Play all audios:
ABSTRACT H-Ras is well known as one of the essential components of Ras/Raf/MEK/ERK cascade, which is a critical prosurvival signaling mechanism in most eukaryotic cells. Ras targets
Raf/MEK/ERK cascade by integrating and transmitting extracellular signals from growth factor receptors to Raf, leading to the propagation of signals to modulate a serious of cellular
survival events. Apoptosis signal-regulating kinase1 (ASK1) serves as a general mediator of cell death because it is responsive to a variety of death signals. In this study, we found that
H-Ras interacted with ASK1 to cause the inhibition of both ASK1 activity and ASK1-induced apoptosis _in vivo_, which was reversed only partially by addition of RafS621A, an antagonist of
Raf, whereas MEK inhibitor, PD98059, and PI3K inhibitor, LY294002, did not disturb the inhibitory effect of H-Ras on ASK-1-induced apoptosis. Furthermore, by means of immunoprecipitate and
kinase assays, we demonstrated that the interaction between H-Ras and ASK1 as well as the inhibition of ASK1 activity were dependent on the binding activity of H-Ras. These results suggest
that a novel mechanism may be involved in H-Ras-mediated cell survival in addition to the well established MEK/ERK and PI3K/Akt kinase-dependent enhancement of cell survival. SIMILAR CONTENT
BEING VIEWED BY OTHERS THE PRO-ONCOGENIC NONCANONICAL ACTIVITY OF A RAS•GTP:RANGAP1 COMPLEX FACILITATES NUCLEAR PROTEIN EXPORT Article Open access 11 November 2024 PLASMA
MEMBRANE-ASSOCIATED ARAF CONDENSATES FUEL RAS-RELATED CANCER DRUG RESISTANCE Article 27 January 2025 ONCOGENIC KRAS IS DEPENDENT UPON AN EFR3A-PI4KA SIGNALING AXIS FOR POTENT TUMORIGENIC
ACTIVITY Article Open access 09 September 2021 INTRODUCTION In response to diverse extracellular stimuli such as peptide growth factors, cytokines, hormones and a variety of stresses, cells
constantly face dual complete opposite selections: survival or death. Diverse signaling pathways, including those mediated by tyrosine kinase receptors and heterotrimeric G protein-coupled
receptors take part in the decision of cell fate 1. Ras/Raf/MEK/ERK cascade is well known as a critical prosurvival signaling mechanism. H-Ras, a small guanosine triphosphate-binding protein
encoded by _H-Ras_ proto-oncogene, is one of the essential components in this pathway2. It is believed that H-Ras is involved centrally in diverse prosurvival signaling transduction
pathways and acts as a key regulator of cell growth in all eukaryotic cells3. H-Ras targets Raf/MEK/ERK cascade by integrating and transmitting extracellular signals from growth factor
receptors to Raf (a MAPKK kinase, MAPKKK). Upon activation, Raf relays to phosphorylate mitogen-activated protein kinase kinase (MEK), which in turn activates mitogen-activated protein
kinase/extracellular signal-regulated kinases (MAPK/ERKs), leading to the propagation of signals to modulate a serious of cellular survival events4, 5. Apoptosis-signal regulating kinase 1
(ASK1) is one of MAP kinase kinase kinases (MAPKKK), which serves as a general mediator of cell death because it is responsive to a variety of death signals6, including oxidative stresses,
treatment of TNFα7, methylglyoxal8 (a physiological metabolite), and the chemotherapeutic drugs cisplatin and paclitaxal9, 10. As an upstream activator of the stress responsive protein
kinases, ASK1 activates the JNK and p38 MAP kinase cascades and induce apoptosis during exposure to oxidative stress and other stressful stimuli. Regulation of ASK1 by both pro-apoptotic and
