Play all audios:
ABSTRACT Amyotrophic lateral sclerosis (ALS) is a late-onset progressive neurodegenerative disease characterized by the loss of motor neurons in the spinal cord and brain. Mutations in Cu/Zn
superoxide dismutase 1 (SOD1) are known to induce ALS. Although many research models have been developed, the exact pathological mechanism of ALS remains unknown. The recently developed
induced pluripotent stem (iPS) cell technology is expected to illuminate the pathological mechanisms and new means of treatment for neurodegenerative diseases. To determine the pathological
mechanism of ALS, we generated mouse iPS (miPS) cells from experimental ALS transgenic mice and control mice and characterized the cells using molecular biological methods. The generated
miPS cells expressed many pluripotent genes and differentiated into three germ layers _in vitro_ and _in vivo_. Motor neurons derived from ALS-related miPS cells recapitulated the
pathological features of ALS. The ALS-model motor neurons showed SOD1 aggregates, as well as decreased cell survival rate and neurite length compared with wild-type motor neurons. Our study
will be helpful in revealing the mechanism of motor neuronal cell death in ALS. SIMILAR CONTENT BEING VIEWED BY OTHERS NEURONAL MITOCHONDRIAL DYSFUNCTION IN SPORADIC AMYOTROPHIC LATERAL
SCLEROSIS IS DEVELOPMENTALLY REGULATED Article Open access 23 September 2021 CELLULAR ANALYSIS OF SOD1 PROTEIN-AGGREGATION PROPENSITY AND TOXICITY: A CASE OF ALS WITH SLOW PROGRESSION
HARBORING HOMOZYGOUS _SOD1-D92G_ MUTATION Article Open access 25 July 2022 HETEROZYGOUS KNOCKOUT OF SYNAPTOTAGMIN13 PHENOCOPIES ALS FEATURES AND TP53 ACTIVATION IN HUMAN MOTOR NEURONS
Article Open access 03 August 2024 INTRODUCTION Amyotrophic lateral sclerosis (ALS) is a late-onset neurodegenerative disease characterized by the loss of motor neurons in the spinal cord
and brain.1 Progressive paralysis of voluntary muscles and progressive spread of symptoms are typical features of ALS. Respiratory failure with denervation of the respiratory muscles and
diaphragm is the last symptom in ALS. Most cases of ALS (~90%) are sporadic, and the remaining cases (~10%) are familial.2 Mutations of Cu/Zn superoxide dismutase 1 (SOD1) are related to the
development of ~20% of familial ALS cases.3 SOD1 is a predominantly cytoplasmic protein that consists of 153 amino acids. SOD1 converts superoxide anion to hydrogen peroxide to protect
cells. It was reported that while SOD1 null mice do not develop motor neuron death, mutant SOD1 transgenic mice recapitulate ALS symptoms.4 It is thought that mutant SOD1 induces cell death
by a gain of function, although the precise pathologic mechanism remains unknown. There are many theories about the cause of motor neuronal cell death in ALS. These include genetic factors,5
oxidative stress,6 mitochondrial dysfunction,7 ER stress,8 excitotoxicity,9 proteasome inhibition,10 axonal transport defeat,11 dysregulation of RNA processing12 and formation of protein
aggregates.13 ALS transgenic mice carrying the human mutant _SOD1 (G93A)_ gene provide a common research model for ALS.14 These mice present a pathology similar to that of human ALS
patients, such as motor neuronal loss in the brain and spinal cord, the presence of aggregates, inflammation and death.15 In particular, these mice present hind limb weakness and tremor
around postnatal day 90 and then die at approximately postnatal day 120. Degenerative processes in the motor neurons are observed in the early stages of the development of symptoms, and
degeneration of neuromuscular junctions may precede the loss of motor neurons. Pathological characteristics, such as mitochondrial vacuolization, Golgi fragmentation or
neurofilament-positive inclusions, are present in the motor neurons of ALS transgenic mice. Motor neurons of these mice are also affected by inflammation that causes astrocytosis and
microgliosis. Recently, somatic reprogramming technology was used to produce induced pluripotent stem (iPS) cells by applying four pluripotent genes, namely, Oct4, Sox2, Klf4 and c-Myc.16
Researchers discovered these key pluripotent genes using differentiated mouse embryonic fibroblasts and tested the expression of these genes and the differentiation ability of iPS cells.
