Play all audios:
ABSTRACT In monogenic diseases, the presence of several sequence variations in the same allele may complicate our understanding of genotype–phenotype relationships. We described new
alterations identified in a cystic fibrosis (CF) patient harboring a 48C>G promoter sequence variation associated in _cis_ of a 3532AC>GTA mutation and in _trans_ with the F508del
mutation. Functional analyses including _in vitro_ experiments confirmed the deleterious effect of the 3532GTA frameshift mutation through the creation of a premature termination codon. The
analyses also revealed that the 48G promoter variant has a negative effect on both transcription and mRNA level, thus demonstrating the importance of analyzing all mutations or sequence
variations with potential impact on CF transmembrane conductance regulator processing, even when the two known disease-causing mutations have already been detected. Our results emphasize the
need to perform, wherever possible, functional studies that may greatly assist the interpretation of the disease-causing potential of rare mutation-associated sequence variations. SIMILAR
CONTENT BEING VIEWED BY OTHERS SPLICING MUTATIONS IN THE _CFTR_ GENE AS THERAPEUTIC TARGETS Article Open access 02 June 2022 CHARACTERISING THE LOSS-OF-FUNCTION IMPACT OF 5’ UNTRANSLATED
REGION VARIANTS IN 15,708 INDIVIDUALS Article Open access 27 May 2020 CFTR FUNCTION, PATHOLOGY AND PHARMACOLOGY AT SINGLE-MOLECULE RESOLUTION Article Open access 22 March 2023 INTRODUCTION
Cystic fibrosis (CF, MIM# 219700) is the most common lethal autosomal recessive disease affecting the Caucasian population, with an average prevalence of 1 in 3000 births. Birth prevalence
varies from country to country and with ethnic background.1 Although in its classical form, CF affects the physiology of several organs including the pancreas and the gastrointestinal and
reproductive tracts, the morbidity is mainly due to pulmonary defects.2 It is caused by mutations in the CF transmembrane conductance regulator (_CFTR_; MIM# 602421) gene, for which over
1850 sequence variations have been reported to date (http://www.genet.sickkids.on.ca/cftr/). The pathogenicity of several types of mutations such as nonsense, frameshift and splicing
mutations are obvious, whereas the interpretation of the molecular consequences of others, such as silent mutations3, 4 or those identified in non-coding regions,5, 6 is arduous. In
addition, the _CFTR_ gene is characterized by the presence of a number of complex alleles, that is, at least two mutations in _cis_ on the same chromosome that generally hinder the
establishment of genotype–phenotype correlations. Mutations found in _cis_ can modulate the effect of the principle mutation. The mutation R117H occurring in _cis_ with the 5-thymidine (5T)
tract variant in intron 8 generally results in pancreatic sufficient CF and as such, is considered as a mild mutation, whereas R117H in _cis_ with the 7T is mainly considered as a
CFTR-related disease-associated mutation with low penetrance.7 Complex alleles may also involve alterations in the _CFTR_ promoter region, as is the case for the (−102T>A;S549R)+(F508del)
genotype for which the promoter sequence variation is associated with an increase in _CFTR_ expression and a moderate clinical phenotype.8 Here we report the functional analysis of a
promoter variant associated in _cis_ with a frameshift mutation (48C>G;3532AC>GTA)+(F508del) identified in a patient with a classic form of CF as characterized by a positive sweat
test, pulmonary symptoms, digestive manifestations and pancreatic insufficiency. The two alterations 48C>G and 3532AC>GTA are rare, as they have been identified in only one patient and
none have been found to occur alone elsewhere. In our continuing efforts to assist in the interpretation of uncharacterized mutations, we assessed the contribution of each alteration
towards the disease phenotype. Although the molecular defect induced by the 3532AC>GTA mutation (frameshift mutation) seems clear, we also evaluated whether the 48C>G promoter sequence