anti-apoptotic signals may provide a critical point of control for cell death and cell survival. In recent years, the functional “cross-talk” between the prosurvival signaling pathway and
pro-apoptosis signaling pathway and the related biological significance to decide cell fate have been understood. For example, it was proved that Raf-1 could bind to the N-terminal
regulatory fragment of ASK1 and decreased the activity of ASK1, which resulted in inhibiting apoptosis mediated by ASK1 signaling pathway and promoting cell survival11. Since H-Ras acts as a
direct upstream regulator of Raf and plays the key role in triggering cellular survival signaling, we were interested in inquiring whether ASK1 was also a potential target of H-Ras. In this
report, we have demonstrated that ASK1 really acted as a potential H-Ras target and its activity was suppressed by H-Ras, which leaded to selectively inhibition of ASK1-mediated
pro-apoptotic signal pathway, suggesting a novel prosurvival mechanism of action for H-Ras. MATERIALS AND METHODS ANTIBODIES AND CHEMICAL REAGENTS Anti-MKK3, anti-ERK1, anti-p38, anti-HA,
anti-H-Ras antibodies, anti−β-actin and Protein A/G plus-agarose were purchased from Santa Cruz Biotechnology. Inc.(Santa Cruz, CA). Anti-phospho-MKK3 and anti-phospho-ERK1/2 antibodies,
PD98059, LY294002, ATF2 peptide and full length JNK1 protein were purchased from New England Biolabs, Inc. (Beverly, MA). GST-MKK4 protein was from Transduction Lab (Lexington, KY). FUGENETM
6 Transfection Reagent was from Roche Molecular Biochemicals (Mannheim, Germany). PLASMIDS The constructed pCMV vectors respectively expressing the wild-type H-Ras protein (H-Ras WT),
constitutively active form of the H-Ras protein that contains a glycine-to-valine mutation at residue 12 (H-Ras V12) and dominant-negative form of the H-Ras protein that contains a
serine-to-asparagine mutation at residue 17 (H-Ras N17), dominant-negative form of the Raf protein that contains a serine-to-alanine mutation at residue 621 (Raf S621A), were purchased from
Clontech Laboratories Inc. (Palo Alto, CA). pcDNA3-HA-ASK1/WT and pcDNA3-HA-ASK1/KM plasmids expressing wild-type or dominant negative ASK1 kinases were a generous gift from Dr. H Ichijo
(Tokyo Medical and Dental University). CELL CULTURE, TREATMENT AND TRANSFECTION A human embryonic kidney cell line (HEK293) was maintained in Dulbecco's modified Eagle's medium
(DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics at 37°C in a CO2 incubator. For transfection, cells were plated at a density of 1×104 cells/cm2. After 24 h of culture,
the cells were transfected with plasmid DNA (0.1 μg of DNA/cm2 per plasmid) and FUGENE6 reagent according to the instructions of the manufacturer. Assays were performed at 36 h after
transfection. At the end of all treatments, the cells were washed twice with ice-cold PBS and then lysed for whole cell lysate preparations. IMMUNOPRECIPITATION AND IMMUNOBLOT ANALYSIS Cells
were lysed in lysis buffer containing 1% Nonidet P-40, 20 mM Tris-HCl (pH 7.6), 0.15 M NaCl, 3 mM EDTA, 3 mM EGTA, 1 mM phenylmethylsulfonyl fluoride, 2 mM sodium vanadate, 20 mg/ml
aprotinin and 5 mg/ml leupeptin. Insoluble material was removed by centrifugation (12,000 rpm 30 min, 4°C). Immunoprecipitation was performed by a rat anti-H-Ras monoclonal antibody. The
immuno-complexes were collected following incubation with protein A/G plus-agarose beads. For Immunoblotting, the cell lysates or immunoprecipitates were separated on 10% or 12%
SDS-polyacrylamide gels, and blotted onto a nitrocellulose membrane. Nonspecific binding was blocked by incubating the membrane with 0.05% Tween-20/PBS containing 5% non-fat dry milk for 1 h
