Play all audios:
The widespread use of thin films in a range of applications and industries, from coatings, inks and lithography to nano-imprinting, optoelectronics and memory devices,1 has made the
understanding of thin films, particularly the changes induced by structural relaxation and solvent evaporation, very important. There is a need to know whether a film will change in
dimensions after its deposition and how fast these changes will occur. Physical aging of thin polymer systems is, more generally, an important phenomenon in polymer physics: when a polymer
is cooled quickly below its glass transition temperature (_T_g), at which point freezing of the matrix takes place,2 it is in a non-equilibrium state. When it is aged at temperatures below
_T_g, a structural relaxation toward the equilibrium volume can take place.2, 3 (The equilibrium volume represents the volume obtained through infinitely slow cooling.) Recent works have
shown that the structure, wetting and mobility of polymers in thin films can differ greatly from those of the bulk.3, 4, 5 For example, a well-documented property of thin polymer films that
exhibits thickness dependence is its _T_g.6, 7 In many applications, thin films are prepared by depositing a dilute solution of polymer onto a substrate; thereafter, the substrate is spun at
high velocity, at which the solution spreads, thereby ejecting any excess off the substrate.8 Subsequently during this process, as the solvent evaporates, the polymer volume fraction,
_φ_pol, increases while at the same time the _T_g of the solution also increases and approaches the experimental temperature. When the solution's _T_g reaches the experimental
temperature, the solution forms a glass. The film is then in a state of non-equilibrium, the structure having been ‘frozen in.’3, 9 This current work is with regard to the relaxation that
occurs in polymers that have undergone such a ‘solvent quench,’ rather than a thermal quench, through glass transition. Two poly(butyl methacrylate) polymers have been used in our study:
P(normal-BMA) and P(tertiary-BMA). These isomers have a large difference in their _T_g values, whereas elemental compositions of their repeat unit are identical and therefore differences in
interactions with solvent or silicon substrate are not expected to affect the experiments. P(norm-BMA) has a _T_g of 20 °C,10 which is below the experimental temperature of ∼23 °C;
therefore, it remains rubbery throughout the experiments. In contrast, P(_tert_-BMA) has a _T_g of 107 °C, below which the polymer forms a glass and is subject to relaxation. Hence,
P(_tert_-BMA) and P(norm-BMA) films were used to explore the differences between rubbery and glassy behavior. The polymers were provided by Polymer Source Inc. (Quebec, CA, USA) and used as
received. The number-average molecular weights (_M_n) of the two polymers were comparable, being 182 kg mol−1 for P(norm-BMA) (polydispersity (_M_w/_M_n) of 1.02) and 138 kg mol−1 for
P(_tert_-BMA), with _M_w/_M_n=1.06. In all experiments, thin films were deposited by spin-casting solutions in toluene at a rate of 2000 r.p.m. for 30 s. The substrates were single-crystal
(100) silicon wafers, used as received such that the native oxide was on the surface. Film thickness was kept constant at 130 nm to minimize thickness effects. Films were aged at room
temperature (23 °C) under vacuum for different periods up to 24 h, to allow the solvent to leave the film and for the structures to relax. Ellipsometry measurements (VASE, J.A. Woollam Co.,
Lincoln, NE, USA) were taken as a function of time over multiple wavelengths, selected to provide the greatest sensitivity. The film samples were placed on the ellipsometer stage previously
fitted with a heating stage (Linkam, Surrey, UK). During the measurements, films were heated in air at a constant temperature increase of 2 °C min−1 from room temperature (∼23 °C) up to a
maximum of 150 °C (chosen to avoid any thermal decomposition of the polymer11). The films were then cooled at an equal but opposite rate of −2 °C min−1. The ellipsometric angles Ψ and Δ were
measured as a function of time. Fitting of data using a Marquardt algorithm was performed to obtain the thickness, _h_, and refractive index, _n_, of the film as a function of time. Time
was correlated to temperature through the heating rate. The fitting parameters were thickness, _h_, and parameter _A_ of the refractive index in the Cauchy dispersion model,8, 12
_n_(_λ_)=_A_+(_B_/_λ_2), where _B_ is a material constant and _λ_ is the wavelength of the incident light. Relaxation processes are normally observed in bulk polymers by measuring the volume
of the glass, _V_t, at a given time. The relative departure from the equilibrium volume, _δ_V, is then defined as _δ_V=(_V_t−_V_∞)/_V_∞, where _V_∞ represents the equilibrium volume
obtained after long relaxation times.2 In an analogy to bulk behavior, the relaxation in thin films can be monitored by measuring the thickness of the glass, _h_, at a given time (and
temperature). A final equilibrium value for thickness, _h_∞, is obtained from a film after heating and cooling to room temperature. Its value is obtained from ellipsometry in a full