variation modulates the effect of the 3532AC>GTA mutation. MATERIALS AND METHODS NOMENCLATURE OF MUTATIONS For convenience to readers, we used legacy nomenclature as previously reported
to the International Consortium Mutation Database (http://www.genet.sickkids.on.ca/cftr/). According to the Human Genome Variation Society nomenclature, the two described alterations
48C>G and 3532AC>GTA correspond to c.-85C>G and c.3400_3401delACinsGTA, respectively (GenBank NM_000492.3), where nucleotide number 1 corresponds to the A of the ATG translation
initiation codon (previously the A of the ATG was numbered 133). PLASMID CONSTRUCTS To study the impact of the mutations, several _CFTR_ constructions were created by direct mutagenesis
(QuickChange II site directed mutagenesis kit, Agilent, Massy, France). The 48C>G sequence variation was introduced either in the _CFTR_ promoter upstream of the _Luciferase_ cDNA (pGL3b
vector) or in the _CFTR_ promoter upstream of the wild-type (WT) _CFTR_ cDNA (pcDNA3-1 vector). The 3532AC>GTA was introduced in the _CFTR_ cDNA under the control of either the _CFTR_
promoter (WT or altered) or the CMV promoter in the pcDNA3-1 vector. The 3532AC>GTA was also introduced in a pSPL3-hybrid minigene containing WT _CFTR_ exon 18 and flanking introns (150
bp). All constructs were verified by sequencing. All the oligonucleotides used for generating the constructs are available on demand. TRANSFECTION ASSAYS The A549 pulmonary epithelial cell
line was grown in DMEM/F12, and the Cos7 simian fibroblast cell line in DMEM, at 37 °C under 5% CO2. Each medium was completed with 5% SVF (Eurobio, Courtaboeuf, France), 1% Ultroser G
(Pall, Saint Germain en Laye, France), 1% antibiotics (Invitrogen, Villebon sur Yvette, France) and 1% L-glutamine (Invitrogen). All transient transfections were realized with Polyfect
(Qiagen, Courtaboeuf, France) in 6-well plates (BD falcon, Le Pont de Claix, France), with 1500 ng of indicated vector. For the reporter assay, cells were transfected in 96-well plates (BD
falcon) using Fugene6 (Roche Applied Science, Meylan, France), with 60 ng of indicated reporter vector and 6 ng of pRL-SV40 (Renilla Luciferase) to normalize for transfection efficiency.
Samples were harvested 48 h after transfection. A stable A549 cell line, containing WT or 48G-altered _CFTR_ promoter was generated as previously described.6 PROTEIN TRUNCATION TEST In the
first step, the _CFTR_ fragment of interest was amplified from _CFTR_ cDNA, using specific primers with the addition of the T7 promoter at the 5′-end.9 To generate WT and mutated peptides
encompassing the 3532 position labeled with 35S-methionine, we used the TNT T7 Quick Coupled Transcription/Translation System (Promega, Charbonnieres, France). Produced peptides were then
separated by migration on a 12% denaturing acrylamide gel. After fixation and amplification, the signal was exposed on a Maximum Resolution film (Kodak, VWR, Pessac, France) during 12 h.
Molecular weights were calculated with compute pl/Mw in ExPASy tools (Expasy Bioinformatics Resource portal, “Compute pl/Mw tool”, http://web.expasy.org/compute_pi/). WESTERN BLOT Whole
protein extracts were directly collected in Laemmli buffer and sonicated before their loading on a 7% SDS-polyacrylamide gel. After migration, proteins were transferred onto a PVDF membrane
(Westran Clear Signal, Whatman, Dominique Dutscher, Issy les Moulineaux, France), which was blocked with 5% skimmed milk in PBS–Tween for 1 h. Membranes were incubated overnight with
anti-CFTR primary antibody (1:400, clone MM13-4 recognizing an N-terminus epitope, Millipore, Molsheim, France) or anti-Lamin A/C primary antibody (1:10 000, Upstate, Millipore). After 1 h
of incubation with the anti-mouse secondary antibody, proteins were revealed with Immobilon Western Chemiluminescent HRP Substrate (Millipore) on a chemiluminescence detection film (Kodak).
