Play all audios:
ABSTRACT Heterozygous missense mutations in the serine-threonine kinase receptor BMPR1B result typically in brachydactyly type A2 (BDA2), whereas mutations in the corresponding ligand GDF5
cause brachydactyly type C (BDC). Mutations in the GDF inhibitor Noggin (NOG) or activating mutations in GDF5 cause proximal symphalangism (SYM1). Here, we describe a novel mutation in
BMPR1B (R486Q) that is associated with either BDA2 or a BDC/SYM1-like phenotype. Functional investigations of the R486Q mutation were performed and compared with the previously reported
BDA2-causing mutation R486W and WT BMPR1B. Overexpression of the mutant receptors in chicken micromass cultures resulted in a strong inhibition of chondrogenesis with the R486Q mutant,
showing a stronger effect than the R486W mutant. To investigate the consequences of the BMPR1B mutations on the intracellular signal transduction, we used stably transfected C2C12 cells and
measured the activity of SMAD-dependent and SMAD-independent pathways. SMAD activation after stimulation with GDF5 was suppressed in both mutants. Alkaline phosphatase induction showed an
almost complete loss of activation by both mutants. Our data extend the previously known mutational and phenotypic spectrum associated with mutations in BMPR1B. Disturbances of
NOG-GDF5-BMPR1B signaling cascade can result in similar clinical manifestations depending on the quantitative effect and mode of action of the specific mutations within the same functional
pathway. SIMILAR CONTENT BEING VIEWED BY OTHERS SPECIFIC HETEROZYGOUS VARIANTS IN _MGP_ LEAD TO ENDOPLASMIC RETICULUM STRESS AND CAUSE SPONDYLOEPIPHYSEAL DYSPLASIA Article Open access 03
November 2023 _SMAD6_-DEFICIENCY IN HUMAN GENETIC DISORDERS Article Open access 21 November 2022 DEFICIENCY OF TMEM53 CAUSES A PREVIOUSLY UNKNOWN SCLEROSING BONE DISORDER BY DYSREGULATION OF
BMP-SMAD SIGNALING Article Open access 06 April 2021 INTRODUCTION Isolated brachydactylies are a group of inherited hand and foot malformations that are characterized by shortening of the
phalanges and/or metacarpal bones. Brachydactylies usually follow an autosomal-dominant inheritance. In 1951, Julia Bell classified them clinically into five groups (A–E) and three subgroups
(A1–A3).1 In recent years, some isolated brachydactylies have been characterized on a molecular level providing new insight into the mechanisms of joint and digit development.2 Heterozygous
missense mutations in bone morphogenetic protein receptor 1B (_BMPR1B)_ were shown to cause brachydactyly type A2 (BDA2), a condition characterized by brachymesophalangy of fingers II and V
resulting in shortening and medial deviation.3 Heterozygous carriers of growth differentiation factor 5 (_GDF5_) loss-of-function mutations are typically affected by brachydactyly type C
(BDC).4, 5 The BDC phenotype includes brachymesophalangy of fingers II, III and V. Finger IV is usually unaffected and thus appears as the longest finger of the hand. Shortening of
metacarpals I and hyperphalangy in fingers II and III may occur and can be considered as relatively characteristic signs. BDC can be highly variable ranging from severely affected hands with
very short fingers to mildly affected cases with only moderate brachydactyly most often affecting the middle and proximal phalanges of fingers II and III. Typical features of BDA2 and BDC
are summarized in Table 1. Thus, brachydactylies C and A2 are clearly distinct entities but have certain overlapping features such as the hypoplasia of middle phalanges. GDF5 and BMPR1B play
an essential role in chondrocyte differentiation and bone formation.6, 7 As a secreted protein, the ligand GDF5 binds to BMP receptors (BMPR types 1 and 2) resulting in the activation of
BMPR1B by transphosphorylation through BMPR2. The transmembrane serine/threonine kinase receptor BMPR1B subsequently activates the SMAD-dependent pathway and/or the p38 MAP kinase pathway.