There are many advantages to using iPS cells. For example, they are easy to create, can be applied in patient-specific cell therapy and research, and require no special ethical
considerations. In particular, iPS cells are expected to help identify drugs for the treatment of patients with neurodegenerative disease.17 For these reasons, many iPS cell lines have been
produced, using human or animal models, for research on ALS.18, 19 In the present study, we report pathological differences between iPS cell-derived motor neurons from ALS mice and those
from control mice; these differences include neural dendrites, aggregates and cell death. Our results demonstrate that motor neurons derived from ALS-related mouse iPS cells recapitulate the
pathological features of ALS. MATERIALS AND METHODS ANIMALS ALS transgenic mice expressing the human mutant _SOD1 (G93A)_ gene (B6SJL-Tg[SOD1-G93A]1Gur/J) and their non-transgenic
littermates (B6SJLF1/J)—the latter used as controls—were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). All mouse care and experiments were agreed upon by the Institutional
Animal Care and Use Committee of Korea University. TAIL-TIP FIBROBLAST CULTURE FROM MOUSE Tail-tip fibroblasts (TTFs) were prepared from the transgenic and control mice as previously
described.20 The TTFs were maintained in Dulbecco’s modified minimal essential medium (DMEM; Gibco, Life Technologies, Grand Island, NY, USA) supplemented with 10% FBS (Gibco). RETROVIRAL
PRODUCTION AND TITRATION Retroviral production and titration were conducted as described elsewhere.21 GENERATION OF MOUSE IPS CELLS To generate the mouse iPS cells, 5 × 104 mouse TTFs were
seeded on a six-well tissue culture plate, and retroviral infection was performed for 3 days on the mouse TTFs with polybrene (4 μg ml−1). After 3 days, the infected TTFs were transferred to
Mouse Embryonic Fibroblast (MEF) feeder cells to produce mouse iPS cells with mouse iPS medium (DMEM containing 10% horse serum (Sigma-Aldrich, St Louis, MO, USA), 2 mM L-glutamine (Gibco),
0.1 mM MEM NEAA (Gibco), 10 mM HEPES (Gibco), 10 mM β-mercaptoethanol (Gibco), 500 U ml−1 LIF (Millipore, Billerica, MA, USA) and penicillin/streptomycin (Gibco)). ALKALINE PHOSPHATASE
STAINING For the alkaline phosphatase staining, we used the Alkaline Phosphatase Staining Kit (Stemgent, Lexington, MA, USA) according to the manufacturer’s protocol. RT-PCR Total RNA was
extracted from the established mouse iPS cells with the easy-Blue Total RNA Extraction Kit (Intron, Seongnam, Korea) and then reverse-transcribed into first-strand cDNA using the RevertAid H
Minus First Strand cDNA Synthesis Kit (Fermentas, Thermo Fischer Scientific, Carlsbad, CA, USA). The primers against each of the pluripotent genes were described.20 IMMUNOFLUORESCENCE The
mouse iPS colonies were fixed with 4% formaldehyde for 15 min and permeabilized with 0.1% Triton X-100. After incubation with 2% BSA, the cells were incubated overnight at 4 °C with a
primary antibody against Oct4 (Abcam, Cambridge, MA, USA), Sox2 (Millipore), Nanog (Millipore) and SSEA-1 (Santa Cruz, Dallas, TX, USA). The cells were then washed three times in
phosphate-buffered saline and incubated with Alexa Fluor 488 and 594 (Life Technologies, Thermo Fisher Scientific, Carlsbad, CA, USA) for 1 h at room temperature. After washing, the nuclei