QUANTIFICATION OF TRANSCRIPT LEVEL Total mRNAs were purified with RNeasy plus Mini Kit (Qiagen) as recommended by the manufacturer. Genomic DNA was removed with DNase I amplification grade
(Invitrogen). After treatment with RNasin (Promega), RT-PCR was performed on 1 _μ_g of RNA with random primers and MML-V reverse transcriptase (Invitrogen). Quantitative PCR was realized
with 1:50 diluted cDNA and amplified with the LightCycler 480 SYBR Green I Master (Roche Applied Science). _CFTR_ cDNA was quantified and normalized by the quantification of _GAPDH_ and
_β-_actin housekeeping genes. Controls with no template or reverse transcriptase were also included. All PCR reactions were performed in triplicate in at least three independent experiments.
MINIGENE ASSAY Total mRNA extraction and RT-PCR were performed as described above for the quantification of transcript level. PCR was then performed using specific primers of pSPL3 plasmid
and PCR Master Mix (Promega). Fragments were separated on a 1.5% agarose gel. REPORTER ASSAY The experiment was performed as previously described.6 Firefly and Renilla Luciferase activities
were measured 48 h after transfection by the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's recommendations. Luciferase activities are representative of
at least three independent experiments, with each construct tested in triplicate per experiment. QUANTITATIVE CHROMATIN IMMUNOPRECIPITATION ASSAYS (Q-CHIP) Q-ChIP was performed as
previously described, except for minor modifications.6 Purified crosslinked chromatin was immunoprecipitated using 3 _μ_g of anti-E2F, anti-MZF1 (myeloid zinc finger 1) or anti-Sp1 (specific
protein 1) antibodies. As a control for non-specific binding of DNA, 3 _μ_g of anti-HA, anti-goat or anti-mouse antibodies were used. All antibodies were obtained from Santa Cruz,
Clinisciences (Montrouge, France), except for anti-HA, which was bought from Roche Applied Science. Input and immunoprecipitated DNAs were analyzed after their amplification on the
LightCycler 480 real-time PCR system using the LightCycler 480 Probes Master, specific primers for the _CFTR_ minimal promoter region encompassing the 48C>G sequence variation and probe
#62 (Roche Applied Science). All PCR reactions were performed in triplicate. Experiments were performed at least three times and expressed relative to the input signal and to non-specific
immunoprecipitated chromatin. STATISTICAL ANALYSES Data are expressed as the mean±SE. Paired comparisons were made using Student's _t_-test with InStat (GraphPad Software, version 3.0,
Instat 3 folder, San Diego, CA, USA). Data were considered statistically significant at _P_<0.05 and extremely significant at _P_<0.0001. RESULTS CONFIRMATION OF THE DELETERIOUS EFFECT
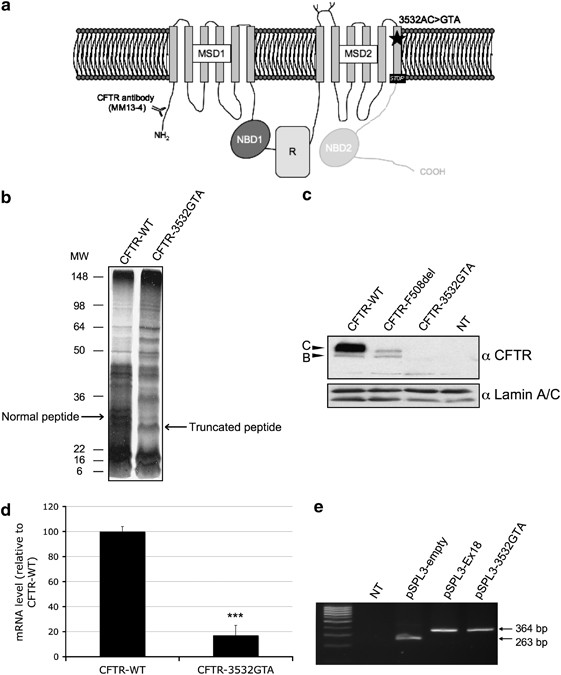
OF THE 3532AC>GTA FRAMESHIFT MUTATION We firstly hypothesized that the probable change in the open reading frame induced by the 3532GTA frameshift mutation generates a premature