at room temperature. Membranes were incubated with the primary antibody overnight at 4°C and then with a horseradish peroxidase-conjugated secondary antibody, and the specific immune
complexes were detected using Western Blot Plus Chemiluminescence Reagent (Life Science, Inc, Boston, MA). FLOW CYTOMETRIC ANALYSIS For analysis of DNA fragmentation, HEK293 cells were
plated onto a 24-well tissue culture plate and transfected with indicated plasmids for 36 h. The cells were starved in a FBS free medium. Twenty-four hours after starvation, total cells were
harvested, re-suspended in 500 μl of propidium iodide buffer (0.1% Triton X-100, 0.1% Trisodium citrate) containing 50 μg/ml propidium iodide, and incubated for 15 min on ice. DNA
fragmentation analysis was carried out using a FACScalibur flow cytometer (Becton Dickinson), as described previously12. Collected data were processed by the Modfit software, and apoptosis
was scored by the percentage of cells appearing in the area below the G1/G0 peak. ASK1 KINASE ASSAY The ASK1 kinase assay was performed as described previously 8. Briefly, the cell extracts
were clarified by centrifugation, and the supernatants were immunoprecipitated with HA antibody. The immunocomplexes were bound to protein A/G plus-agarose beads. The beads were washed twice
with kinase buffer [20 mM Tris-HCl (pH 7.4), 20 mM MgCl2], and subjected to the ASK1 kinase assay. The immunocomplexes were incubated first with 0.1 μg of GST-MKK4 for 15 min at 30°C in a
final volume of 25 μl kinase buffer containing 100 μM of ATP, and subsequently with 1 μg of JNK1 for 15 min at 30 °C. Thereafter, the activated complex was incubated with 0.3 μCi of
[γ-32P]ATP and 1 μg of ATF2 peptide in the same solution. The samples were resolved by SDS-PAGE, and the phosphorylation of ATF2 was detected. RESULTS H-RAS INHIBITED ASK1-INDUCED APOPTOSIS
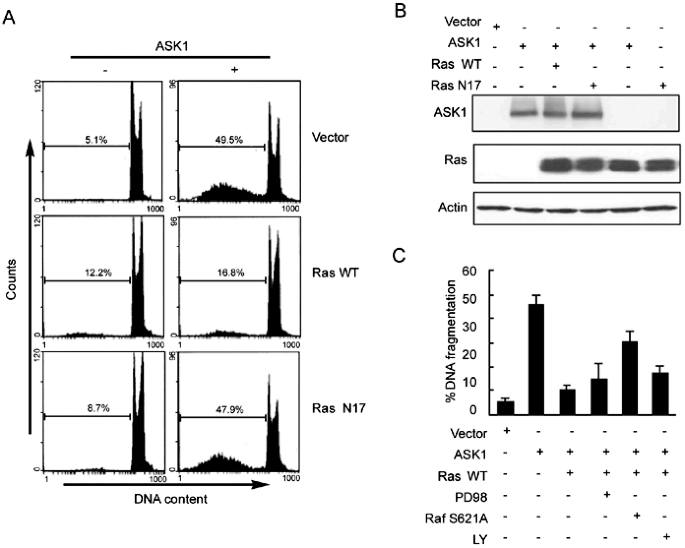
To investigate the role of the H-Ras in suppressing ASK1-dependent apoptotic signaling, we tested the effect of H-Ras on ASK1-induced apoptosis. HEK293 cells were transiently transfected
with human ASK1 alone or together with H-Ras or dominant negative H-Ras N17, in which Ser 17 was replaced by Asn. The cells were stained with PI and DNA fragmentations were analyzed by
flowcy-tometer; and at the same condition, both the expression of ASK1 and H-Ras were detected by immunoblotting with anti-HA and anti-H-Ras antibody respectively. The data showed that the
overexpression of ASK1 alone sufficiently induced cell apoptosis. When cells were co-transfected with _ASK1_ and _H-Ras_, however, apoptotic cell death induced by overexpression of ASK1 was
largely reduced, and the DNA fragment approximated to the level of the cells transfected with _H-Ras_ alone (Fig 1A). This inhibitory effect of H-Ras on ASK1-induced cell apoptosis did not
appear to be caused by the reduction of ASK1 expression level, because co-transfection with H-Ras did not change the expression amount of ASK1 compared to the cells transfected ASK1 alone,
as assessed by immunoblotting assay (Fig 1B). H-Ras-mediated activation of Raf/MEK/ERK and PI3K/Akt cascades were well known as critical prosurvival signaling mechanisms, and some reports
indicated that Raf might promote cell survival by antagonizing ASK1 in a Raf-1 protein kinase activity-independent manner11. To confirm the hypothesis that whether H-Ras inhibits
ASK1-induced apoptosis through Raf/MEK or PI3K/Akt pathway, a negative mutant of Raf (RafS621A), PD98059 (a specific MEK inhibitor) and LY294002 (a specific PI3K inhibitor) were used to