were stained with DAPI (Sigma-Aldrich). The stained mouse iPS colonies were mounted on glass slides using Fluorescent Mounting Medium (Dako, Agilent Technologies, Denmark). The fluorescent
images were captured using a LSM-700 confocal microscope (Carl Zeiss, Oberkochen, Germany). _IN VITRO_ DIFFERENTIATION Mouse iPS cells were directly differentiated into three germ layers on
a gelatin-coated dish for 10 days in a medium without a leukemia inhibitory factor. TERATOMA FORMATION For the teratoma formation, we harvested mouse iPS cells and subcutaneously injected 1
× 106 mouse iPS cells into SCID mice. After 6 weeks, the formed teratomas were analyzed by hematoxylin and eosin (H&E) staining. BISULFITE GENOMIC DNA SEQUENCING Genomic DNA was isolated
from mouse embryonic stem cells (mESs), MEFs and miPS cells using the QIAmp DNA Mini Kit (Qiagen, Valencia, CA, USA) for bisulfite modification. Bisulfite modification was performed using a
CpGenome Modification Kit (Millipore) according to the manufacturer’s protocol. Modified genomic DNA was amplified by PCR using a mouse Nanog gene promoter primer.16 The amplified PCR
products were cloned into a T&A Cloning Vector Kit (RBC, New Taipei City, Taiwan) and sequenced using the BIQ Analyzer software (Max Planck Institute, Saarbrucken, Germany). MOTOR NEURON
DIFFERENTIATION OF MIPS CELLS For the motor neuron differentiation, a procedure modified from a previous study was used.22 First, embryoid bodies (EBs) were formed in a suspension culture
for 4 days using an EB medium (DMEM containing 10% FBS (Gibco), 2 mM L-glutamine (Gibco), 0.1 mM MEM NEAA (Gibco), 10 mM HEPES (Gibco), 10 mM β-mercaptoethanol (Gibco) and
penicillin/streptomycin (Gibco)). After 4 days, the EBs were cultured for ~10 days in a tissue culture dish using an insulin-transferrin-selenium (ITS) medium (DMEM/F12 (Gibco) containing
ITS supplement (Gibco) and penicillin/streptomycin (Gibco)) to select neural precursor cells. The ITS medium was replaced every 2 or 3 days. To promote the proliferation of neural precursor
cells, selected neural precursor cells were detached by Trypsin-EDTA and maintained in an N2 medium (DMEM/F12 (Gibco) containing N2 supplement (Gibco) and penicillin/streptomycin (Gibco))
with bFGF (10 ng ml−1). Enriched neural precursor cells were differentiated into motor neurons using retinoic acid (1 μM) and Shh (200 ng ml−1) in an N2 medium. After differentiation, the
motor neurons were cultured in an N2 medium containing ascorbic acid for their maturation and survival. TUNEL ASSAY For the terminal deoxynucleotidyl transferase dUTP nick end labeling
(TUNEL) assay, the DeadEnd Fluorometric TUNEL System Kit (Promega, Madison, WI, USA) was used according to the manufacturer’s protocol. MEASUREMENT OF NEURITE LENGTH To measure the neurites
of differentiated motor neurons, differentiated motor neurons were immunostained with Tuj1 and Islet-1 antibodies. As Tuj1 was expressed in the cytosol, we measured neurite length by
immunostaining Tuj1 from cell body to end of neurite in co-immunostained motor neurons. The length of the neurite was calculated using the measurement function (using ‘Open Bezier’) of the
ZEN analysis program (Carl Zeiss, Oberkochen, Germany). STATISTICAL ANALYSIS The differences between the various experimental groups were calculated using Student’s two-tailed _t_-test.