termination codon (PTC; Figure 1a). We therefore performed a protein truncation test (PTT) by transcribing and translating _in vitro_ the WT and mutated _CFTR_ fragments. Results of
electrophoresis showed a shift in the molecular weight of the synthesized peptides between CFTR-WT and CFTR-3532GTA conditions (Figure 1b). The presence of a band with the expected molecular
weight confirmed the creation of a PTC, 21 codons after the mutation, at the end of the last transmembrane segment. To investigate further the several additional bands in the CFTR-3532GTA
compared with the WT conditions revealed by our PTT, we assessed the effect of the 3532GTA mutation on CFTR maturation by western blot. Total proteins from cells transfected with an
expression vector containing WT or mutated _CFTR_ cDNA, downstream of the CMV promoter, were used for immunoblot assay. Total proteins from cells transfected with the construction containing
WT _CFTR_ cDNA resulted in typical bands including band C (mature, fully glycosylated protein, 170 kDa) and band B (immature, core-glycosylated protein, 140 kDa; Figure 1c). As a control,
we used the F508del mutation, known to induce protein misconformation and in turn, its premature degradation. No bands were detected, however, for the 3532GTA mutation as in the
non-transfected condition, whereas protein was present, as confirmed using anti-Lamin A/C primary antibody. The mutated sequence therefore prevents protein synthesis, suggesting that
transcripts bearing this PTC are potential targets for degradation through nonsense-mediated mRNA decay (NMD) and/or for exon skipping through nonsense-associated alternative splicing (NAS).
We next assessed the impact of the 3532GTA allele on the mRNA level or splicing process. Evaluation of mRNA amounts revealed that the 3532GTA mutation induces a decrease in _CFTR_
transcript levels of more than 80%, compared with the WT condition in Cos-7 cells (Figure 1d). The same results were obtained in A549 lung-specific cells (data not shown). Because this
frameshift mutation might also produce both correctly and aberrantly spliced mRNA, we used a minigene assay to investigate whether the second control mechanism NAS could account for the
observed inhibited synthesis. Hybrid minigene assay showed no difference between WT and mutated contexts (Figure 1e). As alternative splicing events have been proposed to differ between cell
lines,10 we verified that the same results were obtained in the other tested cell line A549 (data not shown). These results indicate that degradation of the transcript bearing the PTC in
exon 18 is the result of NMD rather than NAS. CHARACTERIZATION OF THE PATHOGENICITY OF THE 48C>G PROMOTER SEQUENCE VARIATION To evaluate the effect of the 48G variant on transcriptional
regulation, we performed a reporter assay. Luciferase activity, reflecting _CFTR_ promoter control, indicated a decrease of about 40% in the 48G variant context compared with WT (Figure 2a).