block the down-stream effectors of H-Ras in the cells expressing ASK1. As shown in Fig 1C, blocking Raf activity by co-expression of RafS621A, which was believed to weaken the binding
ability of Raf, reversed only partially the inhibitory effect of H-Ras. Whereas blocking MEK activity by addition of PD98059 had no obviously change on the inhibitory effect of H-Ras.
Moreover, no such inhibition was also observed when cells were treated with LY294002, at a concentration known to abolish PI3K activity. These data suggested that H-Ras inhibited
ASK1-induced apoptosis by a MEK/ERK and PI3K/Akt-independent mechanism, and Raf might partially contribute to the inhibitory process. H-RAS DID NOT ACTIVATE THE MKK3/P38 KINASE PATHWAY In
the pro-apoptosis signaling cascade, p38 MAPK is an important down-stream component of ASK1 in response to diverse extracellular stresses stimuli. Therefore, it is necessary to investigate
whether MKK3/p38 pathway is also the target of H-Ras. HEK293 cells were transiently transfected with wild-type (H-Ras WT), dominant negative (H-Ras N17) and active form (H-Ras V12) of H-Ras,
wild-type (ASK1 WT) and kinase-inactive form (ASK1 KM, in which Lys 709 was replaced by Met) of ASK1, respectively. The MKK3 and p38 kinase activity was measured by western blotting with
specific antibodies. Unlike wild-type ASK1, which could markedly increase the phosphorylation of both MKK3 and p38 MAPK, the expression of either wild H-Ras or mutated H-Ras failed to
stimulate the phosphorylation of MKK3 and p38 MAPK (Fig 2), demonstrating that downstream signaling pathways of H-Ras were insufficient to induce MKK3 and p38 activation. In other terms,
both MKK3 and p38 were not the direct targets of H-Ras. H-RAS SUPPRESSED ASK1-MEDIATED ACTIVATION OF P38 MAPK We next examined whether H-Ras activation is sufficient to affect ASK1-mediated
activation of p38 MAPK _in vivo_. Wild type and mutated H-Ras were co-transfected with ASK1 WT, and phosphorylation of the p38 under serum-free conditions was determined by Western blotting.
While H-Ras WT moderately inhibited the phosphorylation of p38 MAPK in response to ASK1 expression, the active form of H-Ras, H-Ras V12, led to clear inhibition of phosphorylation of p38,
whereas co-expression of H-Ras N17 with ASK1 failed to suppress ASK1-mediated p38 phosphorylation (Fig 3). In contrast, co-expression of ASK1 did not alter ERK1/2 phosphorylation state
induced by expression of wild-type and active form of H-Ras (Fig 3). Similar results were also observed in the treatment with H2O2 that has been believed to activate p38 MAPK through
ASK1-mediated signaling. It was found that the active H-Ras but not binding-defective mutant H-Ras N17 could oppose the effect of H2O2 on increasing the ASK1-mediated phosphorylation of p38
MAPK (Fig 4). These data imply that H-Ras-inhibited signaling from ASK1 to p38 may be through a target binding mechanism. INHIBITION OF ASK1 SIGNALING WAS RELATIVE TO THE BINDING ACTIVITY OF
H-RAS Since binding to its targets is one of the mechanisms for H-Ras to affect a number of downstream signaling states, one possible mechanism for H-Ras to inhibit apoptotic activity of
ASK1 is through direct interaction between the two proteins. To test this hypothesis, the plasmids encoding H-Ras WT, H-Ras N17 and H-Ras V12 were respectively co-transfected into HEK293
cells with HA-tagged ASK1, and the binding was determined by co-im-munoprecipitation from serum-starved cells. As shown in Fig 5, ASK1 was present mainly in H-Ras V12 immunoprecipitate and
moderately in H-Ras WT immunoprecipitate but absent in H-Ras N17 immunoprecipitate, indicating that the association between H-Ras and ASK1 was dependent on the binding activity of H-Ras.
Furthermore, we examined whether H-Ras and ASK1 interaction was sufficient for inhibiting activity of ASK1. The activity of ASK1 in various immune complexes was measured by a couple kinase
assay. The result indicated that co-expression of either H-Ras WT or H-Ras V12 with ASK1 appeared to strongly inhibit the activation of ASK1. In contrast, co-expression of binding-defective
form of H-Ras, H-Ras N17, had no effect on the activity of ASK1 (Fig 6). Taken together, these findings imply that the binding activity of H-Ras is necessary for negatively regulating the
function of ASK1. DISCUSSION Recently, growing evidences have shown that the functional “cross-talk” frequently take place between the prosurvival signaling pathway and pro-apoptosis
signaling pathway. The relative mechanisms appear to be complex because it is possible that a variety of signaling cascades from these two functional antagonistic signaling pathways get
involved in the processes. It has been postulated that apoptotic cell death is the default program of metazoan cells, which must be suppressed continuously by survival mechanisms13. In most
eukaryotes, H-Ras is a vital com-ponent of a variety of growth factor-induced signaling pathways and plays an important role in integrating and transmitting extracellular signals to cellular
prosurvival signaling pathways. Generally, H-Ras functions as a positive regulator of the ERK/MAPK signal transduction cascade by activating MEKKs, including Raf-1 and MEKK1. Our findings
indicated that there existed another mechanism in which H-Ras promoted cell survival through a downstream kinases activity- independent fashion. We found that ASK1, a critical component of
the cellular pro-apoptotic machinery, also acted as a potential target of H-Ras and ASK1-mediated cell death signal was blocked by the interaction of wild-type and active form of H-Ras
protein, suggesting a novel prosurvival mechanism for H-Ras. This finding supports the observation that ASK1-mediated cell death is suppressed in response to growth factors, which dominantly
activate Ras14. It has been reported that Raf-1 could bind to the N-terminal regulatory fragment of ASK1 and decreased the activity of ASK1, which resulted in inhibiting the apoptosis
mediated by ASK1 and promoting cell survival mediated by Raf-111. Therefore, Raf plays the key role not only in triggering cellular survival signaling mediated by Raf/MEK/ERK cascade but
also in inhibiting directly ASK1-mediated pro-apoptotic pathway. Since H-Ras acts as a direct upstream activator of Raf, the activation of MEK/ERK and Raf induced by expression of H-Ras
might be responsible for the inhibitory action of H-Ras in ASK1-induced apoptosis. In other terms, the regulative effect of H-Ras on ASK1-mediated pro-apoptotic signaling pathway might be
dependent on Raf or Raf/MEK/ERK signaling cascade. We provided evidence contradicting this view. Addition of MEK inhibitor, PD98059, at a concentration known to abolish MEK activity, did not
block the effect of H-Ras on the inhibition of ASK-1-induced apoptosis. This result indicated that the activation of MEK/ERK, which caused by over-expression of wild-type and active form of
H-Ras, is not sufficient for inhibiting ASK-1-induced apoptosis. In addition, inhibition of PI3K, an established downstream target of H-Ras, did not limit the effect of H-Ras the on the
inhibition of ASK-1-induced apoptosis. Our results also showed that the co-expression of a dominant negative form of Raf-1 only weakly reversed the action of H-Ras, suggesting that Raf-1 was
involved partially in the inhibition of ASK-1-induced apoptosis. There must be other other signaling pathways, which are closely relative to H-Ras binding activity, take part in the
process. In this study, we found that ASK1 was presented in wild-type and active form of H-Ras immuno-complexes, but not in that of the binding activity-defective form of H-Ras, H-Ras N17.
Moreover, ASK1 activity was reduced in the same manner. These results indicated that physical association of H-Ras might be required for inhibition of ASK1 activity, and subsequent to block
ASK1-induced apoptosis. How the interaction of H-Ras-ASK1 causes the down regulation of ASK1 activity remains unclear. Previous studies indicated that ASK1 directly interacted with its
regulators and the ASK1 activity decreased with the separating of ASK1 homo-oligomerization. Homo-oligomerization and association with death receptor associated proteins such as TRAF2 are
important process for ASK1 activation15. ASK1 interacts physically with the reduced form of thiore-doxin, a redox-sensitive protein, and is sequestered in an inactive form16. Oxidation of
thioredoxin by intracellular ROS results in the activation of ASK1, involving homo-oligomerization and its association with TRAF2. 14-3-3, a phosphoserine-binding molecule, specifically
binds to ASK1 via Ser-967 of ASK1 and results in inhibition of ASK1-induced apoptosis. Binding to ASK1 by co-expression of a ligand-binding-defective mutant of 14-3-3 causes an increase in
homo-oligomerization of ASK117. CDC25, a cell cycle-dependent expression of protein phosphatase, has been reported to bind to C-terminal domain of ASK1 and inhibit its kinase activity. This
inhibitory action of CDC25 on ASK1 activity is closely related to diminishing the homo-oligomerization of ASK118. Thus, it seems that the physical association of H-Ras may be inhibitory to
this series of events leading to full activation of ASK1. It is possible that H-Ras induces an inactive conformation due to disturbing homo-oligomerization of ASK1. And it is also possible
that membrane localization of ASK1 by H-Ras binding will lead to phosphorylation or dephosphorylation of some amino residue(s) on kinases, which might disrupt the interaction of ASK1 with
its effectors, causing inactivation of ASK1. Further studies are needed to clarify the exact mechanism of how the activity of ASK1 is inhibited through its physical interaction with H-Ras.
REFERENCES * Chang L, Karin M . Mammalian MAP kinase signalling cascades. _Nature_ 2001; 410(6824):37–40. Article CAS Google Scholar * Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ,
Der CJ . Increasing complexity of Ras signaling. _ Oncogene_ 1998; 17(11):1395–413. Article CAS Google Scholar * Bonni A, Brunet A, West AE, et al. Cell survival promoted by the Ras-MAPK
signaling pathway by transcription-dependent and -independent mechanisms. _Science_ 286(5443):1358–62. Article CAS Google Scholar * Widmann C, Gibson S, Jarpe MB, Johnson GL .
Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. _Physiol Rev_ 1999; 79(1):143–80. Article CAS Google Scholar * English J, Pearson G,
Wilsbacher J, et al. New insights into the control of MAP kinase pathways. _Exp Cell Res_ 1999; 253(1):255–70. Article CAS Google Scholar * Ichijo H, Nishida E, Irie K, et al. Induction
of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. _Science_ 1997; 275(5296):90–4. Article CAS Google Scholar * Nishitoh H, Saitoh M, Mochida Y,
et al. ASK1 is essential for JNK/SAPK activation by TRAF2. _Mol Cell_ 1998; 2(3):389–95. Article CAS Google Scholar * Du J, Suzuki H, Nagase F, et al. Superoxide-mediated early oxidation
and activation of ASK1 are important for initiating methylglyoxal-induced apoptosis process. _Free Radic Biol Med_ 2001; 31(4):469–78. Article CAS Google Scholar * Chen Z, Seimiya H,
Naito M, et al. ASK1 mediates apoptotic cell death induced by genotoxic stress. _Oncogene_ 1999; 18(1):173–80. Article CAS Google Scholar * Wang TH, Wang HS, Ichijo H, et al.
Microtubule-interfering agents activate c-Jun N-terminal kinase/stress-activated protein kinase through both Ras and apoptosis signal-regulating kinase pathways. _J Biol Chem_ 1998;
273(9):4928–36. Article CAS Google Scholar * Chen J, Fujii K, Zhang L, Roberts T, Fu H . Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a
MEK-ERK independent mechanism. _Proc Natl Acad Sci USA_ 2001; 98(14):7783–8. Article CAS Google Scholar * Du J, Cai S, Suzuki H, et al. Involvement of MEKK1/ERK/P21Waf1/Cip1 signal
transduction pathway in inhibition of IGF-I-mediated cell growth response by methylglyoxal. _J Cell Biochem_ 2003; 88(6):1235–46. Article CAS Google Scholar * Santen RJ, Song RX,
McPherson R, et al. The role of mitogen-activated protein (MAP) kinase in breast cancer. _J Steroid Biochem Mol Biol_ 2002; 80(2):239–56. Article CAS Google Scholar * Galvan V, Logvinova
A, Sperandio S, et al. IGF-IR signaling inhibits apoptosis signal-regulating kinase 1 (ASK1). _J Biol Chem_ 2003; 278(15):13325–32. Article CAS Google Scholar * Liu H, Nishitoh H, Ichijo
H, Kyriakis JM . Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor associated factor 2 requires prior dissociation of the ASK1 inhibitor
thioredoxin. _Mol Cell Biol_ 2000; 20(6):2198–208. Article CAS Google Scholar * Saitoh, M . Nishitoh H, Fujii M, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis
signal-regulating kinase (ASK) 1. _EMBO J_ 1998; 17(9):2596–606. Article CAS Google Scholar * Liu Y, Yin G, Surapisitchat J, Berk BC, Min W . Laminar flow inhibits TNF-induced ASK1
activation by preventing dissociation of ASK1 from its inhibitor 14-3-3. _J Clin Invest_ 2001; 107(7):917–23. Article CAS Google Scholar * Zou X, Tsutsui T, Ray D, et al. The cell
cycle-regulatory CDC25A phosphatase inhibits apoptosis signal-regulating kinase 1. _Mol Cell Biol_ 2001; 21(14):4818–28. Article CAS Google Scholar Download references AUTHOR INFORMATION
AUTHORS AND AFFILIATIONS * Department of Medical Technology, Nagoya University School of Health Sciences, Aichi, 461-8673, Japan Jun DU & Fumihiko NAGASE * School of Pharmaceutical
Science, Sun Yat-Sen University, Guangzhou, 510089, China Jun DU * Department of Immunology, Nagoya University Graduate and Faculty School of Medicine, Aichi, 466-8550, Japan Shao Hui CAI
& Zhe SHI * College of Pharmacy, Jinan University, Guangzhou, 510632, China Shao Hui CAI Authors * Jun DU View author publications You can also search for this author inPubMed Google
Scholar * Shao Hui CAI View author publications You can also search for this author inPubMed Google Scholar * Zhe SHI View author publications You can also search for this author inPubMed
Google Scholar * Fumihiko NAGASE View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Jun DU. RIGHTS AND PERMISSIONS
Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE DU, J., CAI, S., SHI, Z. _et al._ Binding activity of H-Ras is necessary for _in vivo_ inhibition of ASK1 activity. _Cell Res_
14, 148–154 (2004). https://doi.org/10.1038/sj.cr.7290214 Download citation * Received: 23 June 2003 * Revised: 31 December 2003 * Accepted: 02 February 2004 * Issue Date: 01 April 2004 *
DOI: https://doi.org/10.1038/sj.cr.7290214 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * H-Ras * ASK1 * apoptosis * binding activity