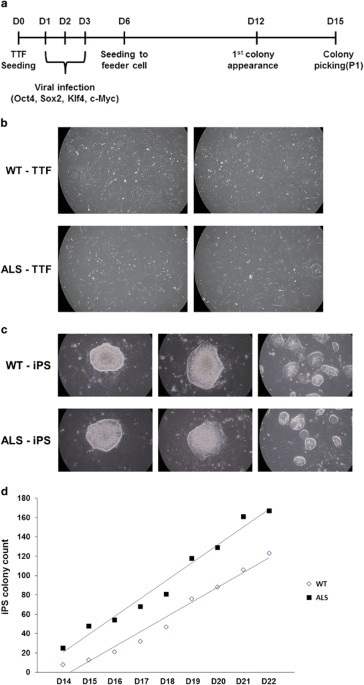
_P_-values of <0.05 were considered to be statistically significant. RESULTS ESTABLISHMENT OF MIPS CELLS FROM ALS-RELATED TRANSGENIC MICE TTFS We produced mouse iPS cells from primary
mouse TTFs derived from ALS transgenic mice and control mice. First, we obtained fibroblasts from mouse tail-tip tissue and cultured them for 2 weeks for sufficient viral infection (Figure
1b). We reprogrammed the mouse TTFs by infection with four pluripotent genes (Oct4, Sox2, Klf4 and c-Myc) using a retrovirus for 3 days. We then seeded the infected TTFs with MEF feeder
cells to maintain the generation of miPS cells. After 6 days, the first miPS colony was observed in the infected ALS-TTFs (Figure 1a). On day 15 (D15), we picked multiple miPS colonies and
transferred them to other MEF feeder cells (Figure 1c). Next, miPS colonies were counted during D14 to D22 to measure the differences between WT-iPS and ALS-iPS cell lines. Although there
were a few differences in the total number of colonies on a single day, the rate of increase between WT- and ALS-iPS cells was almost equal during the counting period (WT-iPS; 18 and
ALS-iPS; 15 colonies per day) (Figure 1d). Thus, we can conclude that the mutant SOD1 gene did not affect the production of miPS cells. ESTABLISHED MIPS CELLS FROM ALS-MODEL MICE HAD
PLURIPOTENT PROPERTIES Many molecular biological experiments were conducted to characterize the established WT-iPS and ALS-iPS cell lines. To evaluate the iPS properties of these established
miPS cells, we investigated alkaline phosphatase activity (Figure 2a). Alkaline phosphatase staining experiments revealed that the WT-iPS and ALS-iPS cell lines were positive for alkaline
phosphatase activity. Next, we analyzed the mRNA expression of pluripotent genes, such as Ecat1, Nanog, Gdf3, Oct4, Sox2, Rex1 and Zfp296 by RT-PCR (Figure 2b). All cell lines of WT-iPS and
ALS-iPS expressed these genes. Moreover, all cell lines showed immunostaining for pluripotent markers, such as Oct4, Sox2, Nanog and SSEA-1 (Figures 2c and d). To test the _in
vitro_-differentiation ability of the established miPS cells, WT-iPS and ALS-iPS were differentiated in a LIF-free medium. The differentiated WT-iPS and ALS-iPS cells were immunostained with
Tuj-1 (ectoderm), AFP (endoderm) and Desmin (mesoderm) antibodies. The results demonstrate that the established miPS cells differentiated to three germ layers _in vitro_ (Figure 2e).
Furthermore, we also tested the _in vivo_-differentiation potential of the established miPS cells by observing teratoma formation. Teratomas formed within 6 weeks of the injection of the
miPS cells. The teratomas were analyzed by H&E staining (Figure 2f). The H&E staining revealed that the teratomas included three germ layers: epidermis and neural tissue (ectoderm),
muscle and blood (mesoderm), and intestine and gland (endoderm). These results demonstrate that the established miPS cells have the potential to differentiate into three germ layers _in
vivo_. Finally, we investigated epigenetic changes in the Nanog promoter in the established miPS cells (Figure 2g). Bisulfite sequencing analysis showed that the methylation patterns of
WT-iPS and ALS-iPS were similar to the mES cell pattern. Taken together, the results demonstrate that the established miPS had properties of general iPS cells and that mutant SOD1 gene did
not affect pluripotency, differentiation ability and epigenetic change. ALS-IPS CELLS DIFFERENTIATED INTO MOTOR NEURONS _IN VITRO_ Because the features of ALS were confirmed in motor
neurons, we differentiated WT-iPS and ALS-iPS cells into motor neurons using the five-stage differentiation method with minor modifications (Figure 3a). First, we induced EB formation to
differentiate the miPS cells without LIF for 4 days. Well-shaped EBs were observed in the culture dish with ITS medium for 10 days. Selected neural precursor cells were cultured with bFGF to
promote the proliferation of neural precursor cells. Finally, enriched neural precursor cells were differentiated into motor neurons with RA and Shh. We monitored all of the differentiation
steps by observing the differentiating cells (Figure 3b). To test the differentiation of the motor neurons, Islet-1 and Tuj-1 immunostaining experiments were performed. Figure 3c shows
successfully differentiated motor neurons. Moreover, other motor neuron markers, such as HB9 and ChAT, were also observed in the differentiated motor neurons (Figure 3d). We measured the