spectroscopic scan from 400 to 800 nm. It was found that the thickness changes occurring over time after a heating–cooling cycle were negligible in comparison with that found during the
cycle. The relative departure from equilibrium, defined as the excess thickness normalized with respect to _h_∞ at room temperature, was calculated as _δ_RT=(_h_−_h_∞)/_h_∞. In these
experiments in which temperature is not constant, _δ_RT also contains information on the thickness increase arising from thermal expansion. Using a measurement of the initial film thickness
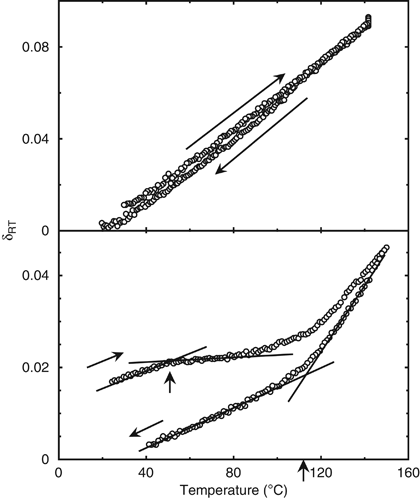
after spin-casting, _h_0, the _initial_ departure from equilibrium thickness, _δ_0, can be defined as _δ_0=(_h_0−_h_∞)/_h_∞. Figure 1 shows the variation of _δ_RT as a function of
temperature for the P(norm-BMA) isomer (top figure), which is a rubbery polymer at room temperature. The linear thermal expansion coefficient _α_=1/_h_(∂_h_/∂_T_)_P_,_t_ was calculated from
a linear fit to measurements of thickness as a function of temperature. A value of _α_=7.3 × 10−4 K−1 was obtained, which agrees with the literature value10 of 7.0 × 10−4 K−1. (Note that if
thermal expansion is restricted in the plane of the film, one would expect to see a higher expansivity in the direction normal to the plane.13 Thus, in this materials system, there is no
evidence that an attraction between the polymer and substrate is preventing slippage of the polymer along the substrate.) It was found that the thickness dependence on temperature was
essentially identical during the heating and cooling stages. There is no evidence for structural relaxation, as is expected for a rubbery polymer. The superposition of the heating and
cooling curves supports the conclusion that the rubbery polymer retains no solvent, which would lead to a volume change upon its liberation. This result clearly agrees with the ideas
proposed by Leibler and Sekimoto11 that an important factor in solvent retention in polymers must be the increase in bulk modulus of the polymer as it undergoes transition from the rubbery
to the glassy phase. They proposed that the chemical potential of the solvent in a polymer solution should be modified from the standard Flory–Huggins expression when the polymer becomes a
glass, adding an additional term, which is a function of the polymer bulk modulus. On the other hand, Figure 1 (bottom part) shows temperature-dependent data obtained from P(_tert_-BMA),
having a _T_g of 107 °C, that is well above room temperature. Initially, as the temperature rises, the film exhibits a thermal expansion that is broadly consistent with the literature values
for a polymer glass expansion coefficient. If the film contained no solvent, we would expect the rising trend to continue until it approached the _T_g of the neat polymer glass, at which
point the expansivity would increase toward that of the rubbery state. However, this is clearly not the case, as the film's expansion is arrested when reaching 47±3 °C, which is
interpreted as being an apparent glass transition temperature (arrow in the figure) of the glassy polymer matrix. If we consider that there is some solvent trapped in the polymer matrix,
then, below this temperature, the solvent is unable to leave the film as this would require a compression of the matrix (assuming no voids are allowed to be created because of the associated
rise in surface energy). According to the Kelley–Bueche polymer/solvent description,14 the presence of solvent (with a low _T_g) in a polymer depresses its _T_g as a function of the amount
of solvent present.14, 15 When this lower glass transition temperature is reached, the distortion energy2, 15 is overcome and trapped solvent is released, allowing the film to decrease in
thickness while at the same time raising the polymer/solvent _T_g further. During this period, both phenomena compete: thermal expansion increases the thickness, whereas solvent loss
decreases it. On reheating the film in a second cycle (data not shown), the thermal expansivity follows the same paths for the glassy and rubbery states that were observed in the first
cooling curve. This result indicates that relaxation and the accompanying solvent loss are complete after the initial heating. These differences in relaxation and expansion behavior observed
between glassy and rubbery polymers have not been demonstrated explicitly in thin films in any previous work that we have seen. Figure 1 clearly shows this period as expansion is arrested
until temperatures approach the bulk _T_g of the polymer. Cooling data shown in Figure 1 can accurately be used to calculate values such as the _T_g of the neat polymer (112±3 °C calculated,
107 °C literature value10), and thermal expansion coefficient of the glassy phase (_α_g=2.0 × 10−4 K−1 calculated, _α_g=2.80 × 10−4 K−1 literature value) and the rubbery phase (_α_r=7.4 ×
10−4 K−1 calculated, _α_r=7.20 × 10−4 K−1 literature value).10 Sufficiently good agreement with the literature values contributes to our confidence in interpreting the data obtained.