The oligomerization mode of the BMP receptors at the cell surface determines the activation of the different downstream pathways. The formation of ligand-induced signaling complexes of BMP
receptors leads to the activation of the MAP kinase pathway and, as a consequence, to the induction of alkaline phosphatase (ALP). Ligand binding to already preformed hetero-oligomeric BMP
receptor complexes stimulates the SMAD pathway. Both signal cascades regulate the transcription of specific target genes that are involved in bone formation.8, 9, 10 Extracellular inhibitors
such as Noggin (NOG) prevent BMP receptor activation by binding to GDFs and BMPs. Inactivation of nog in the mouse results in a massive overproduction of cartilage, whereas NOG mutations in
humans cause fusion of joints as observed in proximal symphalangism (SYM1) and multiple synostosis syndrome. Interestingly, a very similar phenotype can be caused by mutations in GDF5 that
result in an activation of the protein through an alteration of the receptor binding affinitiy.11 Here, we present a new mutation in BMPR1B (R486Q) that is located in a highly conserved
region of the receptor. The mutation can either result in a typical BDA2 phenotype, or a combination of BDC and SYM1. Functional assays demonstrate a strong inhibitory effect of this
mutation on chondrogenesis and a loss of intracellular SMAD activation and induction of ALP. MATERIALS AND METHODS CASE REPORT OF PATIENTS (1) The proband, a 34-year-old German woman,
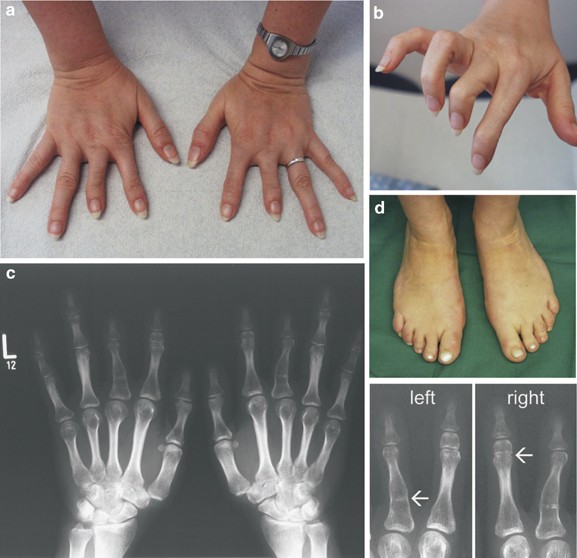
presented with isolated brachydactyly. Fingers II and III were severely shortened and appeared slightly thinner than the others (Figure 1a). The flexion of fingers I, IV and V was
unremarkable. Fingers II had only one interphalangeal joint, and in fingers III the flexion was limited owing to stiffened proximal interphalangeal joints (Figure 1b). All fingernails were
present. Hand radiographs (Figure 1c; left, right) showed hypoplastic middle phalanges of fingers II with a symphalangism between the proximal and middle phalanx on the right hand (arrow in
Figure 1, right). Fingers III consisted of only two phalanges: the proximal phalanges were abnormally formed and showed lateral bowing and distal narrowing of both diaphyses. Within these
diaphyses, a dense line was visible (arrow in Figure 1, left), possibly corresponding to a rudimentary joint or a segmented epiphysis between the original proximal phalanges and the middle
phalanges. Fingers I, IV, V, metacarpal and carpal bones appeared normal. Overall, the hand phenotype resembled BDC with additional features of SYM1. A mild involvement of the feet with a
lateral deviation of the first and second toes, a slight cutaneous syndactyly between the toes II and III, and a moderate shortening of toes III to V with camptodactyly was noted (Figure
1d). Body proportions, height, weight and head circumference were normal. Neither of her parents was affected. (2) The affected boy is the first child of unrelated healthy parents. After
birth, an unusual bilateral shortening and deviation of the index fingers with only one flexion crease was noticed. The finger movements in all existing joints were unremarkable. All
fingernails were present. Radiographs of the hands at the age of 26 months confirmed the index fingers malformation with an absent middle phalanx on the left and a very small middle phalanx