Islet-1 positive/Tuj-1 positive cells to evaluate the differentiation rate of neural precursor cells into motor neurons. Approximately 86.1 and 87.3% of the neural precursor cells
differentiated into motor neurons among the WT- and ALS-iPS cells, respectively (Figure 3e). Therefore, we can conclude that there was no difference in the motor neuron differentiation
between WT- and ALS-iPS cells and that the mutant SOD1 gene does not affect motor neuron differentiation. ALS-MOTOR NEURONS RECAPITULATE PATHOLOGICAL FEATURES OF ALS Considering that a
decrease in motor neuron neurite length is one of the major characteristics of ALS pathology, we evaluated the motor neuron neurite lengths of induced WT- and ALS-MNs. We immunostained the
cells with Islet-1 and Tuj-1 antibodies to distinguish motor neurons from other cells. To estimate the differences in neurite length between the induced WT- and ALS-MNs, we selected Islet-1
and Tuj-1 double-positive cells and measured neurite lengths using the Tuj-1 immunostaining (Figure 4a). A week after differentiation, the relative neurite length of ALS-MNs (0.68) decreased
more than the neurites of WT-MNs (1) (Figure 4b). Moreover, 3 weeks after differentiation, the relative neurite length of ALS-MNs (0.56) decreased to a greater extent (Figure 4c). These
results demonstrate that relative neurite lengths of ALS-motor neurons continuously decrease in the process of motor neuron maturation. The mutant SOD1 aggregate is the principal
pathological feature of ALS. Intracellular SOD1 aggregates were observed in ALS-MNs (Figure 4d), showing that the mutant SOD1 gene affects the physiology of the motor neuron state. Taken
together, these results let us conclude that the induced ALS-motor neurons recapitulate the pathological features of ALS. CELL SURVIVAL OF ALS MOTOR NEURONS IS DECREASED The most important
characteristic of ALS is death of motor neurons. We investigated motor neuron death using the TUNEL assay. We counted cells that were double positive for Islet-1 and TUNEL to distinguish
motor neuron cells and determine motor neuronal death (Figure 5a). The results showed that the cell survival rate of ALS-MNs was ~47.9% compared with that of WT-MNs (Figure 5b). Furthermore,
we assessed cell death in the iPS stage using the TUNEL assay (Figure 5c). There were 3.4 and 4.1 dead cells per iPS colony in the WT- and ALS-iPS colonies, respectively; thus, WT- and
ALS-iPS colonies appeared to have similar proportions of cell death (Figure 5d). Thus, we can conclude that induced ALS-MNs and WT-MNs did not show any difference of cell death rate in the
embryonic stage, but showed a drastic difference in the motor neuron stage. DISCUSSION The pathological mechanism and medical treatment of many neurodegenerative diseases, including ALS, are
uncertain. Because neurodegenerative diseases have a late onset, the investigation of their causes and processes is very difficult. Although many animal models have been developed, there is
no system for studying the development of ALS from the embryonic to the adult stage. The use of iPS cells has many advantages in neurodegenerative disease research, such as avoiding ethical
considerations and providing patient-specific therapy.17 In the present study, we used iPS cells derived from a transgenic mouse model to reveal the pathological mechanism of ALS. The
results of our study will be helpful in revealing the mechanism of motor neuronal cell death in ALS. We produced mouse iPS cells using TTFs derived from ALS transgenic mice (Figure 1). The
generated miPS cells developed a rounded and brightened appearance on the MEF feeder cells. In particular, we investigated the number of miPS colonies produced after viral infection to
reveal the relationship between the transduced mutant SOD1 gene and miPS-cell production. The results demonstrate that overexpressed mutant SOD1 did not affect the production of the miPS
cells. Moreover, the characterization of the miPS cells suggests that overexpressed mutant SOD1 does not affect the miPS cells. No differences were found between WT-iPS and ALS-iPS cells in
alkaline phosphatase staining, RT-PCR, immunostaining, _in vitro_ differentiation, or _in vivo_ differentiation (Figure 2). Therefore, we can assume that the pathological symptoms of ALS
would not occur in the embryonic stage. To elucidate the differences in the developmental stage of the ALS model, we used a five-stage method to differentiate motor neuron cells from the
miPS cells (Figure 3). We divided motor neuron differentiation into the iPS stage, the neural precursor stage, and the motor neuron stage, similar to that of embryonic development _in vivo_.