Consideration of the relative departure from equilibrium, _δ_, is best suited for isothermal experiments. To gain an insight into the temperature dependence of the fractional ‘excess
volume,’ beyond the equilibrium volume, a better approach is to calculate the departure from equilibrium thickness at a given temperature, relative to the equilibrium thickness at that same
temperature, as _δ_(_T_)=[_h_(T)−_h_∞(_T_)]/_h_∞(_T_). In this case, the _h_∞ values were taken from the cooling curve. When _δ_(_T_) is plotted as in Figure 2, it is observed that it is
unchanged until a certain temperature (47 °C) is reached, at which point the film is able to relax. This finding is consistent with the idea that the concentration of solvent in the film
lowers its _T_g from that of the neat polymer. Only at the apparent glass transition temperature does _δ_(_T_) start to decrease. We can use the apparent glass transition temperature to
obtain the solvent concentration required in the film to depress the temperature from the bulk value. To lower the glass transition temperature to 47±3 °C, the solvent volume fraction needs
to be 0.140±0.011. (This calculation assumes a value for the _T_g of toluene taken as 2/3 of the crystal melting temperature and uses the Kelley–Bueche polymer/solvent equation.14) Follow-up
experiments were carried out to examine the relaxation processes in blends of P(_tert_-BMA) and poly(methyl methacrylate) (PMMA). Three different P(_tert_-BMA)/PMMA blend ratios of 9:1, 1:1
and 1:9 were measured. Samples were aged in vacuum for different times at room temperature before analysis. The departure from room temperature equilibrium, _δ_RT, was measured with
ellipsometry when the system was heated at a constant rate of 2 °C min−1 for blends with aging times ranging from 0 to 96 h. Figure 3 shows, as an example, _δ_RT as a function of temperature
for the P(_tert_-BMA)/PMMA blend with a ratio of 1:9 for a range of times aged in vacuum at room temperature. When the films are heated above an apparent _T_g, there is a structural
relaxation leading to a decrease in film thickness. Once the film has fully relaxed and reached equilibrium at the experimental temperature, the thickness starts to increase again, as is
expected according to its thermal expansivity. When the film is aged for longer, the departure from equilibrium and the amount of relaxation decrease. Little or no change is appreciable as
the aging time is increased both for pure P(_tert_-BMA) and for P(_tert_-BMA)/PMMA with a ratio of 9:1 (data not shown). The results differ strongly for PMMA-rich films with a
P(_tert_-BMA)/PMMA ratio of 1:1 and 1:9. We observe in freshly cast (non-aged) films that, as the fraction of PMMA is increased in the blend, the film exhibits a higher initial departure
from its equilibrium. When a film is aged at room temperature, structural relaxation occurs—albeit very slowly—before the heating of the film. Hence, the amount of relaxation observed as the
film is heated decreases with increasing aging time. This concept explains why volume relaxation is greatly affected by the temperature of the system16 and the time of aging. Similar
behavior has been observed in many studies in literature on bulk polymers,2, 16, 17 but few reports have explored these phenomena in thin film systems.18 The data seem to suggest that
P(_tert_-BMA)-rich films relax very fast during the deposition procedure, such that they obtain an equilibrium thickness soon after deposition. In contrast, PMMA films undergo a significant
amount of slow relaxation. Further analysis was used to identify differences between the polymers. A volume relaxation rate, _β_, can be defined in terms of the slope of isothermal
measurements of _δ_ as16 _β_=d_δ_/dlog(_t__i_−_t_0), where _t_0 is initial time, _t__i_ is the time at point _i_ and _δ_ is as defined previously. This relaxation rate can be compared for in
the different systems to obtain some insight into the relation between the isothermal relaxation rate and the compositions of the different polymer blends. For each of the compositions, the
gradient of _δ_ (obtained isothermally at room temperature) plotted against the logarithm of time was used to obtain a value of the isothermal relaxation rate, _β_, at room temperature. The
measurements of _β_ and _δ_0 for these different systems are reported in Table 1. On considering the trend in _β_, it is apparent that PMMA-rich films exhibit the largest values compared
with other compositions. As the ratio of P(_tert_-BMA) increases in the blend, the relaxation rate decreases considerably, reaching the point at which the film is pure P(_tert_-BMA) and at
which the relaxation rate is the lowest. The initial departure from equilibrium is lowest for P(_tert_-BMA) and increases with increasing PMMA fraction. The fact that P(_tert_-BMA) has the
lowest _δ_0 and also exhibits almost no relaxation for over a period of several days suggests that P(_tert_-BMA) films hold much less solvent after spin-casting from dilute solution than do
PMMA-rich films. These differences are surprising considering the fact that PMMA and P(_tert_-BMA) have similar glass transition temperatures and bulk moduli. This difference in relaxation
characteristics between polymers may stem from differences in the selective character of the solvent and not from the mechanical properties of the polymers, which are similar. The χ
parameter determines how the chemical potential varies with _φ_pol.17 The χ parameter for the P(_tert_-BMA)/toluene system, for a polymer volume fraction of 0.8, is around 0.25–0.3,19, 20,
21 similar to that of the PS/toluene system, whereas for PMMA/toluene the value is higher, at around 0.45.10, 18, 19 One can argue that if the chemical potential is higher (as is the case
for toluene/PMMA) and when there is some solvent in the vapor phase—such as during the spin-casting process—it will be thermodynamically favorable to have more solvent in the polymer
solution. Our results are consistent with this expectation for the toluene/PMMA case in comparison with toluene/P(_tert_-BMA) or toluene/PS (H. Richardson, private communication). Despite
its non-selective character, toluene is a better solvent for P(_tert_-BMA) than for PMMA. In conclusion, the film's initial conformation after a solvent quench was explored for
different polymers and their blends. In spin-cast rubbery P(norm-BMA) films, there is no evidence for structural relaxation on heating and no evidence for remnant solvent. On the other hand,
spin-cast glassy P(_tert_-BMA) films undergo structural relaxation above an apparent _T_g. Comparisons between glassy P(_tert_-BMA) and PMMA/P(_tert_-BMA) with different blend ratios have
shown a strong dependence of both the initial departure from equilibrium thickness, _δ_0, and the relaxation rate, _β_, on the P(_tert_-BMA) content of the film. These results indicate that
P(_tert_-BMA)-rich films could be losing solvent and forming dense films much faster than PMMA-rich films, such that they are closer to their equilibrium thickness when first deposited. On
the other hand, the rate of structural relaxation in the solid P(_tert_-BMA) film is significantly slower than in PMMA. This result shows that P(_tert_-BMA) forms films with a high thickness
stability, something that is attractive in practical applications, such as lithography and emerging nano-imprinted coatings.22 REFERENCES * Binnig, G., Despont, M., Drechsler, U., Häberle,
W., Lutwyche, M., Vettiger, P., Mamin, H. J., Chui, B. W. & Kenny, T. W. Ultrahigh-density atomic force microscopy data storage with erase capability. _Appl. Phys. Lett._ 74, 1329
(1999). Article CAS Google Scholar * Kovacs, A. J. Transition vitreuse dans les polymères amorphes. _Etude. phénoménologique. Fortschr. Hochpolym.-Forsch._ 3, 394 (1964). Article CAS
Google Scholar * Frank, C. W., Rao, V. V., Despotopoulou, M. M., Pease, R. F. W., Hinsberg, W. D., Miller, R. D. & Rabolt, J. F. Structure in thin and ultrathin spin-cast polymer films.
_Science_ 273, 912 (1996). Article CAS Google Scholar * Reiter, G., Sharma, A., Khanna, R., Casoli, A. & David, M. O. The strength of long-range forces across thin liquid films. _J.
Colloid Interface Sci._ 214, 126 (1999). Article CAS Google Scholar * Forrest, J. A., Svanberg, C., Révész, K., Rodahl, M., Torell, L. M. & Kasemo, B. Relaxation dynamics in ultrathin
polymer films. _Phys. Rev. E_ 58, R1226 (1998). Article CAS Google Scholar * Fukao, K. & Miyamoto, Y. Glass transition and dynamics in thin polymer films: dielectric relaxation of
thin film of polystyrene. _Phys. Rev. E_ 61, 1743 (2000). Article CAS Google Scholar * Keddie, J. L., Jones, R. A. L. & Cory, R. A. Interface and surface effects on the
glass-transition temperature in thin polymer-films. _Faraday Discuss._ 98, 219 (1994). Article CAS Google Scholar * Richardson, H., López-García, I., Sferrazza, M. & Keddie, J. L.
Thickness dependence of structural relaxation in spin-cast, glassy polymer thin films. _Phys. Rev. E_ 70, 051805 (2004). Article CAS Google Scholar * Strobl, G. _The Physics of Polymers_
(Springer Verlag, Berlin Heidelberg, Germany, 1997). Book Google Scholar * _Polymer Handbook_ (eds Brandrup, J. _et al._) (Wiley & Sons, 2005). * Leibler, L. & Sekimoto, K. On the
sorption of gases and liquids in glassy-polymers. _Macromolecules_ 26, 6937 (1993). Article CAS Google Scholar * Parbhoo, B., Izrael, S., Salamanca, J. M. & Keddie, J. L. Use of
ellipsometry and gravimetry to develop calibration standards for measuring silicone coat weight and thickness with X-ray fluorescence spectroscopy. _Surf. Interface Anal._ 29, 341 (2000).
Article CAS Google Scholar * Worajittiphon, P., Jurewicz, I., King, A. A. K., Keddie, J. L. & Dalton, A. B. Thermal actuation in thin polymer films through particle nano-squeezing by
carbon nanotube belts. _Adv. Mater._ doi:10.1002/adma.201003145 (2010). * Kelley, F. N. & Bueche, F. Viscosity and glass temperature relations for polymer-diluents systems. _J. Polym.
Sci._ L 549 (1961). * Reiter, G. & de Gennes, P. G. Spin-cast, thin glassy polymer films: highly metastable forms of matter. _Eur. Phys. J. E_ 6, 25 (2001). Article CAS Google Scholar
* Hutchinson, J. M. Physical aging of polymers. _Prog. Polym. Sci._ 20, 703 (1995). Article CAS Google Scholar * Simon, S. L., Bernazzini, P. & McKenna, G. B. Effects of
freeze-drying on the glass temperature of cyclic polystyrenes. _Polymer_ 44, 8025 (2003). Article CAS Google Scholar * Young, R. J. & Lovell, P. A. _Introduction to Polymers_ (CRC
Press, London, 1991). Book Google Scholar * Elbs, H. & Krausch, G. Ellipsometric determination of Flory-Huggins interaction parameter in solution. _Polymer_ 45, 7935 (2004). Article
CAS Google Scholar * Wulf, M., Grundke, K., Kwok, D. Y. & Neumann, A. W. Influence of different alkyl side chains on solid surfaces of polymethacrylate. _J. Appl. Polym. Sci._ 77, 2493
(2000). Article CAS Google Scholar * Liang Cui, L., Ding, Y., Li, X., Wang, Z. & Han, Y. Solvent and polymer concentration effects on the surface morphology evolution of immiscible
polysterene/poly(methyl methacrylate) blends. _Thin Solid Films_ 2038, 515 (2006). Google Scholar * Gourgon, C., Tortai, J. H., Lazzarino, F., Perret, C., Micouin, G., Joubert, O. &
Landis, S. Influence of residual solvent in polymers patterned by nanoimprint lithography. _J. Vac. Sci. Tech._ 22, 602 (2004). Article CAS Google Scholar Download references
ACKNOWLEDGEMENTS We gratefully acknowledge funding from the EPSRC (UK), FNRS (Belgium) and Sun Chemical, Orpington, Kent. We benefited from useful discussions with Drs H Richardson (Surrey)
and D Illsley (Sun Chemical). We thank Dr de Silva (ULB) for his help with some figures. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Physics, University of Surrey, Surrey, UK
Izaro López García & Joseph L Keddie * Département de Physique, Université Libre de Bruxelles, Bruxelles, Belgium Michele Sferrazza Authors * Izaro López García View author publications
You can also search for this author inPubMed Google Scholar * Joseph L Keddie View author publications You can also search for this author inPubMed Google Scholar * Michele Sferrazza View
author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Michele Sferrazza. RIGHTS AND PERMISSIONS Reprints and permissions
ABOUT THIS ARTICLE CITE THIS ARTICLE López García, I., Keddie, J. & Sferrazza, M. Some insights into the structural relaxation of spin-cast, glassy polymer thin films. _Polym J_ 43,
214–217 (2011). https://doi.org/10.1038/pj.2010.123 Download citation * Received: 31 August 2010 * Revised: 27 October 2010 * Accepted: 30 October 2010 * Published: 08 December 2010 * Issue
Date: February 2011 * DOI: https://doi.org/10.1038/pj.2010.123 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * non-equilibrium * polymer thin
films * relaxation * solvent loss * spin-casting * thermal expansion