We next assessed differences in _CFTR_ mRNA amounts and found that the 48G allele induces a decrease of 75% in the _CFTR_ transcript level when compared with the WT (Figure 2b). These
results suggest that the 48G promoter variant, in addition to its deleterious effect on transcription, may also disrupt the stability of the _CFTR_ transcript. When associated in _cis_ of
the 3532GTA frameshift mutation, the 48G variant did not, however, increase the impact of 3532GTA alone on the mRNA level. Finally, we investigated which regulatory motifs, encompassing the
48C>G promoter sequence variation, were involved. Bioinformatics analyses predicted a change in binding capability of E2F, MZF1 and Sp1 transcription factors. We therefore performed
Q-ChIP on WT or sequence-varied promoters, using specific anti-E2F, anti-MZF1 and anti-Sp1 antibodies. Results show that none of the three transcription factors bound _in vivo_ to the tested
region of the WT promoter (Figure 2c), whereas E2F, MZF1 and Sp1 proteins bound to the sequence-varied promoter with high affinity (14, 15 and 23-fold enrichment relative to input,
respectively), suggesting their putative involvement in the 48G promoter variant effect on transcriptional control. DISCUSSION Here we have described the contribution of each alteration
(48C>G and 3532AC>GTA) to CFTR function. The creation of a PTC with the 3532GTA frameshift mutation has been confirmed using PTT assay. This mutation was shown to have a deleterious
effect at both the mRNA and protein levels with results, suggesting the involvement of NMD in the recognition and degradation of aberrant transcripts. Although no band C was observed by
western blot, indicating 100% degradation, residual CFTR mRNA was obtained. Previous studies have shown the concomitant nature of the translation and folding of CFTR.11, 12 Our data suggest
the role of an additional quality control mechanism that rapidly directs the elimination of the truncated protein (lacking NBD2) by ER-quality control systems. We next explored the
functional impact of the 48C>G sequence variation found in the promoter in _cis_ of the frameshift mutation. Study of the 48G variant shows its deleterious effect at the transcriptional
and mRNA levels. This outcome could be, in part, explained by the higher affinity of the altered sequence for the three transcription factors E2F, MZF1 and Sp1. MZF1 is well known for its
role in regulating cell proliferation,13 but along with Sp1, it is also involved in regulating the expression of the human _MUC1_ gene, expressed in several epithelial tissues.14
Furthermore, MZF1 has previously been identified as a putative inhibitor of the _Cftr_ gene, through its binding in the −834 to −524 region of the mouse promoter.15 Here we have shown that
MZF1 only binds to the altered _CFTR_ allele (48G), suggesting its partial role in decreasing promoter activity. E2F transcription factors are largely involved in cell cycle regulation,16,
17 and along with MZF1, regulate genes that are developmentally regulated during cystogenesis.18 Moreover, E2F and Sp1 can control the initiation of transcription in TATA-less promoters19,
20 through their interaction with other proteins. For instance, E2F could superactivate Sp1-dependant transcription in _DHFR_ TATA-less promoter.21 Transcription factors, through their
binding to regulatory regions, either activate or repress the transcription of their target genes by promoting or inhibiting the recruitment of RNA polymerase II, through TSS use. Indeed,
E2F and Sp1 have been shown to interact with several proteins of the general transcription machinery, such as TFIIH and TFIID.22, 23, 24, 25 These data suggest that the binding of these
factors putatively modifies the use of the major TSS essential for _CFTR_ transcription during lung development.26, 27 The identification of alterations in non-coding regions and the