on the right side. In addition, the middle phalanges of fingers V were hypoplastic. The feet showed slightly shortened toes II. This clinical manifestation represents a typical BDA2
phenotype.3 At 26 months of age, the patient's development was unremarkable and height, weight and head circumference were normal. Neither of his parents was affected. MOLECULAR TESTING
Genomic DNA was extracted from blood samples by standard methods. The coding sequence, cds, (10 exons) of _BMPR1B_ was amplified by standard PCR protocols as reported previously.3 The cds
of _GDF5, NOG_ and _IHH_ was also amplified and sequenced in a similar manner. Exact PCR conditions are reported elsewhere (for _GDF5_12; for _NOG_13; for _IHH_14). MICROMASS CULTURES
Micromass cultures from chicken limb buds were prepared as described previously.11 Infection was performed with 1 _μ_l of replication competent avian sarcoma (RCAS) viral supernatants
containing the cds encoding WT-chicken Bmpr1b (WT-chBmpr1b) or R486W-chBmpr1b as described previously.3 Additional _in vitro_ mutagenesis was performed to introduce the mutation R486Q into
the WT-chBmpr1b using the Quick Change site-directed Mutagenesis kit (Stratagene, La Jolla, CA, USA). Culture medium (DMEM-F12, 2% chicken serum, 4 mM L-glutamine, 1000 U/ml penicillin and
100 _μ_g/ml streptomycin) was replaced every 2 days. For each condition, four replicates were performed in parallel. Micromass cultures were fixed and stained with Alcian blue at days 5, 6
and 7. After 10 and 12 days in culture, micromass cultures were fixed and detection of ALP activity was achieved by incubation with NBT-BCIP. Quantification of Alcian blue and ALP staining
were achieved by histomorphometric analyses using the Axio-Vision software tool AutMess (Zeiss, Carl Zeiss MicroImaging GmbH, Göttingen). GENERATION OF C2C12 CELLS STABLY EXPRESSING WT
BMPR1B, R486Q-BMPR1B OR R486W-BMPR1B Site-directed mutagenesis on the Bmpr1b-retroviral construct (Bmpr1b-EGIRT) was performed using the Quick Change site-directed Mutagenesis kit
(Stratagene). C2C12 cells were transduced by infection with helper-free VSV-G-pseudotyped retroviruses as described previously.15 Briefly, 293T cells were used as packaging cells and
transfected with the retroviral construct (carrying the Bmpr1b sequence) and plasmids coding for gag-pol and VSV-G. Twenty-four hours post-transfection, 293T cells were treated with 10 mM
sodium-butyrate for 10 h. C2C12 cells were infected with retroviral supernatant 48 h and 72 h post-transfection. Transduction efficiencies were controlled by GFP, which was located on the
same retroviral construct. Expression of WT Bmpr1b, R486Q-Bmpr1b or R486W-Bmpr1b was controlled by Western blotting using monoclonal mouse anti-HA antibody (7HA Sigma-Aldrich Chemie gmbH,
Taufkirchen). The HA epitope was integrated at the N-terminus (extracellular) of each receptor.9 LUCIFERASE REPORTER GENE ASSAY C2C12 cells stably expressing WT Bmpr1b, R486Q-Bmpr1b or
R486W-Bmpr1b were transfected with BRE luciferase reporter construct (p(BRE)4-luc),16 renilla luciferase (pRLTK) (Promega GmbH, Mannheim) as an internal control for transfection efficiency
using Lipofectamine™ (Invitrogen GmbH, Karlsruhe, Germany). Cells were starved for 5 h in DMEM containing 0.5% FCS and stimulated for 16–24 h with increasing amounts of recombinant GDF5
under starving conditions. Cell lysis and luciferase measurements were carried out according to manufacturers’ instructions of the Dual Luciferase Assay System (Promega) in triplicate.
