262–25272 Download references ACKNOWLEDGEMENTS We thank Dr. Peter Griač for helpful discussions. This work was supported by grants from the NIH to MF
(GM035759) and WD (GM056389). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Vollum Institute, Oregon Health & Sciences University, 3181 SW Sam Jackson Park Road, Portland, OR, 97239, USA
P Polčic, J Fowlkes, E Blachly-Dyson & M Forte * Department of Biochemistry & Molecular Biology, University of Texas-Houston, Medical School, 6431 Fannin, Suite 6.200, Houston, TX,
77030, USA X Su & W Dowhan Authors * P Polčic View author publications You can also search for this author inPubMed Google Scholar * X Su View author publications You can also search for
this author inPubMed Google Scholar * J Fowlkes View author publications You can also search for this author inPubMed Google Scholar * E Blachly-Dyson View author publications You can also
search for this author inPubMed Google Scholar * W Dowhan View author publications You can also search for this author inPubMed Google Scholar * M Forte View author publications You can also
search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to M Forte. RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Polčic,
P., Su, X., Fowlkes, J. _et al._ Cardiolipin and phosphatidylglycerol are not required for the _in vivo_ action of Bcl-2 family proteins. _Cell Death Differ_ 12, 310–312 (2005).
https://doi.org/10.1038/sj.cdd.4401566 Download citation * Published: 14 January 2005 * Issue Date: 01 March 2005 * DOI: https://doi.org/10.1038/sj.cdd.4401566 SHARE THIS ARTICLE Anyone you
share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the
Springer Nature SharedIt content-sharing initiative