determination of the impact of each allele component have several important consequences for recessive disorders such as CF. They permit a better understanding of the complex
genotype–phenotype relationships and the opportunity to offer personalized genetic counseling or adapted therapy. These data reinforce the necessity to screen, wherever possible, for
additional alterations even when two allelic mutations known to be disease causing are found, and especially when a discrepancy between phenotype and genotype is observed. REFERENCES *
O’Sullivan BP, Freedman SD : Cystic fibrosis. _Lancet_ 2009; 373: 1891–1904. Article Google Scholar * Chmiel JF, Davis PB : State of the art: why do the lungs of patients with cystic
fibrosis become infected and why can’t they clear the infection? _Respir Res_ 2003; 4: 8. Article Google Scholar * Bartoszewski RA, Jablonsky M, Bartoszewska S _et al_: A synonymous single
nucleotide polymorphism in DeltaF508 CFTR alters the secondary structure of the mRNA and the expression of the mutant protein. _J Biol Chem_ 2010; 285: 28741–28748. Article CAS Google
Scholar * Pagani F, Raponi M, Baralle FE : Synonymous mutations in CFTR exon 12 affect splicing and are not neutral in evolution. _Proc Natl Acad Sci USA_ 2005; 102: 6368–6372. Article CAS
Google Scholar * Taulan M, Lopez E, Guittard C _et al_: First functional polymorphism in CFTR promoter that results in decreased transcriptional activity and Sp1/USF binding. _Biochem
Biophys Res Commun_ 2007; 361: 775–781. Article CAS Google Scholar * Lopez E, Viart V, Guittard C _et al_: Variants in CFTR untranslated regions are associated with congenital bilateral
absence of the vas deferens. _J Med Genet_ 2011; 48: 152–159. Article CAS Google Scholar * Kiesewetter S, Macek Jr M, Davis C _et al_: A mutation in CFTR produces different phenotypes
depending on chromosomal background. _Nat Genet_ 1993; 5: 274–278. Article CAS Google Scholar * Romey MC, Guittard C, Carles S, Demaille J, Claustres M, Ramsay M : First putative sequence
alterations in the minimal CFTR promoter region. _J Med Genet_ 1999; 36: 263–264. CAS PubMed PubMed Central Google Scholar * Romey MC, Tuffery S, Desgeorges M, Bienvenu T, Demaille J,
Claustres M : Transcript analysis of CFTR frameshift mutations in lymphocytes using the reverse transcription-polymerase chain reaction technique and the protein truncation test. _Hum Genet_
1996; 98: 328–332. Article CAS Google Scholar * Disset A, Michot C, Harris A, Buratti E, Claustres M, Tuffery-Giraud S : A T3 allele in the CFTR gene exacerbates exon 9 skipping in vas
deferens and epididymal cell lines and is associated with Congenital Bilateral Absence of Vas Deferens (CBAVD). _Hum Mutat_ 2005; 25: 72–81. Article CAS Google Scholar * Du K, Sharma M,
Lukacs GL : The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. _Nat Struct Mol Biol_ 2005; 12: 17–25. Article CAS
Google Scholar * Kleizen B, van Vlijmen T, de Jonge HR, Braakman I : Folding of CFTR is predominantly cotranslational. _Mol Cell_ 2005; 20: 277–287. Article CAS Google Scholar * Gaboli
M, Kotsi PA, Gurrieri C _et al_: Mzf1 controls cell proliferation and tumorigenesis. _Genes Dev_ 2001; 15: 1625–1630. Article CAS Google Scholar * Shiraga T, Winpenny JP, Carter EJ,
McCarthy VA, Hollingsworth MA, Harris A : MZF-1 and DbpA interact with DNase I hypersensitive sites that correlate with expression of the human MUC1 mucin gene. _Exp Cell Res_ 2005; 308:
41–52. Article CAS Google Scholar * Ulatowski LM, Whitmore KL, Romigh T, VanderWyden AS, Satinover SM, Drumm ML : Strain-specific variants of the mouse Cftr promoter region reveal
transcriptional regulatory elements. _Hum Mol Genet_ 2004; 13: 1933–1941. Article CAS Google Scholar * van den Heuvel S, Dyson NJ : Conserved functions of the pRB and E2F families. _Nat
Rev Mol Cell Biol_ 2008; 9: 713–724. Article CAS Google Scholar * Chen HZ, Tsai SY, Leone G : Emerging roles of E2Fs in cancer: an exit from cell cycle control. _Nat Rev Cancer_ 2009; 9:
785–797. Article CAS Google Scholar * Lantinga-van Leeuwen IS, Leonhard WN, Dauwerse H _et al_: Common regulatory elements in the polycystic kidney disease 1 and 2 promoter regions. _Eur
J Hum Genet_ 2005; 13: 649–659. Article CAS Google Scholar * Azizkhan JC, Jensen DE, Pierce AJ, Wade M : Transcription from TATA-less promoters. dihydrofolate reductase as a model. _Crit
Rev Eukaryot Gene Expr_ 1993; 3: 229–254. CAS PubMed Google Scholar * Li H, Liu H, Wang Z _et al_: The role of transcription factors Sp1 and YY1 in proximal promoter region in initiation
of transcription of the mu opioid receptor gene in human lymphocytes. _J Cell Biochem_ 2008; 104: 237–250. Article CAS Google Scholar * Lin SY, Black AR, Kostic D, Pajovic S, Hoover CN,
Azizkhan JC : Cell cycle-regulated association of E2F1 and Sp1 is related to their functional interaction. _Mol Cell Biol_ 1996; 16: 1668–1675. Article CAS Google Scholar * Gill G, Pascal
E, Tseng ZH, Tjian R : A glutamine-rich hydrophobic patch in transcription factor Sp1 contacts the dTAFII110 component of the Drosophila TFIID complex and mediates transcriptional
activation. _Proc Natl Acad Sci USA_ 1994; 91: 192–196. Article CAS Google Scholar * Pearson A, Greenblatt J : Modular organization of the E2F1 activation domain and its interaction with
general transcription factors TBP and TFIIH. _Oncogene_ 1997; 15: 2643–2658. Article CAS Google Scholar * Smale ST : Core promoters: active contributors to combinatorial gene regulation.
_Genes Dev_ 2001; 15: 2503–2508. Article CAS Google Scholar * Liu WL, Coleman RA, Ma E _et al_: Structures of three distinct activator-TFIID complexes. _Genes Dev_ 2009; 23: 1510–1521.
Article CAS Google Scholar * Riordan JR, Rommens JM, Kerem B _et al_: Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. _Science_ 1989; 245:
1066–1073. Article CAS Google Scholar * White NL, Higgins CF, Trezise AE : Tissue-specific _in vivo_ transcription start sites of the human and murine cystic fibrosis genes. _Hum Mol
Genet_ 1998; 7: 363–369. Article CAS Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by the French association Vaincre La Mucoviscidose, by PhD fellowships
grants to VV (G0804), the Hospital of Montpellier (Centre Hospitalo-Universitaire) and the Institut National de la Santé et de la Recherche Médicale. AUTHOR INFORMATION AUTHORS AND
AFFILIATIONS * INSERM U827, Laboratoire de Génétique de Maladies Rares, Montpellier, France Victoria Viart, Marie Des Georges, Mireille Claustres & Magali Taulan * Université Montpellier
I, UFR de Médecine, Montpellier, France Victoria Viart, Mireille Claustres & Magali Taulan * CHU Montpellier, Hôpital Arnaud de Villeneuve, Laboratoire de Génétique Moléculaire,
Montpellier, France Marie Des Georges & Mireille Claustres Authors * Victoria Viart View author publications You can also search for this author inPubMed Google Scholar * Marie Des
Georges View author publications You can also search for this author inPubMed Google Scholar * Mireille Claustres View author publications You can also search for this author inPubMed Google
Scholar * Magali Taulan View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Magali Taulan. ETHICS DECLARATIONS
COMPETING INTERESTS The authors declare no conflict of interest. RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Viart, V., Georges, M., Claustres, M.
_et al._ Functional analysis of a promoter variant identified in the _CFTR_ gene in _cis_ of a frameshift mutation. _Eur J Hum Genet_ 20, 180–184 (2012).
https://doi.org/10.1038/ejhg.2011.161 Download citation * Received: 20 April 2011 * Revised: 29 June 2011 * Accepted: 15 July 2011 * Published: 17 August 2011 * Issue Date: February 2012 *
DOI: https://doi.org/10.1038/ejhg.2011.161 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * CFTR * promoter sequence variation * frameshift
mutation * functional analysis