Studies of motor neuron differentiation showed that the mutant SOD1 gene did not affect the rate of differentiation of neural precursor cells into motor neurons derived from ALS-iPS. The
major pathology of ALS includes motor neuronal cell death, a decrease in motor neuron neurites, and the presence of mutant SOD1 aggregates.23, 24, 25 In previous studies, these ALS
pathologies were presented in G93A SOD1 transgenic mice, an ALS model. Therefore, we investigated WT- and ALS-motor neurons derived from the miPS cells to confirm whether these pathologies
were reproducible (Figure 4). In this study, the length of ALS-MN neurites decreased more than the neurites from the WT-MNs, and mutant SOD1 aggregates were present in ALS-MNs but not in
WT-MNs. Moreover, the cell survival rate of ALS-MNs also decreased (Figure 5). In particular, the difference in the cell death rate between WT and ALS was present in the motor neuron stage.
Thus, ALS-motor neurons derived from the miPS cells recapitulated various pathologies found in the G93A transgenic mouse model of ALS. Although G93A mutant SOD1 retains native enzyme
activity, oxidative stress may accelerate the aggregation of G93A SOD1.26 Since SOD1 is an anti-oxidant enzyme and converts superoxide to hydrogen peroxide, G93A SOD1 suffers from oxidative
damage more easily than other proteins.6 Moreover, oxidation of SOD1 is caused by hydrogen peroxide, and mutant SOD1 has a more increased affinity for hydrogen peroxide than wild-type
SOD1.27, 28 Thus, oxidized G93A SOD1 proteins have conformational changes that are strongly prone to becoming insoluble aggregates.29 These insoluble aggregates may be the reason for the
fragmented neural fibers observed in the differentiated motor neurons from ALS-iPS cells (Figure 4d). In a previous study,30 researchers generated motor neurons from ALS transgenic
mouse-derived miPS cells, but they did not use the miPS-derived motor neurons in further studies. In the present study, we carried out functional studies of the properties of miPS-derived
motor neurons, showing that miPS cells recapitulate various pathologies in the ALS model. Therefore, ALS-MNs derived from miPS cells could provide an effective model for drug screening or
experimental subjects, although our study was limited to molecular biological research. Further studies should investigate genetic changes of iPS cells and motor neurons. Our study results
imply that motor neurons derived from miPS cells could have many advantages for the study of the molecular pathogenesis of other neurodegenerative diseases. REFERENCES * Mulder DW . Clinical
limits of amyotrophic lateral sclerosis. _Adv Neurol_ 1982; 36: 15–22. CAS PubMed Google Scholar * Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ . Molecular pathways of motor
neuron injury in amyotrophic lateral sclerosis. _Nat Rev Neurol_ 2011; 7: 616–630. Article CAS PubMed Google Scholar * Andersen PM, Sims KB, Xin WW, Kiely R, O’Neill G, Ravits J _et al_.
Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: a decade of discoveries, defects and disputes. _Amyotroph Lateral Scler Other Motor Neuron
Disord_ 2003; 4: 62–73. Article CAS PubMed Google Scholar * Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF _et al_. Motor neurons in Cu/Zn superoxide
dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. _Nat Genet_ 1996; 13: 43–47. Article CAS PubMed Google Scholar * Rosen DR . Mutations in
Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. _Nature_ 1993; 364: 362. Article CAS PubMed Google Scholar * Andrus PK, Fleck TJ, Gurney ME,
Hall ED . Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. _J Neurochem_ 1998; 71: 2041–2048. Article CAS PubMed Google Scholar * Liu J,
Lillo C, Jonsson PA, Vande Velde C, Ward CM, Miller TM _et al_. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. _Neuron_ 2004; 43: 5–17.
Article CAS PubMed Google Scholar * Nishitoh H, Kadowaki H, Nagai A, Maruyama T, Yokota T, Fukutomi H _et al_. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron
death by targeting Derlin-1. _Genes Dev_ 2008; 22: 1451–1464. Article CAS PubMed PubMed Central Google Scholar * Van Damme P, Dewil M, Robberecht W, Van Den Bosch L . Excitotoxicity and
amyotrophic lateral sclerosis. _Neurodegener Dis_ 2005; 2: 147–159. Article CAS PubMed Google Scholar * Cheroni C, Marino M, Tortarolo M, Veglianese P, De Biasi S, Fontana E _et al_.
Functional alterations of the ubiquitin-proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis. _Hum Mol Genet_ 2009; 18: 82–96. Article CAS PubMed
Google Scholar * Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G . Deficits in axonal transport precede ALS symptoms _in vivo_. _Proc Natl Acad Sci USA_ 2010; 107:
20523–20528. Article CAS PubMed PubMed Central Google Scholar * Kirby J, Halligan E, Baptista MJ, Allen S, Heath PR, Holden H _et al_. Mutant SOD1 alters the motor neuronal
transcriptome: implications for familial ALS. _Brain_ 2005; 128 (Pt 7): 1686–1706. Article PubMed Google Scholar * Johnston JA, Dalton MJ, Gurney ME, Kopito RR . Formation of high
molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. _Proc Natl Acad Sci USA_ 2000; 97: 12571–12576. Article CAS
PubMed PubMed Central Google Scholar * Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD _et al_. Motor neuron degeneration in mice that express a human Cu,Zn superoxide
dismutase mutation. _Science_ 1994; 264: 1772–1775. Article CAS PubMed Google Scholar * Turner BJ, Talbot K . Transgenics, toxicity and therapeutics in rodent models of mutant
SOD1-mediated familial ALS. _Prog Neurobiol_ 2008; 85: 94–134. Article CAS PubMed Google Scholar * Takahashi K, Yamanaka S . Induction of pluripotent stem cells from mouse embryonic and
adult fibroblast cultures by defined factors. _Cell_ 2006; 126: 663–676. Article CAS PubMed Google Scholar * Robinton DA, Daley GQ . The promise of induced pluripotent stem cells in
research and therapy. _Nature_ 2012; 481: 295–305. Article CAS PubMed PubMed Central Google Scholar * Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W _et al_.
Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. _Science_ 2008; 321: 1218–1221. Article CAS PubMed Google Scholar * Bilican B,
Serio A, Barmada SJ, Nishimura AL, Sullivan GJ, Carrasco M _et al_. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific
vulnerability. _Proc Natl Acad Sci USA_ 2012; 109: 5803–5808. Article CAS PubMed PubMed Central Google Scholar * Takahashi K, Okita K, Nakagawa M, Yamanaka S . Induction of pluripotent
stem cells from fibroblast cultures. _Nat Protoc_ 2007; 2: 3081–3089. Article CAS PubMed Google Scholar * Park HS, Hwang I, Choi KA, Jeong H, Lee JY, Hong S . Generation of induced
pluripotent stem cells without genetic defects by small molecules. _Biomaterials_ 2015; 39: 47–58. Article CAS PubMed Google Scholar * Okabe S, Forsberg-Nilsson K, Spiro AC, Segal M,
McKay RD . Development of neuronal precursor cells and functional postmitotic neurons from embryonic stem cells _in vitro_. _Mech Dev_ 1996; 59: 89–102. Article CAS PubMed Google Scholar
* Ince PG, Tomkins J, Slade JY, Thatcher NM, Shaw PJ . Amyotrophic lateral sclerosis associated with genetic abnormalities in the gene encoding Cu/Zn superoxide dismutase: molecular
pathology of five new cases, and comparison with previous reports and 73 sporadic cases of ALS. _J Neuropathol Exp Neurol_ 1998; 57: 895–904. Article CAS PubMed Google Scholar *
Karumbayaram S, Kelly TK, Paucar AA, Roe AJ, Umbach JA, Charles A _et al_. Human embryonic stem cell-derived motor neurons expressing SOD1 mutants exhibit typical signs of motor neuron
degeneration linked to ALS. _Dis Model Mech_ 2009; 2: 189–195. Article CAS PubMed PubMed Central Google Scholar * Chen H, Qian K, Du Z, Cao J, Petersen A, Liu H _et al_. Modeling ALS
with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. _Cell Stem Cell_ 2014; 14: 796–809. Article CAS PubMed PubMed Central Google Scholar * Rakhit R,
Cunningham P, Furtos-Matei A, Dahan S, Qi XF, Crow JP _et al_. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis.