Luciferase activity was recorded in relative light units by calculating the quotient between firefly and renilla activity. ALP ASSAY Quantitative ALP assays were performed as described
previously.11 C2C12 cells stably expressing WT Bmpr1b, R486Q- or R486W-Bmpr1b were seeded at a density of 1 × 104 cells/96-well plates in growth media (high-glucose DMEM, 10% FCS and 2 mM
L-glutamine in 10% CO2). After 24 h, stimulation with recombinant GDF5 was performed in media with reduced FCS (high-glucose DMEM, 2% FCS and 2 mM L-glutamine in 10% CO2) for 3 days. ALP
activity was measured in triplicate for C2C12 cells in 96-well plates by lysing cells in ALP-buffer1 (0.1 M glycine, pH 9.6, 1% Nonidet P-40, 1 mM MgCl2 and 1 mM ZnCl2). After the addition
of ALP substrate through 1 volume of ALP-buffer2 (5 mM _p_-nitrophenyl phosphate (_p_-NPP), 0.1 M glycine (pH 9.6), 1 mM MgCl2 and 1 mM ZnCl2), and subsequent termination of the reaction by
adding 0.25 volumes 2 M NaOH ALP activity was calculated by the released amount of _p_-NP from the substrate _p_-NPP, which was determined spectrophotometrically at 405 nm. RESULTS AND
DISCUSSION IDENTIFICATION OF A R486Q MUTATION IN BMPR1B THAT RESULTS IN A BDC/SYM1 PHENOTYPE OR BDA2 As patient 1 showed phenotypic similarities with BDC and SYM1, we first sequenced the
coding region of _GDF5_ and _NOG_, but no mutation was found. Subsequently, we performed mutation screening in _BMPR1B_. We identified a new c.1457G>A missense mutation which results in
an arginine to glutamine substitution at the position 486 (R486Q). This mutation is located in a highly conserved region of BMPR1B within a C-terminal motif also termed
nonactivating-non-downregulating box. Previous work has shown that this region is important for endocytosis of the TGF_β_ type 1 receptor and for the regulation of the activation process by
transphosphorylation between TGF_β_ receptors.17 Because of the similarities between the highly homologous TGF_β_ and BMP receptors, it is likely that this region has a similar function in
BMPR1B. The identical mutation was also found in patient 2 who showed, in contrast to patient 1, typical BDA2. A possible modifying IHH mutation (causing brachydactyly type A1) was excluded
in both patients. The mutational screening of the parents confirmed that both mutations had occurred _de novo_. The middle phalanges of fingers II were hypoplastic or absent in both
patients. However, the severe shortening and abnormal configuration of finger III together with symphalangism in patient 1 was not observed in case 2 and are not part of the BDA2 spectrum.
The more severe phenotype in patient 1 could be owing to the stronger functional effect of the R486Q mutation when compared with the previously described BDA2-associated mutations I200K and
R486W. The phenotype in the investigated patient group with these latter mutations shows only a slight clinical variability. The clinical features seen in patient 1, with BDC-similar
phenotype and symphalangism, has never been observed in these patients. The clinical difference between cases 1 and 2 can only be explained by phenotypic variability owing to unknown
modifiers and/or stochastic effects at present. Parts of the observed phenotype in patient 1 have been described to be caused by mutations in GDF5 (BDC) or NOG (SYM1). The phenotypic overlap
of case 1 with BDC and SYM1 reflects the interaction of the major players within the BMP/GDF pathway, that is, the ligand (GDF5), its receptor (BMPR1B) and the inhibitor (NOG). Furthermore,
it shows that mutations within one pathway can result in overlapping or even identical phenotypes, as recently also demonstrated for activating mutations in GDF5 that cause SYM1.11 Several
_in vitro_ and _in vivo_ assays were performed previously to investigate the functional consequences of the I200K and R486W mutations in Bmpr1b.3 It was demonstrated that the mutation I200K
acts in a dominant-negative manner potentially by a disturbance of the proper transphosphorylation process between the BMP receptors and the loss of Bmpr1b kinase function. In contrast, the
mutation at the highly conserved amino-acid position 486 showed a normal kinase activity and thus differs in its mode of action, but the exact functional mechanism of the R486W mutation
remained unclear. It is conceivable that a special amino-acid change at such a conserved region of presently unexplained function may cause disturbances of protein–protein interactions
between the BMP receptors and other molecules, or alters the activity of the receptor itself. In addition, modifier genes could have an important role concerning the severity of the
manifestation. FUNCTIONAL ANALYSIS OF BMPR1B MUTANTS IN MICROMASS CULTURES INDICATES A STRONGER INHIBITING EFFECT ON CHONDROCYTE DIFFERENTIATION BY THE R486Q MUTANT THAN THE R486W MUTANT WT
and mutant Bmpr1b cds constructs were expressed in chicken limb micromass cultures using an RCAS virus. This system is an _in vitro_ model for chondrogenic differentiation. Cell
differentiation and cartilagenous matrix production were measured by Alcian blue and ALP. We investigated chondrogenesis induced by both mutants (R486Q and R486W) in comparison to WT Bmpr1b.
Cells infected with WT Bmpr1b were not distinguishable from non-infected control cultures. As demonstrated in Figure 2a and b, transfection with mutant Bmpr1b resulted in a strong
inhibition of cartilage differentiation at days 5, 6 and 7, whereas the R486Q mutant shows a stronger effect than the R486W mutant. This result was also confirmed by ALP staining of
micromass cultures at days 10 and 12. As shown in Figure 2c, uninfected control and WT Bmpr1b cultures are clearly ALP positive, whereas cultures infected with the mutant Bmpr1b showed only
little induction of ALP. Again, the R486Q mutation had a stronger effect than the R486W mutant. BMPR1B MUTATIONS AT THE POSITION 486 FAIL TO INDUCE INTRACELLULAR SMAD SIGNALING To identify
possible functional differences between the two Bmpr1b mutants and WT Bmpr1b, we treated C2C12 cells stably expressing Bmpr1b (WT or mutant) with recombinant GDF5. C2C12 cells are
mesenchymal progenitor cells that can differentiate into different cells (eg osteoblasts). The binding of a ligand to preformed BMP receptor complexes at the cell surface results in the
phosphorylation and activation of Bmpr1b, which in turn results in the activation of the intracellular SMAD signaling cascade.9 To further characterize the functional effect caused by the
mutations, we used a C2C12 cell assay with a SMAD-dependent reporter construct. The quantity of measurable luciferase activity reflects the degree of activated SMAD signaling. In general,
GDF5 has a very high affinity to BMPR1B but only a minimal signal transduction efficiency via other BMP receptors.19 As demonstrated in Figure 3a, C2C12 cells stably transfected with WT
Bmpr1b showed a dose-dependent increase in GDF5-mediated reporter gene activity. Saturation was reached at 500 pM. Both mutants are not able to transduce SMAD activation when treated with
variable amounts of recombinant GDF5. It seemed that the R486Q mutant had a stronger effect than the R486W mutant. The control culture showed some minimal luciferase activity. This effect
can be explained by minimal endogenous SMAD signaling activated by other pathways. FUNCTIONAL ANALYSIS OF BMPR1B MUTANTS IN C2C12 CELLS DEMONSTRATES A COMPLETE LOSS OF OSTEOBLASTIC
DIFFERENTIATION In this assay, the degree of ALP activity reflects the activation of p38 MAP kinase signaling as the transcription of the ALP gene depends mainly on this pathway. C2C12 cells
do not express significant amounts of Bmpr1b and therefore show no ALP activity when treated with GDF5. Figure 3b illustrates a dose-dependent increase of ALP induction upon GDF5 treatment
in stably WT Bmpr1b-expressing C2C12 cells with a saturation at about 10 nM. The level of ALP activity corresponded to the GDF5 concentrations used for treatment up to 5 nM. Higher GDF5
concentrations did not increase the ALP activity presumably owing to saturation of the BMP receptors capacity. The Bmpr1b mutations were tested with the same dose range of recombinant GDF5.
Both the R486Q and R486W mutant showed no osteoblastic differentiation as demonstrated by the lack of ALP activity under 2.5–30 nM GDF5 concentrations. A quantitative difference between the
two mutants was not detectable. Our results demonstrate that both BMPR1B mutations at position 486 have a strong inhibitory effect on chondrogenesis and osteoblastic differentiation and
cause a disturbance in the intracellular SMAD and MAP kinase signaling. Our functional assays indicate a stronger effect of the R486Q mutant when compared with the R486W mutant. This may
explain the phenotypic variability observed in the two affected individuals described here. Whereas patient 2 had classical BDA2, patient 1 showed a combination of BDC and SYM1 and thus a
more severe phenotype. Neither of the latter phenotypes has previously been observed in individuals with the R486W mutation, indicating that the R486Q mutation shifts the spectrum to the
more severe side. Furthermore, SYM1 is not regarded to be part of the BDC spectrum and has not been reported in association with GDF5 loss-of-function mutations. The occurrence of
symphalangism caused by mutations in the BMP inhibitor NOG suggests that an upregulation of BMP signaling causes abnormal joint formation. In fact, gain-of-function mutations in GDF5 cause
SYM1 most likely through the activation of the BMPR1A, the receptor normally activated by BMP2.11 The association of symphalangism and BDC with the BMPR1B receptor mutation R486Q suggests a
twofold effect of this mutation. First, it results in a partial inactivation of the GDF5 pathway causing BDC. Second, the downregulation of BMPR1B signaling leads to an relatively enhanced
activation of BMPR1A and thus BMP2 signaling which then causes symphalangism similar to the GDF5 gain-of-function mutations as shown in our previous work.11 Our observations indicate that
hand disorders like BDC, BDA2 or SYM1 belong to a molecular disease family consisting of the essential parts of the BMP pathway: the ligands, the receptors and their inhibitors. Mutations in
any of these components may imitate phenotypes classically associated with mutations in another component by dysregulating the intricate equilibrium between them. REFERENCES * Bell J :
_Treasury of Human Inheritance_. London: Cambridge University Press, 1951, vol 5, pp 1–31. Google Scholar * Kornak U, Mundlos S : Genetic disorders of the skeleton: a developmental
approach. _Am J Hum Genet_ 2003; 73: 447–474. Article CAS Google Scholar * Lehmann K, Seemann P, Stricker S _et al_: Mutations in bone morphogenetic protein receptor 1B cause
brachydactyly type A2. _Proc Natl Acad Sci USA_ 2003; 100: 12277–12282. Article CAS Google Scholar * Everman DB, Bartels CF, Yang Y _et al_: The mutational spectrum of brachydactyly type
C. _Am J Med Genet_ 2002; 112: 291–296. Article Google Scholar * Polinkovsky A, Robin NH, Thomas JT _et al_: Mutations in CDMP1 cause autosomal dominant brachydactyly type C. _Nat Genet_
1997; 17: 18–19. Article CAS Google Scholar * Storm EE, Kingsley DM : GDF5 coordinates bone and joint formation during digit development. _Dev Biol_ 1999; 209: 11–27. Article CAS Google
Scholar * Baur ST, Mai JJ, Dymecki SM : Combinatorial signaling through BMP receptor IB and GDF5: shaping of the distal mouse limb and the genetics of distal limb diversity. _Development_
2000; 127: 605–619. CAS Google Scholar * Gilboa L, Nohe A, Geissendorfer T, Sebald W, Henis YI, Knaus P : Bone morphogenetic protein receptor complexes on the surface of live cells: a new
oligomerization mode for serine/threonine kinase receptors. _Mol Biol Cell_ 2000; 11: 1023–1035. Article CAS Google Scholar * Nohe A, Hassel S, Ehrlich M _et al_: The mode of bone
morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. _J Biol Chem_ 2002; 277: 5330–5338. Article CAS Google Scholar * von Bubnoff A, Cho K :
Intracellular BMP signaling regulation in vertebrates: pathway or network? _Dev Biol_ 2001; 239: 1–14. Article CAS Google Scholar * Seemann P, Schwappacher R, Kjaer KW _et al_:
Activating and deactivating mutations in the receptor interaction site of GDF5 cause symphalangism or brachydactyly type A2. _J Clin Invest_ 2005; 115: 2373–2381. Article CAS Google
Scholar * Schwabe GC, Turkmen S, Leschik G _et al_: Brachydactyly type C caused by a homozygous missense mutation in the prodomain of CDMP1. _Am J Med Genet A_ 2004; 124: 356–363. Article
Google Scholar * Gong Y, Krakow D, Marcelino J _et al_: Heterozygous mutations in the gene encoding noggin affect human joint morphogenesis. _Nat Genet_ 1999; 21: 302–304. Article CAS
Google Scholar * Gao B, Guo J, She C _et al_: Mutations in IHH, encoding Indian hedgehog, cause brachydactyly type A-1. _Nat Genet_ 2001; 28: 386–388. Article CAS Google Scholar * Lutz
M, Krieglstein K, Schmitt S _et al_: Nerve growth factor mediates activation of the Smad pathway in PC12 cells. _Eur J Biochem_ 2004; 271: 920–931. Article CAS Google Scholar *
Korchynskyi O, ten Dijke P : Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. _J
Biol Chem_ 2002; 277: 4883–4891. Article CAS Google Scholar * Garamszegi N, Dore Jr JJ, Penheiter SG, Edens M, Yao D, Leof EB : Transforming growth factor beta receptor signaling and
endocytosis are linked through a COOH terminal activation motif in the type I receptor. _Mol Biol Cell_ 2001; 12: 2881–2893. Article CAS Google Scholar * Wieser R, Wrana JL, Massague J :
GS domain mutations that constitutively activate T beta R-I, the downstream signaling component in the TGF-beta receptor complex. _EMBO J_ 1995; 14: 2199–2208. Article CAS Google Scholar
* Sammar M, Stricker S, Schwabe GC _et al_: Modulation of GDF5/BRI-b signalling through interaction with the tyrosine kinase receptor Ror2. _Genes Cells_ 2004; 9: 1227–1238. Article CAS
Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by a grant to KL (LE1851) by the Deutsche Forschungsgemeinschaft. We thank Dr Lindemann for the retroviral
construct EGIRT. Work in PK's laboratory was supported by BMBF (0312117). Recombinant GDF5 was a kind gift from BIOPHARM GmbH. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Institut für
Medizinische Genetik, Charité, Universitätsmedizin Berlin, Berlin, Germany Katarina Lehmann & Stefan Mundlos * Max-Planck-Institut für Molekulare Genetik, Berlin, Germany Petra Seemann
& Stefan Mundlos * Institut für Chemie-Biochemie, Freie Universität Berlin, Berlin, Germany Jan Boergermann & Petra Knaus * Département de Pédiatrie, Unité de Génétique Clinique,
Amiens, France Gilles Morin * Institut für Humangenetik und Medizinische Biologie, Universität Halle/Saale, Germany Silke Reif Authors * Katarina Lehmann View author publications You can
also search for this author inPubMed Google Scholar * Petra Seemann View author publications You can also search for this author inPubMed Google Scholar * Jan Boergermann View author
publications You can also search for this author inPubMed Google Scholar * Gilles Morin View author publications You can also search for this author inPubMed Google Scholar * Silke Reif View
author publications You can also search for this author inPubMed Google Scholar * Petra Knaus View author publications You can also search for this author inPubMed Google Scholar * Stefan
Mundlos View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Katarina Lehmann. RIGHTS AND PERMISSIONS Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Lehmann, K., Seemann, P., Boergermann, J. _et al._ A novel R486Q mutation in BMPR1B resulting in either a brachydactyly type
C/symphalangism-like phenotype or brachydactyly type A2. _Eur J Hum Genet_ 14, 1248–1254 (2006). https://doi.org/10.1038/sj.ejhg.5201708 Download citation * Received: 15 May 2006 * Revised:
20 July 2006 * Accepted: 20 July 2006 * Published: 06 September 2006 * Issue Date: 01 December 2006 * DOI: https://doi.org/10.1038/sj.ejhg.5201708 SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative KEYWORDS * bone morphogenetic protein receptors * growth differentiation factor 5 * noggin * brachydactyly * symphalangism