_J Biol Chem_ 2002; 277: 47551–47556. Article CAS PubMed Google Scholar * Yim HS, Kang JH, Chock PB, Stadtman ER, Yim MB . A familial amyotrophic lateral sclerosis-associated A4V Cu,
Zn-superoxide dismutase mutant has a lower Km for hydrogen peroxide. Correlation between clinical severity and the Km value. _J Biol Chem_ 1997; 272: 8861–8863. Article CAS PubMed Google
Scholar * Wiedau-Pazos M, Goto JJ, Rabizadeh S, Gralla EB, Roe JA, Lee MK _et al_. Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. _Science_ 1996; 271:
515–518. Article CAS PubMed Google Scholar * Valentine JS, Hart PJ . Misfolded CuZnSOD and amyotrophic lateral sclerosis. _Proc Natl Acad Sci USA_ 2003; 100: 3617–3622. Article CAS
PubMed PubMed Central Google Scholar * Yao XL, Ye CH, Liu Q, Wan JB, Zhen J, Xiang AP _et al_. Motoneuron differentiation of induced pluripotent stem cells from SOD1G93A mice. _PLoS ONE_
2013; 8: e64720. Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by the Korea Healthcare Technology R&D Project,
Ministry for Health, Welfare & Family Affairs, Republic of Korea (no. A120340), a grant from the National R&D Program for Cancer Control, the Ministry of Health & Welfare,
Republic of Korea (no. 1320010), the National Research Foundation of Korea Grants, the Ministry of Science, ICT and Future Planning, Republic of Korea (NRF-2015R1A4A1041919) and the National
Research Foundation of Korea (NRF) grant (MEST) (NRF-2015R1A2A2A01003516). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Division of Life Sciences, College of Life Sciences and
Biotechnology, Korea University, Seoul, Republic of Korea Ju-Hwang Park & Seongman Kang * Department of Integrated Biomedical and Life Science, College of Health Science, Korea
University, Seoul, Republic of Korea Hang-Soo Park & Sunghoi Hong Authors * Ju-Hwang Park View author publications You can also search for this author inPubMed Google Scholar * Hang-Soo
Park View author publications You can also search for this author inPubMed Google Scholar * Sunghoi Hong View author publications You can also search for this author inPubMed Google Scholar
* Seongman Kang View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Seongman Kang. ETHICS DECLARATIONS COMPETING
INTERESTS The authors declare no conflict of interest. RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not
included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit
http://creativecommons.org/licenses/by-nc-sa/4.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Park, JH., Park, HS., Hong, S. _et al._ Motor neurons derived from ALS-related
mouse iPS cells recapitulate pathological features of ALS. _Exp Mol Med_ 48, e276 (2016). https://doi.org/10.1038/emm.2016.113 Download citation * Received: 07 March 2016 * Revised: 06 July
2016 * Accepted: 01 August 2016 * Published: 09 December 2016 * Issue Date: December 2016 * DOI: https://doi.org/10.1038/emm.2016.113 SHARE THIS ARTICLE Anyone you share the following link
with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative