Play all audios:
ABSTRACT DNA methylation is an epigenetic modification that results in dynamic changes during ontogenesis and cell differentiation. DNA methylation patterns regulate gene expression and have
been widely researched. While tools for DNA methylation analysis have been developed, most of them have focused on intergroup comparative analysis within a dataset; therefore, it is
difficult to conduct cross-dataset studies, such as rare disease studies or cross-institutional studies. This study describes a novel method for DNA methylation analysis, namely, methPLIER,
which enables interdataset comparative analyses. methPLIER combines Pathway Level Information Extractor (PLIER), which is a non-negative matrix factorization (NMF) method, with
regularization by a knowledge matrix and transfer learning. methPLIER can be used to perform intersample and interdataset comparative analysis based on latent feature matrices, which are
obtained via matrix factorization of large-scale data, and factor-loading matrices, which are obtained through matrix factorization of the data to be analyzed. We used methPLIER to analyze a
lung cancer dataset and confirmed that the data decomposition reflected sample characteristics for recurrence-free survival. Moreover, methPLIER can analyze data obtained via different
preprocessing methods, thereby reducing distributional bias among datasets due to preprocessing. Furthermore, methPLIER can be employed for comparative analyses of methylation data obtained
from different platforms, thereby reducing bias in data distribution due to platform differences. methPLIER is expected to facilitate cross-sectional DNA methylation data analysis and
enhance DNA methylation data resources. SIMILAR CONTENT BEING VIEWED BY OTHERS REFERENCE-FREE DECONVOLUTION, VISUALIZATION AND INTERPRETATION OF COMPLEX DNA METHYLATION DATA USING
DECOMPPIPELINE, MEDECOM AND FACTORVIZ Article 25 September 2020 CHARTING DIFFERENTIALLY METHYLATED REGIONS IN CANCER WITH ROCKER-METH Article Open access 02 November 2021 FROM MULTI-OMICS
INTEGRATION TOWARDS NOVEL GENOMIC INTERACTION NETWORKS TO IDENTIFY KEY CANCER CELL LINE CHARACTERISTICS Article Open access 18 May 2021 INTRODUCTION DNA methylation – i.e., the addition of a
methyl group to the 5’ end of cytosine at CpG sites – is a type of epigenetic modification conserved across prokaryotes and eukaryotes1. In eukaryotes, DNA hypermethylation near
transcription start sites (TSSs) and around promoters has been reported to suppress gene expression by preventing the binding of transcription factors and other transcriptional regulators,
thus contributing to the formation of cell- and tissue-specific gene expression patterns during embryogenesis and cell differentiation2. The relationship between gene expression and DNA
methylation patterns has also been extensively explored in the cancer research field3. Comprehensive DNA methylation analysis methods include methylated DNA immunoprecipitation (MeDIP), the
Illumina HumanMethylation BeadChip, reduced-representation bisulfite sequencing (RRBS), and whole-genome bisulfite sequencing (WGBS)4,5. The methylated DNA data acquired on these platforms
are registered in the Gene Expression Omnibus (GEO) and Sequence Read Archive (SRA), with Illumina HumanMethylation BeadChip analysis data being the most commonly registered and published.
As of April 15, 2023, the HumanMethylation450 BeadChip (HM450) has been used to generate 114,799 samples registered in the GEO database, making it a widely used tool in the fields of
developmental biology and cancer research. In HM450 data analysis, raw data are generally normalized by preprocessing, followed by comparative and clustering analyses. Several preprocessing
methods have been proposed to date, with different researchers and projects adopting different preprocessing methods6,7,8. Therefore, when performing DNA methylation analysis using public
data, it is necessary to preprocess the raw data first. However, preprocessing DNA methylation data from hundreds to thousands of samples requires considerable computational power, thus
making cross-dataset analysis difficult. In addition, some datasets registered in GEO are available in the form of raw data, while others are available only in the form of preprocessed data,
thus requiring comparative analysis between data processed via different preprocessing methods. Importantly, HM450 data have been reported to have different data distributions depending on
the preprocessing method used9,10,11, which may compromise the reproducibility and scientific reproducibility of dataset comparison studies and meta-analyses using these data. The problem of
preprocessing-derived bias in data distribution has been established not only for DNA methylation data but also for transcriptomics data. For example, the platform recount2
(https://jhubiostatistics.shinyapps.io/recount/)12, which provides transcriptome data processed through a unified pipeline, and MultiPLIER13, an analysis method based on latent features
generated from large datasets, were developed to perform comparative analysis between datasets with data distribution bias. MultiPLIER first obtains a latent matrix via non-negative matrix
factorization (NMF)14 of large gene expression data obtained from recount2 with regularization to sparsely include knowledge matrices from gene sets for various pathways and ontologies.
Then, through matrix decomposition of the dataset to be analyzed using a latent matrix, datasets from different domains are represented by the same latent feature vector to achieve
interdataset comparative analysis. In this study, we applied the MultiPLIER architecture to DNA methylation data to develop a novel DNA methylation analysis architecture with high biological
semantic interpretability, thus enabling easy interdataset comparative analysis. We named this analysis architecture “methPLIER” and evaluated its data analysis capacity and biological
interpretability. methPLIER is a method for performing NMF under regularization conditions that sparsely includes knowledge matrices composed of gene sets such as pathways and ontologies.
Since multiple analysis probes are designed for a single gene or transcript in HM450 data, it is necessary to compress the data from probe-wise data to gene-wise data for regularized NMF
with a knowledge matrix. Since the analysis of DNA methylation patterns near the transcription start sites of transcripts is essential for DNA methylation analysis, we compressed the data
into principal component scores for the analysis probes located 1500 bps from the TSSs, transforming them into gene-wise data for each transcript. In addition, mapping DNA methylation probe
data near significant genes onto the genome made it possible to observe the spatial relationship between DNA methylation and expression patterns (Fig. 1). Using methPLIER, we analyzed a lung
cancer dataset obtained from the GEO and obtained latent variables (LVs) related to relapse-free survival (RFS). The gene sets included in the dataset were easy to interpret biologically.
Furthermore, comparative analysis between different preprocessing methods and different platforms using methPLIER suggested that distributional differences between the datasets could be
reduced. Based on the above findings, we believe that DNA methylation analysis using methPLIER is useful for interdataset comparisons and for understanding the biological significance of DNA
methylation based on available data. MATERIALS AND METHODS DATASET USED IN THIS STUDY For the development of methPLIER, DNA methylation data from 9756 samples of different cancers (lung
squamous cell carcinoma; lung adenocarcinoma; ovarian cancer; uterine corpus endometrial carcinoma; glioblastoma multiforme; kidney renal clear cell carcinoma; invasive breast carcinoma;
thyroid cancer; low-grade glioma; skin cutaneous melanoma; kidney renal papillary cell carcinoma; cervical squamous cell carcinoma and endocervical adenocarcinoma; liver hepatocellular
carcinoma; sarcoma; kidney chromophobe; acute myeloid leukemia; adrenocortical carcinoma; mesothelioma; lymphoid neoplasm diffuse large B-cell lymphoma; esophageal carcinoma; head and neck
squamous cell carcinoma; prostate adenocarcinoma; colon adenocarcinoma; pheochromocytoma and paraganglioma; uterine carcinosarcoma; uveal melanoma; rectum adenocarcinoma; cholangiocarcinoma;
testicular germ cell tumors; thymoma; pancreatic adenocarcinoma; bladder urothelial carcinoma; stomach adenocarcinoma) obtained from The Cancer Genome Atlas (TCGA; and
https://portal.gdc.gov) were used. To demonstrate the use of methPLIER, we used a LUAD dataset (GSE39279)15 obtained from the Gene Expression Omnibus (GEO;
https://www.ncbi.nlm.nih.gov/geo/)16. For preprocessing and comparative analysis, we used a clear cell renal cell carcinoma dataset (GSE61441)17 obtained from the GEO. For platform
comparative analysis, we used HM450, WGBS, and RRBS data from 4 cell lines—GM12878, H1-hESC, HepG2, and SK-N-SH—obtained from the Encyclopedia of DNA Elements (ENCODE;
https://www.encodeproject.org)18. The Public/Private R&D Investment Strategic Expansion PrograM (PRISM) database is one of the largest lung cancer databases in the world (in-house
database of NCC Japan) and contains clinical information from 1714 lung cancer patients, whole-exome analysis data from 1559 lung cancer patients, and RNA-seq data from 1682 lung cancer
patients. In addition to clinical information, total exome analysis, and RNA-seq analysis, the PRISM database features whole-genome analysis data for 413 patients, DNA methylation analysis
data for 402 patients, and ChIP-seq analysis (H3K27Ac) data for 222 patients (as of April 15, 2023). COMPARISON OF DNA METHYLATION DATA The HM450 data are expressed as numerical data of DNA
methylation status from 0 to 1 for 485,577 probes. To apply the regularization condition via the knowledge matrix, the data were compressed from probe-by-probe data to promoter methylation
pattern data for each transcript. Data compression was performed through the following two steps. First, DNA methylation data for analysis probes located within 1500 bps of the transcription
start site (TSS) for each transcript were extracted. These analysis probes were subsequently used to perform principal component analysis (PCA) to compress the obtained principal component
scores (PCSs) down to the point where the cumulative contribution rate exceeded 80%. The eigenvectors for each principal component axis were used for data compression of the new data. In the
feasibility study for compressing gene-wise data, probes located in the 1st exon were used. The compression of gene-wise data from probewise data was conducted using the previously
described method. CURATION OF KNOWLEDGE MATRICES The knowledge matrix used for methPLIER consisted of 817 gene sets obtained from the Molecular Signatures Database (MSigDB,
https://www.gsea-msigdb.org/gsea/msigdb/)19,20,21. For the knowledge matrix, we used four integrated datasets, namely, bloodCellMarkersIRISDMAP, svmMarkers, canonicalPathways, and
oncogenicPathways, which were incorporated into the R package PLIER22. The knowledge matrix included 545 biological pathways23, 83 cell- and tissue-specific genetic markers24, 189
cancer-related pathways25, and 817 gene sets. The methPLIER-CTD was constructed using a gene set for 2545 adenocarcinoma-related chemicals obtained from the Comparative Toxicogenomics
Database (CTD). These gene sets were subsequently transformed into a one-hot matrix consisting of genes and gene set names for use as a knowledge matrix. CONVERSION OF WGBS AND RRBS DATA TO
HM450 PROBEWISE DATA WGBS and RRBS data were converted to HM450 probewise data for comparative analysis with HM450 data using methyLiftover
(https://github.com/Christensen-Lab-Dartmouth/methyLiftover)26 to compare with the HM450 probewise data. The original methyLiftover converts WGBS and RRBS data mapped to hg19 to HM450
probe-wise data; however, since the WGBS and RRBS data obtained from ENCODE were mapped to hg38, the program was slightly edited to convert them to HM450 probe-wise data using the hg38
remapped HM450 manifest file (http://zwdzwd.github.io/InfiniumAnnotation)27 created by Zhou et al. NMF WITH KNOWLEDGE MATRIX SPARSE CONSTRAINTS NMF with sparse regularization by a knowledge
matrix was performed using the R PLIER package (https://github.com/wgmao/PLIER)22. With the PLIER package, we aimed to minimize the following equations by searching for matrices _U_, _Z_,
and _B_ when given an input data matrix _D_ and a knowledge matrix \({\boldsymbol{C}}\in 0,{1}^{n\times m}\) consisting of m gene sets containing n genes, where _U_ is the loading matrix for
the knowledge matrix and _B_ is the loading matrix for the latent feature matrix _Z_: $${\Vert {\boldsymbol{D}}-{\boldsymbol{ZB}}\Vert }_{F}^{2}+{\lambda }_{1}{\Vert
{\boldsymbol{Z}}-{\boldsymbol{CU}}\Vert }_{F}^{2}+{\lambda }_{2}{\Vert {\boldsymbol{B}}\Vert }_{F}^{2}+{\lambda }_{3}{\Vert {\boldsymbol{U}}\Vert }_{{L}^{1}}$$
$$subject\,to\,{\boldsymbol{U}}\, >\, 0,\,{\boldsymbol{Z}}\, >\, 0$$ The first term is the degree of approximation index of the matrix product _ZB_ of the latent feature matrix _Z_ and
the factor loading matrix _B_ to the input data matrix _D_, the so-called reconstruction error. The second, third and fourth terms are regularization terms for the matrices _Z_, _U_, and
_B_. Each regularization term is adjusted by the hyperparameters \({\lambda }_{1}\), \({\lambda }_{2}\), and \({\lambda }_{3}\). In addition, both matrices _Z_ and _U_ are non-negative
matrices. In the second term, the matrix product _CU_ of the knowledge matrix _C_ and its factor loading matrix _U_, given as constants, is intended to approximate the latent feature matrix
_Z_. A regularity condition due to the \({L}^{1}\) norm of the factor loading matrix _U_ is given in the fourth term, which optimizes the latent feature matrix _Z_ to be sparser. The third
term is the Frobenius norm of the factor loading matrix _B_ relative to the latent matrix _Z_. The purpose is to ensure that the factor loading matrix _B_ is not too large relative to the
latent feature matrix _Z_. In this study, PLIER was run on data _D_ converted from probe-wise data to gene-wise data with the following hyperparameters: frac = 0.7, max.iter = 350, maxPath =
10, minGenes = 10, glm_alpha = 0.9, and tol = 10−6. UNSUPERVISED CLUSTERING Unsupervised clustering was performed to capture the macroscopic sample characteristics of the DNA methylation
data and loading matrix output from methPLIER. Hierarchical cluster analysis (HCA), principal component analysis (PCA), k-means clustering, and uniform manifold approximation and projection
(UMAP)28 were used for clustering. HCA performed clustering classification using Ward’s method29 based on the Euclidean distance between samples. k-means clustering was performed using the
Hartigan‒Wong method30, with a maximum number of iterations of 100. UMAP was based on the Euclidean distance between samples and included the following parameters: neighbors = 15, components
= 2, and epochs = 200. MOTIF ANALYSIS OF SEQUENCES AROUND DMPS For the motif analysis of sequences surrounding differentially methylated positions (DMPs), we utilized the Analysis of Motif
Enrichment (AME) tool included in the MEME Suite31. We focused on the 50 base pair genomic sequences corresponding to the probe design locations on the HumanMethylation BeadChip. To compare
the hypomethylated and hypermethylated DMPs in pan-negative cases relative to those in _EGFR_ mutation cases, we performed a one-tailed Fisher’s exact test for statistical evaluation. Motifs
exhibiting significant differences (_P_ < 0.05) were extracted for further analysis. AVAILABILITY AND IMPLEMENTATION The code used to construct methPLIER was deposited on GitHub
(https://github.com/hamamoto-lab/methPLIER). Additionally, the data used for building methPLIER and the methPLIER model itself were deposited in Figshare
(https://doi.org/10.6084/m9.figshare.21938528, https://figshare.com/s/e656bd498a660778f988). By building a Docker image from the Dockerfile available on GitHub, users can create an
analytical environment that enables the utilization of methPLIER. SOFTWARE AND PACKAGES The analysis was performed using R 4.1.1 (https://cran.r-project.org), R Studio 1.3
(https://www.rstudio.com), and the following R packages: ChAMP32, minfi33, PLIER (https://github.com/wgmao/PLIER)22, multiPLIER (https://github.com/greenelab/multi-PLIER)13, and umap
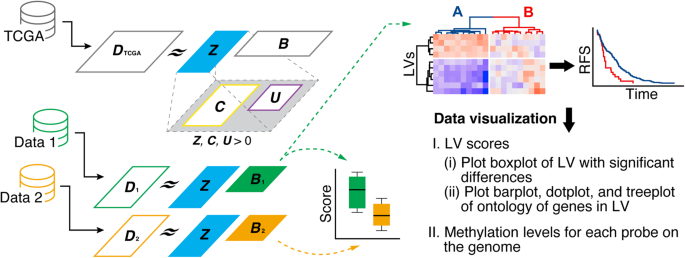
(https://github.com/tkonopka/umap). RESULTS DEVELOPMENT OF METHPLIER To construct methPLIER, HM450 data from 9756 samples obtained from the TCGA were used (Fig. 2). methPLIER is an NMF
performed under regular conditions where the latent matrix sparsely contains the knowledge matrix, which is a matrix consisting of multiple gene lists. The methodology of methPLIER
presupposes a relationship between the input data and the prior knowledge matrix, which is necessary because of its reliance on a matrix decomposition approach based on this prior knowledge.
This knowledge predominantly comprises curated information such as pathway information and ontology data, which are generally more compatible with gene expression data. However, a direct
correlation between DNA methylation data and pathways or ontologies is not always evident, often leading to a lack of applicable prior knowledge. To address this challenge, innovative
strategies for aligning analytical probes using knowledge matrices must be devised. This alignment is crucial for ensuring the relevance and accuracy of the matrix decomposition process in
the context of DNA methylation data. In addition, the knowledge matrix used in our study was exclusively available at the gene level and not at the probe level. Since the knowledge matrix is
a matrix consisting of multiple gene lists, HM450 data expressed as DNA methylation data for a single CpG site were converted into data for each gene to perform matrix decomposition. First,
DNA methylation data for analysis probes located within 1500 bps of the TSS for each transcript were extracted and compressed for each transcript via PCA. Among the principal components
obtained via PCA, the PCSs for the principal components reached the point where the cumulative contribution ratio exceeded 0.8 and were used as the compressed data for each transcript. The
TCGA DNA methylation data were compressed into a matrix of 9756 samples ×26,525 features. NMF was subsequently performed on the compressed data under the regularization condition that the
knowledge matrix was sparsely contained, resulting in a latent matrix _Z_ with 524 LVs (Fig. 2). To use the methPLIER tool, matrix decomposition was performed on new DNA methylation data
using this latent matrix Z to derive a loading matrix _B_. The loading matrix for the feature space was represented by the same latent matrix _Z_, allowing DNA methylation data from
different datasets to be analyzed in a common feature space. Unsupervised clustering classification and intergroup comparative analysis can then be performed using this loading matrix.
METHPLIER ENABLES ANALYSIS THAT REFLECTS SAMPLE CHARACTERISTICS To evaluate the performance of the methPLIER dataset, we analyzed a LUAD dataset (GSE39279)15 obtained from the GEO. The LUAD
dataset was compressed into a matrix decomposable data format using eigenvectors obtained during the compression of TCGA DNA methylation data, followed by latent matrix decomposition. HCA
was performed on the resulting loading matrix (Fig. 3a). Based on the HCA results, the samples were divided into two clusters, and Kaplan‒Meier survival analysis was performed for each
cluster (Fig. 3b). The log-rank test results showed a significant difference in RFS between the two clusters (_p_ = 0.00093). In a previous study using the same dataset, the sample clusters
were divided into two groups based on the HCA results, and the results of the survival response analysis showed that there were two groups, one with a high recurrence risk and the other with
a low recurrence risk. This grouping is consistent with the results of methPLIER analysis (GSE39279)15. FEASIBILITY OF USING 1ST EXON REGION PROBES IN METHPLIER ANALYSIS In the construction
of methPLIER, a critical element is the use of a knowledge matrix primarily derived from gene-wise data, which extensively incorporates curated information on gene expression patterns in
relation to pathways and ontologies. To compensate for the lack of scrutiny of the correlation of the knowledge matrix with DNA methylation patterns, the analysis concentrated on probes near
the TSS, specifically within a 1500 base pair area known for its strong link to gene expression. To expand the scope of methPLIER further, we incorporated data compression for probes
located in the 1st exon region, acknowledging the potential interest of researchers in this field. This decision was based on the assumption that DNA methylation in the 1st exon is likely to
correlate with gene expression, which parallels the rationale applied to the TSS. The methodology was subsequently applied and validated using a lung cancer dataset. The results of the
expanded analysis are discussed. By using factor loading matrix _B_ for hierarchical clustering analysis in methPLIER constructed with 1st exon region data, we observed the formation of two
distinct clusters, mirroring the findings from the TSS1500-based analysis (Supplementary Fig. 1). Crucially, survival time analysis based on cluster labels revealed significant differences
between the two groups, underscoring the robustness of the approach. LVS OBTAINED VIA METHPLIER HAVE HIGH BIOLOGICAL INTERPRETABILITY Comparative analysis of LVs between the two clusters
revealed significant differences in 261 LVs (FDR < 0.05, Supplementary Table 1). The knowledge variables in the top 10 LVs were confirmed by heatmap plotting, which showed that the
knowledge matrices were sparse among the latent variables and included gene sets related to lung cancer and cancer progression, such as those related to KRAS, the p38 MAPK pathway, and the
integrin pathway (Supplementary Fig. 2). A heatmap of the contents of the knowledge variables in the top 10 LVs showed that the knowledge matrices were sparsely included among the latent
variables. LV96 contained a set of genes whose expression was suppressed when the oncogenic form of KRAS (G12V) was overexpressed in four epithelial cell types34, and LV121 contained the p38
MAPK signaling pathway, all of which scored higher in the short-term RFS group. Lung cancer patients with _KRAS_ G12V mutations have been shown to have shorter overall survival than
non-_KRAS_ G12V mutation cases35. In addition, p38 generally functions as a tumor suppressor protein; however, in vitro experiments have shown that it contributes to cell proliferation and
malignant transformation in transgenic lung cancer cell lines harboring the _KRAS_ G12V mutation36. Based on the above, we believe that latent features in methPLIER reflect biological
characteristics and allow DNA methylation data analysis with high explanatory potential. METHPLIER REDUCES PREPROCESSING METHOD-ASSOCIATED DIFFERENCES IN DATA DISTRIBUTION To examine the
effect of methPLIER on data distribution bias due to differences in preprocessing methods, dataset distribution comparisons were performed using a single dataset preprocessed via four
different preprocessing methods. The dataset comprised 92 renal cell carcinoma samples and was preprocessed via four methods: a preprocessing method mimicking the Illumina Genome Studio
preprocessing method (Genome Studio)33, subset-quantile within array normalization (SWAN)37, peak-based correction (PBC)38, and beta-MIxture Quantile dilation (BMIQ)39. The data preprocessed
via each method were then combined into simply integrated data and into data with the loading matrix derived by methPLIER (methPLIER-integrated). Each of these merged datasets was embedded
into a two-dimensional space via UMAP, and a color-coded scatter plot was generated for each preprocessing method. After simple integration, clusters were formed for each preprocessing
method, while after using methPLIER for analyses, no clusters were observed (Fig. 4). The methPLIER-integrated data were then classified into two clusters by k-means clustering and color
coded based on the classification results. In both preprocessing methods, the respective clusters were separated on the UMAP 2 axis, showing a similar pattern regardless of the preprocessing
method (Fig. 4b). Color coding of the UMAP plots by sample type (cancer/noncancer) and sex information for the dataset used in the analysis showed a similar pattern to that of the cluster
information obtained by K-means clustering (Supplementary Fig. 3). These results suggest that methPLIER reduces the data distribution bias caused by the preprocessing method. METHPLIER
REDUCES DIFFERENCES IN DATA DISTRIBUTION ACROSS ANALYSIS PLATFORMS DNA methylation data can be obtained not only via BeadChip analysis but also via NGS-based WGBS. WGBS has been increasingly
used in recent years due to its ability to yield genome-wide methylation data, including data on DNA methylation at non-CpG sites. Therefore, to investigate the potential of using methPLIER
for WGBS data analysis, we analyzed data from four cell lines in the ENCODE project via the HM450K and WGBS databases. Since methPLIER is an analysis method compatible with HM450K probe
data, WGBS data were converted to corresponding HM450K analysis probe data using MhyLiftover26. The converted WGBS data were merged with the HM450K data obtained from the same cell line, and
unsupervised clustering was performed via HCA to form clusters for each analysis platform (Fig. 5a). PCA was performed on the integrated data, and a scatter plot of principal components
(PCs) 1 to 3 was generated, revealing that clusters were formed for each analysis platform, as was the case for HCA (Fig. 5b). This finding suggested that direct integration of WGBS data
with HM450K data is subject to data distribution bias due to the analysis platform. The loading matrix was subsequently obtained using methPLIER for the previously integrated data, with
classification by HCA and PCA showing that clusters were formed based on cell lines (Fig. 5c, d) rather than analysis platforms, as previously observed. These results suggest that the
loading matrix output by methPLIER reduces the data distribution bias caused by differences in analysis platforms and enables an analysis that focuses on sample characteristics. In addition,
we evaluated the suitability of the methPLIER tool for use with RRBS data. The results showed that the RRBS data formed distinct clusters (Supplementary Fig. 4a–d). This pattern was
attributable to the limited alignment of the RRBS data with the HM450K probes, which achieved only an approximately 10% match, in contrast to the higher compatibility observed for WGBS data
(Supplementary Fig. 4e). Despite the use of methPLIER, this discrepancy led to the persistent formation of separate clusters in the RRBS data, highlighting a notable platform-specific bias.
THE KNOWLEDGE MATRIX OF METHPLIER CAN BE FLEXIBLY MODIFIED FOR DIFFERENT PURPOSES Since the latent feature matrix of methPLIER is constrained by the knowledge matrix, the biological
interpretation of latent features is strongly influenced by the knowledge matrix applied in constructing methPLIER. The methPLIER tool constructed thus far used classical pathways and
cancer-related gene sets focused on cell biological functions for general-purpose data analysis. However, in drug response prediction and drug development research, we expect that knowledge
matrices, which include gene sets, such as pathways and ontologies related to compounds, and drugs that can be easily analyzed at a later stage and easily explained to stakeholders, will be
more appropriate. Therefore, we reconstructed methPLIER using the adenocarcinoma-related gene set obtained from the CTD as the knowledge matrix and examined the variability of the knowledge
matrix in methPLIER and its usefulness. The CTD is a database that aggregates the effects of environmental substances, compounds, and drugs on living organisms, with curated toxicogenomic
data available for more than 16,300 compounds. In this study, among adenocarcinoma-related chemicals in the CTD, we extracted the inference network that had more than 10 reference papers,
created a knowledge matrix consisting of 2545 chemicals and 778 genes, and constructed a methPLIER-CTD with 230 LVs. PRISM DNA METHYLATION DATASET ANALYSIS USING THE METHPLIER-CTD The
methPLIER-CTD was used to analyze the DNA methylation dataset of lung cancer patients in the PRISM database. It has been reported that approximately half of Japanese lung adenocarcinoma
patients have _EGFR_ mutations, and approximately 30% of patients have no identified driver genes, such as _KRAS_, _ALK_ fusion, or _RET_ fusion genes (pan-negative)40. A two-group
comparative analysis of _EGFR_-mutated and pan-negative cases was performed on the PRISM DNA methylation dataset generated by using the methPLIER-CTD. A _t_ test for factor loadings on each
latent feature obtained via methPLIER-CTD was performed, and LVs with an FDR of less than 0.05 according to the Benjamini–Hochberg method were extracted. The results revealed significant
differences in 92 LVs, and 64 features contained gene sets related to lung cancer (Supplementary Table 2). To confirm the biological interpretability of these 64 LVs, a heatmap plot of the
factor loadings on the gene set for each latent feature was generated (Supplementary Fig. 5). Each latent feature contained sparse gene sets, including cancer-related gene sets, anticancer
drug-related gene sets, general drug-related gene sets, toxicant-related gene sets, endogenous molecule-related gene sets, and other suggestive gene sets. Focusing on the anticancer
drug-related gene set, 54 LVs were extracted. Comparison of the DNA methylation levels of analytical probes located near genes in the anticancer drug-related gene set in _EGFR_-mutated
patients and pan-negative cases revealed 841 DMPs located near 225 genes (FDR < 0.05, Supplementary Table 3). Among the DMPs, 466 DMPs tended to be hypomethylated in pan-negative cases,
while 375 DMPs tended to be hypermethylated. The genes that exhibited hypomethylation in pan-negative cases included _MMP2_41,42, which has been reported to be involved in metastasis and
local invasion in breast cancer, and _HES1_43, which has been reported to be hypomethylated in colorectal cancer (Fig. 6a, b). Among the genes that showed a trend toward hypermethylation in
the pan-negative cases were _RUNX3_, a known tumor suppressor gene, and _CCNA1_44, which has been reported to correlate with treatment response to doxorubicin and 5-fluorouracil (Fig. 6c,
d). We performed transcription factor binding motif analysis on the 50 surrounding bps of the 466 identified hypomethylated DMPs and 375 hypermethylated DMPs (Fig. 7). Among the
hypomethylated DMPs, we observed enrichment of transcription factor binding motifs for POU Class 6 Homeobox 2 (PO6F2), CCAAT Enhancer Binding Protein Zeta (CEBPZ), Nuclear Transcription
Factor Y Subunit Beta (NFYB), Basic Helix-Loop-Helix Family Member E40 (BHE40), Forkhead Box P2 (FOXP2), Forkhead Box P1 (FOXP1), Forkhead Box O3 (FOXO3), E4F Transcription Factor 1 (E4F1),
Forkhead Box O6 (FOXO6), NFYA, E4F Transcription Factor 1 (E2F4), E2F Transcription Factor 7 (E2F7), Nuclear Transcription Factor Y Subunit Gamma (NFYC), and MYC Associated Zinc Finger
Protein (MAZ). In contrast, hypermethylated DMPs displayed enrichment of transcription factor binding motifs for NK2 homeobox 2 (NKX2-2), NK3 homeobox 1 (NKX3-1), NK2 homeobox 1 (NKX2-1),
TGFB Induced Factor Homeobox 1 (TGIF), Zinc Finger Protein 667 (ZN667), and Homeobox B13 (HXB13). NKX2-1, an airway epithelial-specific transcription factor, has been shown to inhibit SPDEF
expression, effectively preventing ovalbumin-induced goblet cell differentiation and lung inflammation in transgenic overexpression of Nkx2-145. Intriguingly, in the context of regulating
its target genes surfactant protein B and myosin-binding protein H, NKX2-1 has been found to be sensitive to methylation46,47,48. This finding contributes to a deeper understanding of the
molecular mechanisms involved in airway epithelial cell regulation and may have implications for future research in this area. In the pan-negative cases, we observed enrichment of NKX2-1
recognition sequences in the highly methylated DNA regions. Based on these findings, it is plausible that in pan-negative cases, the binding of NKX2-1 and NKX family proteins may be hindered
by high DNA methylation. This could result in insufficient suppression of cancer-related gene expression, potentially leading to the onset and progression of cancer. In summary, data
analysis using methPLIER-CTD constructed with drug-related gene sets revealed highly methylated regions near drug response-related genes and tumor suppressor genes in pan-negative cases.
DISCUSSION DNA methylation patterns are correlated with cell and tissue type as well as with disease and have been applied for diagnosis and therapy response prediction under various
conditions49. In studies of rare and regional diseases, it is important to integrate DNA methylation data from multiple studies, as small sample sizes and limitations inherent to data from a
single institution or study may compromise analysis50,51. However, DNA methylation data in various public databases differ according to the platform employed and processing applied, which
constitutes a barrier to data integration. Therefore, we developed a new DNA methylation data analysis method, namely, methPLIER, which is oriented toward integrated data analysis. Although
methPLIER is inspired by MultiPLIER, it is not a mere adaptation for DNA methylation data analysis. methPLIER has three novel aspects compared to MultiPLIER and other existing methods.
First, methPLIER employs a unique preprocessing method for the input data. The DNA methylation data used by methPLIER represents the methylation rate of each CpG site, resulting in a
“one-to-many relationship”, where multiple methylation information exists for a single gene or transcript. In contrast, the knowledge matrix utilized for regularization comprises
gene-specific matrices. Consequently, transforming CpG site data into gene-specific data is essential. methPLIER compresses the DNA methylation data for each transcript by extracting the
methylation data for analysis probes situated within 1500 bps of the transcript’s transcription start site and compressing them using principal component analysis52,53. Although DNA
methylation is considered an epigenetic modification that represses expression54,55, in cancer cells and stem cells, a hypermethylated state around the promoter contributes to or correlates
with expression activity56,57,58. For example, in the _TERT_ gene, the methylated variable region is located 1000 bps upstream from the TSS, and the 500 bps upstream region near the TSS was
reportedly hypomethylated, regardless of expression56,57. Gu et al. examined the association between DNA methylation and the expression of promoter regions in 265 samples of clear cell renal
cell carcinoma (ccRCC) and 133 adjacent tissues from The Cancer Genome Atlas (TCGA)59. They analyzed TSS200, TSS1500, and 5’UTR sites as promoters and found that of the regions with
ccRCC-specific methylation patterns, 81.81% were TSS200 sites, 6.97% were TSS1500 sites, and 11.22% were 5’UTRs. Nearly 90% of the specific methylation variation CpGs were located within
1500 bases around the TSS. Analysis of the association between specific methylation variations in promoters and gene expression showed that 31.2% of the promoters were associated with
expression variation. Therefore, compressing DNA methylation data down to the 1500-bp upstream region, as a simple mean or median, may lead to distal methylation patterns that are relevant
to expression being overlooked. We employed PCSs for principal components whose cumulative contribution ratio exceeded 0.8 as the compressed data for each transcript. This approach was
chosen for three reasons: many variable DNA methylation regions related to gene expression regulation are located near transcription start sites; gene expression regulation through DNA
methylation is often governed by differences in the genomic patterns of neighboring regions rather than variations in methylation patterns at individual CpG sites; and DNA methylation
patterns near the transcription start site have been reported to modulate transcriptional variant expression patterns. Second, methPLIER can integrate and analyze data acquired from
different platforms or research teams while reducing data distribution bias. BeadChip analysis was developed more than a decade ago and has been analyzed by many research teams, making its
vast datasets valuable research tools. However, there are significant differences in the data distribution bias between WGBS data and other datasets, and the data characteristics can be
masked by this large bias. In our study, we used the latent feature matrix for approximately 10,000 samples to perform matrix factorization of the analysis target sample, reducing the bias
between datasets and platforms. This enabled us to utilize accumulated valuable resources in future analyses without wasting them and could be useful for meta-analyses of rare diseases where
it is difficult to collect sufficient cases at a single facility. Finally, methPLIER is unique in that interpreting the results after data analysis is straightforward. In traditional DNA
methylation data analysis, variable sites or variable regions are obtained by comparative analysis, followed by functional estimation analysis, such as pathway analysis, GO analysis, or gene
set enrichment analysis. On the other hand, methPLIER performs matrix factorization using the latent matrix _Z_, which sparsely includes the knowledge matrix, and the loading matrix _B_
outputted by methPLIER contains information about the knowledge matrix. Therefore, it is possible to perform functional estimation analyses, such as pathway analysis, GO analysis, and data
feature analysis, simultaneously. This broadens the gateway to DNA methylation data analysis and is useful for users unfamiliar with data analysis. In unsupervised clustering analysis using
factor loadings on latent characteristic variables derived via methPLIER, the results reflect sample characteristics, such as progression-free survival and cell characteristics, highlighting
its utility for DNA methylation data analysis. Furthermore, the factor loading matrix derived by methPLIER reduces the data distribution bias for datasets generated with different platforms
and preprocessing methods, which is important for dataset-to-dataset integration analysis. The methPLIER-CTD, which was constructed by modifying the methPLIER knowledge matrix and oriented
toward drug-related DNA methylation analysis, may have demonstrated the usefulness of methPLIER for purpose-oriented analysis. Comparative analysis of methPLIER-CTD between pan-negative and
_EGFR_-mutated lung cancer patients identified 841 lung cancer pan-negative case-specific DMPs associated with the drug-related gene set and genomic regions of interest. _CCNA1_, which is
hypermethylated according to methPLIER, contributes to cell cycle regulation and was previously reported to be hypermethylated around its promoter in cervical and breast cancer tumors44,60.
In addition, DNA methylation patterns around promoters have been reported to be associated with the sensitivity of breast cancer cells to doxorubicin and 5-fluorouracil44. _RUNX3_, which is
hypermethylated in pan-negative cases, is known to function as a tumor suppressor gene that inhibits Wnt signaling by inhibiting β-catenin/Tcfs, and its expression is known to be regulated
by DNA methylation61. Since _RUNX3_ is reportedly hypermethylated in various cancer types, including lung cancer62,63, we speculate that Wnt inhibitors, such as vantictumab and LGK974, which
are currently in clinical trials for other cancer types, may be useful in the treatment of pan-negative cases. Although we believe that the methPLIER tool is useful for analyzing DNA
methylation data, there are several limitations. First, the biological interpretability of the latent feature matrix _Z_ in methPLIER varies greatly depending on the knowledge matrix used to
construct methPLIER. Therefore, when discussing the results of the analysis using the factor loading matrix _B_ derived by methPLIER, it is necessary to consider what knowledge matrix the
methPLIER used in the analysis. On the other hand, this is both a caveat in using methPLIER and an advantage in objective-oriented data analysis, such as the example of PRISM DNA methylation
dataset analysis using methPLIER-CTD, which was reconstructed using CTD. Second, methPLIER uses data from only a portion of the region in the BeadChip. methPLIER compresses probe-wise data
to gene-wise data prior to matrix decomposition. These data were compressed by principal component analysis (PCA) using analysis probes located 1500 bases upstream from the TSS, thus
eliminating DNA methylation data located in introns and intergenic regions. Therefore, when expectedly analyzing DNA methylation status at a distance away from the TSS, it is necessary to
reconstruct the methPLIER gene under different probe conditions for compression. The third limitation is that the results of the analysis obtained by methPLIER do not guarantee the
regulation of gene expression by DNA methylation. As mentioned above, methPLIER analyzes methylation data around promoters, and it is highly likely that the results contribute to the
regulation of gene expression. On the other hand, the regulation of gene expression via DNA methylation is still unclear in many respects, and whether the DNA methylation patterns observed
between samples are directly involved in the regulation of expression must be confirmed separately by gene expression data or wet laboratory experiments. Fourth, the results of the methPLIER
analysis were influenced by variations in tumor content among the samples. This issue arose because methPLIER is designed to analyze preprocessed data without an inherent mechanism for
adjusting for tumor content. Consequently, disparities in tumor content could influence the analysis results. Nonetheless, it is important to consider that the TCGA data used for developing
methPLIER not only encompassed tumor regions but also included benign tissues and represented cancers from various tissues. This diverse composition suggests that the latent features
identified by methPLIER are likely to capture characteristics specific to different tissues and cell types. When the tumor content varied between samples, we predicted that methPLIER would
tend to return results enriched with latent features not directly related to the tumor. This can indirectly indicate potential variations in tumor content across samples. These insights can
direct users toward the application of specialized methods that have been developed to estimate tumor content, allowing for more targeted correction of their data. Despite these limitations,
we believe that methPLIER will increase the interpretability of DNA methylation analysis and enable easy and comprehensive analyses, including dataset comparative analysis and ontology
analysis. The use of methPLIER will also enable cross-dataset analysis of DNA methylation data in medicine while maintaining biological interpretability, facilitate meta-analysis in rare
disease and epidemiological studies, and accelerate the discovery of disease-related DNA methylation patterns and treatment effect-related DNA methylation patterns. The advent of numerous
omics data analysis pipeline tools has significantly advanced the field of bioinformatics by streamlining complex data analysis processes32,33,64,65,66. These tools integrate a variety of
functions, from raw data ingestion to normalization, batch normalization, and comparative analysis, within a single-command framework. The incorporation of graphical user interfaces in some
of these tools has democratized access to data analysis, enabling researchers with limited programming expertise to engage in sophisticated analytical processes65,66. Despite their utility,
these pipeline tools often require substantial computational resources, including multicore CPUs and extensive memory, particularly when processing raw data files, such as FASTQ or IDAT
files. This demand poses a barrier, necessitating significant computational infrastructure and extended processing times. In contrast, methPLIER operates differently, focusing on the
analysis of preprocessed matrix files rather than raw data. Positioned downstream in the analytical pipeline, methPLIER can use the lighter preprocessed data typically available in public
repositories, such as GEO, or as supplementary data in academic publications. This methodological shift offers a pragmatic approach for preliminary data analysis, enabling rapid and
cost-effective insights that are particularly valuable in the early stages of research. Comprehensive integrated comparative analyses generally require high-end computational resources and
standardized data analysis procedures facilitated by established pipeline tools. However, this approach is not without drawbacks, including considerable financial, temporal, and labor costs,
without guaranteed analytical success. Therefore, methPLIER is a strategic alternative. Enabling preliminary integrated analyses allows researchers to acquire initial data quickly and
affordably, thus facilitating a more cost-effective approach to integrated data analysis. In summary, the purpose of using methPLIER in the broader analytical framework is not to replace
more resource-intensive pipeline tools but rather to complement them. By offering a means to conduct efficient preliminary analyses, methPLIER enhances the overall workflow and balances the
depth and breadth of the analysis against the practical constraints of resource allocation. This approach is particularly relevant in the context of high-throughput omics studies, where the
initial screening of vast datasets is as crucial as a detailed analysis. In conclusion, our new DNA methylation data analysis tool, methPLIER, has high biological interpretability, and the
knowledge matrix can be modified according to the purpose of the analysis. Moreover, this approach reduces the data bias between datasets caused by differences in preprocessing methods and
analysis platforms, contributes to highly reproducible data analysis and facilitates integrated analysis between datasets. In the future, the expansion of DNA methylation data analysis using
methPLIER will contribute to the elucidation of the true nature of pathologies associated with DNA methylation abnormalities and the promotion of the search for disease-related markers
supported by biological significance. DATA AVAILABILITY The data are available upon reasonable request. REFERENCES * Zemach, A., McDaniel, I. E., Silva, P. & Zilberman, D. Genome-wide
evolutionary analysis of eukaryotic DNA methylation. _Science_ 328, 916–919 (2010). Article CAS PubMed Google Scholar * Greenberg, M. V. C. & Bourc’his, D. The diverse roles of DNA
methylation in mammalian development and disease. _Nat. Rev. Mol. Cell Biol._ 20, 590–607 (2019). Article CAS PubMed Google Scholar * Kulis, M & Esteller, M. DNA Methylation and
Cancer. _Adv. Genet._ 70, 27–56 (2010). * Plongthongkum, N., Diep, D. H. & Zhang, K. Advances in the profiling of DNA modifications: cytosine methylation and beyond. _Nat. Rev. Genet_
15, 647–661 (2014). Article CAS PubMed Google Scholar * Hamamoto, R., Komatsu, M., Takasawa, K., Asada, K. & Kaneko, S. Epigenetics Analysis and Integrated Analysis of Multiomics
Data, Including Epigenetic Data, Using Artificial Intelligence in the Era of Precision Medicine. _Biomolecules_ 10, 62 (2019). Article PubMed PubMed Central Google Scholar * Naumov, V.
A. et al. Genome-scale analysis of DNA methylation in colorectal cancer using Infinium HumanMethylation450 BeadChips. _Epigenetics_ 8, 921–934 (2013). Article CAS PubMed PubMed Central
Google Scholar * Fasanelli, F. et al. Hypomethylation of smoking-related genes is associated with future lung cancer in four prospective cohorts. _Nat. Commun._ 6, 10192 (2015). Article
CAS PubMed Google Scholar * Wang, Z. et al. Epigenomic analysis of 5-hydroxymethylcytosine (5hmC) reveals novel DNA methylation markers for lung cancers. _Neoplasia_ 22, 154–161 (2020).
Article CAS PubMed PubMed Central Google Scholar * Pidsley, R. et al. A data-driven approach to preprocessing Illumina 450 K methylation array data. _BMC Genomics_ 14, 293 (2013).
Article CAS PubMed PubMed Central Google Scholar * Felling, RJ, Guo, JU & Song, H Neuronal activation and insight into the plasticity of DNA methylation. _Epigenomics_ 4, 125–127
(2012). * McEwen, L. M. et al. Systematic evaluation of DNA methylation age estimation with common preprocessing methods and the Infinium MethylationEPIC BeadChip array. _Clin. Epigenetics_
10, 123 (2018). Article CAS PubMed PubMed Central Google Scholar * Collado-Torres, L et al. Reproducible RNA-seq analysis using recount2. _Nat. Biotechnol._ 35, 319–321 (2017). *
Taroni, J. N. et al. MultiPLIER: A Transfer Learning Framework for Transcriptomics Reveals Systemic Features of Rare Disease. _Cell Syst._ 8, 380–394.e4 (2019). Article CAS PubMed PubMed
Central Google Scholar * Hamamoto, R et al. Application of non-negative matrix factorization in oncology: one approach for establishing precision medicine. _Brief Bioinform_ 23, bbac246
(2022). * Sandoval, J. et al. A prognostic DNA methylation signature for stage I non-small-cell lung cancer. _J. Clin. Oncol._ 31, 4140–4147 (2013). Article PubMed Google Scholar *
Barrett, T. et al. NCBI GEO: archive for functional genomics data sets–update. _Nucleic Acids Res._ 41, D991–D995 (2013). Article CAS PubMed Google Scholar * Wei, J.-H. et al. A
CpG-methylation-based assay to predict survival in clear cell renal cell carcinoma. _Nat. Commun._ 6, 8699 (2015). Article CAS PubMed PubMed Central Google Scholar * Davis, C. A. et al.
The Encyclopedia of DNA elements (ENCODE): data portal update. _Nucleic Acids Res._ 46, D794–D801 (2018). Article CAS PubMed Google Scholar * Subramanian, A. et al. Gene set enrichment
analysis: a knowledge-based approach for interpreting genome-wide expression profiles. _Proc. Natl Acad. Sci. USA_ 102, 15545–15550 (2005). Article CAS PubMed PubMed Central Google
Scholar * Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. _Bioinformatics_ 27, 1739–1740 (2011). Article CAS PubMed PubMed Central Google Scholar * Liberzon, A. et al.
The Molecular Signatures Database (MSigDB) hallmark gene set collection. _Cell Syst._ 1, 417–425 (2015). Article CAS PubMed PubMed Central Google Scholar * Mao, W., Zaslavsky, E.,
Hartmann, B. M., Sealfon, S. C. & Chikina, M. Pathway-level information extractor (PLIER) for gene expression data. _Nat. Methods_ 16, 607–610 (2019). Article CAS PubMed PubMed
Central Google Scholar * Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. _Nucleic Acids Res._ 44,
D457–D462 (2016). Article CAS PubMed Google Scholar * Abbas, A. R., Wolslegel, K., Seshasayee, D., Modrusan, Z. & Clark, H. F. Deconvolution of blood microarray data identifies
cellular activation patterns in systemic lupus erythematosus. _PLoS One_ 4, e6098 (2009). Article PubMed PubMed Central Google Scholar * Sturm, D. et al. New Brain Tumor Entities Emerge
from Molecular Classification of CNS-PNETs. _Cell_ 164, 1060–1072 (2016). Article CAS PubMed PubMed Central Google Scholar * Titus, A. J., Houseman, E. A., Johnson, K. C. &
Christensen, B. C. methyLiftover: cross-platform DNA methylation data integration. _Bioinformatics_ 32, 2517–2519 (2016). Article CAS PubMed PubMed Central Google Scholar * Zhou, W.,
Laird, P. W. & Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. _Nucleic Acids Res._ 45, e22 (2017). PubMed Google
Scholar * McInnes, L., Healy, J., Saul, N. & Großberger, L. UMAP: Uniform Manifold Approximation and Projection. _J. Open Source Softw._ 3, 861 (2018). Article Google Scholar * Ward,
J. H. Hierarchical Grouping to Optimize an Objective Function. _J. Am. Stat. Assoc._ 58, 236–244 (1963). Article Google Scholar * Hartigan, J. A. & Wong, M. A. A k-means clustering
algorithm. _Appl Stat._ 28, 100–108 (1979). Article Google Scholar * McLeay, R. C. & Bailey, T. L. Motif Enrichment Analysis: a unified framework and an evaluation on ChIP data. _BMC
Bioinforma._ 11, 165 (2010). Article Google Scholar * Tian, Y. et al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. _Bioinformatics_ 33, 3982–3984 (2017). Article
CAS PubMed PubMed Central Google Scholar * Aryee, M. J. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays.
_Bioinformatics_ 30, 1363–1369 (2014). Article CAS PubMed PubMed Central Google Scholar * Barbie, D. A. et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers
require TBK1. _Nature_ 462, 108–112 (2009). Article CAS PubMed PubMed Central Google Scholar * Renaud, S. et al. Prognostic value of the KRAS G12V mutation in 841 surgically resected
Caucasian lung adenocarcinoma cases. _Br. J. Cancer_ 113, 1206–1215 (2015). Article CAS PubMed PubMed Central Google Scholar * Vitos-Faleato, J. et al. Requirement for epithelial p38α
in KRAS-driven lung tumor progression. _Proc. Natl Acad. Sci. USA_ 117, 2588–2596 (2020). Article CAS PubMed PubMed Central Google Scholar * Maksimovic, J., Gordon, L. & Oshlack, A.
SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. _Genome Biol._ 13, R44 (2012). Article PubMed PubMed Central Google Scholar *
Dedeurwaerder, S. et al. Evaluation of the Infinium Methylation 450 K technology. _Epigenomics_ 3, 771–784 (2011). Article CAS PubMed Google Scholar * Teschendorff, A. E. et al. A
beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. _Bioinformatics_ 29, 189–196 (2013). Article CAS PubMed Google
Scholar * Shiraishi, K. et al. Association of variations in HLA class II and other loci with susceptibility to EGFR-mutated lung adenocarcinoma. _Nat. Commun._ 7, 12451 (2016). Article
CAS PubMed PubMed Central Google Scholar * Artacho-Cordón, F. et al. Matrix metalloproteinases: potential therapy to prevent the development of second malignancies after breast
radiotherapy. _Surg. Oncol._ 21, e143–e151 (2012). Article PubMed Google Scholar * Pereira, I. T. et al. Fibronectin affects transient MMP2 gene expression through DNA demethylation
changes in non-invasive breast cancer cell lines. _PLoS One_ 9, e105806 (2014). Article PubMed PubMed Central Google Scholar * Wu, Y. et al. The clinicopathological significance of HES1
promoter hypomethylation in patients with colorectal cancer. _Onco Targets Ther._ 10, 5827–5834 (2017). Article PubMed PubMed Central Google Scholar * Klajic, J. et al. DNA methylation
status of key cell-cycle regulators such as CDKNA2/p16 and CCNA1 correlates with treatment response to doxorubicin and 5-fluorouracil in locally advanced breast tumors. _Clin. Cancer Res._
20, 6357–6366 (2014). Article CAS PubMed Google Scholar * Maeda, Y. et al. Airway epithelial transcription factor NK2 homeobox 1 inhibits mucous cell metaplasia and Th2 inflammation.
_Am. J. Respir. Crit. Care Med._ 184, 421–429 (2011). Article CAS PubMed PubMed Central Google Scholar * Cao, Y. et al. Epigenetic mechanisms modulate thyroid transcription factor
1-mediated transcription of the surfactant protein B gene. _J. Biol. Chem._ 285, 2152–2164 (2010). Article CAS PubMed Google Scholar * Hosono, Y. et al. MYBPH, a transcriptional target
of TTF-1, inhibits ROCK1, and reduces cell motility and metastasis. _EMBO J._ 31, 481–493 (2012). Article CAS PubMed Google Scholar * Song, J. et al. Aberrant DNA methylation and
expression of SPDEF and FOXA2 in airway epithelium of patients with COPD. _Clin. Epigenetics_ 9, 42 (2017). Article CAS PubMed PubMed Central Google Scholar * Locke, W. J. et al. DNA
Methylation Cancer Biomarkers: Translation to the Clinic. _Front. Genet._ 10, 1150 (2019). Article CAS PubMed PubMed Central Google Scholar * Sadikovic, B et al. Correction: Clinical
epigenomics: genome-wide DNA methylation analysis for the diagnosis of Mendelian disorders. _Genet. Med._ 23, 2228 (2021). * Paparella, A. et al. Genome-wide DNA methylation profiling and
exome sequencing resolved a long-time misdiagnosed case. _J. Hum. Genet_ 67, 547–551 (2022). Article CAS PubMed Google Scholar * Moore, L. D., Le, T. & Fan, G. DNA methylation and
its basic function. _Neuropsychopharmacology_ 38, 23–38 (2013). Article CAS PubMed Google Scholar * He, X.-J., Chen, T. & Zhu, J.-K. Regulation and function of DNA methylation in
plants and animals. _Cell Res._ 21, 442–465 (2011). Article CAS PubMed PubMed Central Google Scholar * Witte, T., Plass, C. & Gerhauser, C. Pan-cancer patterns of DNA methylation.
_Genome Med._ 6, 66 (2014). Article PubMed PubMed Central Google Scholar * Esteller, M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future.
_Oncogene_ 21, 5427–5440 (2002). Article CAS PubMed Google Scholar * Takasawa, K. et al. DNA hypermethylation enhanced telomerase reverse transcriptase expression in human-induced
pluripotent stem cells. _Hum. Cell_ 31, 78–86 (2018). Article CAS PubMed Google Scholar * Lee, D. D. et al. DNA hypermethylation within TERT promoter upregulates TERT expression in
cancer. _J. Clin. Invest._ 129, 223–229 (2019). Article PubMed Google Scholar * Rauluseviciute, I., Drabløs, F. & Rye, M. B. DNA hypermethylation associated with upregulated gene
expression in prostate cancer demonstrates the diversity of epigenetic regulation. _BMC Med Genom._ 13, 6 (2020). Article CAS Google Scholar * Gu, Y. et al. Promoter DNA methylation
analysis reveals a novel diagnostic CpG-based biomarker and RAB25 hypermethylation in clear cell renel cell carcinoma. _Int J. Biol. Sci. Rep._ 7, 142000 (2017). Google Scholar * Zuo, Q. et
al. Methylation in the promoters of HS3ST2 and CCNA1 genes is associated with cervical cancer in Uygur women in Xinjiang. _Int J. Biol. Markers_ 29, e354–e362 (2014). Article CAS PubMed
Google Scholar * Wang, Y. et al. Association of promoter methylation of RUNX3 gene with the development of esophageal cancer: a meta analysis. _PLoS One_ 9, e107598 (2014). Article PubMed
PubMed Central Google Scholar * Kim, T. Y. et al. Methylation of RUNX3 in various types of human cancers and premalignant stages of gastric carcinoma. _Lab. Invest._ 84, 479–484 (2004).
Article CAS PubMed Google Scholar * Sato, K. et al. Epigenetic inactivation of the RUNX3 gene in lung cancer. _Oncol. Rep._ 15, 129–135 (2006). CAS PubMed Google Scholar * Morris, T.
J. & Beck, S. Analysis pipelines and packages for Infinium HumanMethylation450 BeadChip (450k) data. _Methods_ 72, 3–8 (2015). Article CAS PubMed PubMed Central Google Scholar *
Assenov, Y. et al. Comprehensive analysis of DNA methylation data with RnBeads. _Nat. Methods_ 11, 1138–1140 (2014). Article CAS PubMed PubMed Central Google Scholar * Müller, F. et al.
RnBeads 2.0: comprehensive analysis of DNA methylation data. _Genome biol._ 20, 55 (2019). Article PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We thank
Prof. Hiroyuki Mano and the members of the Division of Medical AI Research and Development at the National Cancer Center Research Institute for their technical support and helpful
discussions. FUNDING This work was supported by the AMED Innovative Cancer Medical Practice Research Project (Grant Number JP22ck0106643), JST CREST (Grant Number JPMJCR1689), JSPS
Grant-in-Aid for Scientific Research on Innovative Areas (Grant Number JP18H04908), JST AIP-PRISM (Grant Number JPMJCR18Y4), and MEXT subsidy for Advanced Integrated Intelligence Platform.
AUTHOR INFORMATION Author notes * These authors contributed equally: Ken Takasawa, Ken Asada, Syuzo Kaneko. AUTHORS AND AFFILIATIONS * Division of Medical AI Research and Development,
National Cancer Center Research Institute, Tokyo, 104-0045, Japan Ken Takasawa, Ken Asada, Syuzo Kaneko, Hidenori Machino, Satoshi Takahashi, Norio Shinkai, Nobuji Kouno, Kazuma Kobayashi,
Masaaki Komatsu & Ryuji Hamamoto * Cancer Translational Research Team, RIKEN Center for Advanced Intelligence Project, Tokyo, 103-0027, Japan Ken Takasawa, Ken Asada, Syuzo Kaneko,
Hidenori Machino, Satoshi Takahashi, Norio Shinkai, Nobuji Kouno, Kazuma Kobayashi, Masaaki Komatsu & Ryuji Hamamoto * Division of Genome Biology, National Cancer Center Research
Institute, Tokyo, 104-0045, Japan Kouya Shiraishi, Takaaki Mizuno & Takashi Kohno * Department of Thoracic Oncology, National Cancer Center Hospital, Tokyo, 104-0045, Japan Takaaki
Mizuno, Tatsuya Yoshida, Hidehito Horinouchi & Yuichiro Ohe * Department of Experimental Therapeutics, National Cancer Center Hospital, Tokyo, 104-0045, Japan Takaaki Mizuno * Department
of Thoracic Surgery, National Cancer Center Hospital, Tokyo, 104-0045, Japan Yu Okubo, Yukihiro Yoshida & Shun-Ichi Watanabe * Division of Medical Informatics, National Cancer Center
Hospital, Tokyo, 104-0045, Japan Masami Mukai * Department of Diagnostic Pathology, National Cancer Center Hospital, Tokyo, 104-0045, Japan Yasushi Yatabe Authors * Ken Takasawa View author
publications You can also search for this author inPubMed Google Scholar * Ken Asada View author publications You can also search for this author inPubMed Google Scholar * Syuzo Kaneko View
author publications You can also search for this author inPubMed Google Scholar * Kouya Shiraishi View author publications You can also search for this author inPubMed Google Scholar *
Hidenori Machino View author publications You can also search for this author inPubMed Google Scholar * Satoshi Takahashi View author publications You can also search for this author
inPubMed Google Scholar * Norio Shinkai View author publications You can also search for this author inPubMed Google Scholar * Nobuji Kouno View author publications You can also search for
this author inPubMed Google Scholar * Kazuma Kobayashi View author publications You can also search for this author inPubMed Google Scholar * Masaaki Komatsu View author publications You can
also search for this author inPubMed Google Scholar * Takaaki Mizuno View author publications You can also search for this author inPubMed Google Scholar * Yu Okubo View author publications
You can also search for this author inPubMed Google Scholar * Masami Mukai View author publications You can also search for this author inPubMed Google Scholar * Tatsuya Yoshida View author
publications You can also search for this author inPubMed Google Scholar * Yukihiro Yoshida View author publications You can also search for this author inPubMed Google Scholar * Hidehito
Horinouchi View author publications You can also search for this author inPubMed Google Scholar * Shun-Ichi Watanabe View author publications You can also search for this author inPubMed
Google Scholar * Yuichiro Ohe View author publications You can also search for this author inPubMed Google Scholar * Yasushi Yatabe View author publications You can also search for this
author inPubMed Google Scholar * Takashi Kohno View author publications You can also search for this author inPubMed Google Scholar * Ryuji Hamamoto View author publications You can also
search for this author inPubMed Google Scholar CONTRIBUTIONS KT and RH conceptualized and designed the study. KT, KA, SK, and KS conducted the experiments and analyzed the data. KT, KA, SK,
KS, HM, ST, NS, NK, KK, MK, TM, YO, MM, TY, YYoshida, HH, SW, YO, YYatabe, TK, and RH contributed to the data analysis and interpretation. KT and RH wrote the manuscript. KT, KA, SK, KS, HM,
ST, NS, NK, KK, MK, TM, YO, MM, TY, YYoshida, HH, SW, YYatabe, and SK edited the manuscript. All the authors have read and approved the final version of the manuscript. The corresponding
authors take full responsibility for the credibility of the descriptions and data presented in this work. CORRESPONDING AUTHORS Correspondence to Ken Takasawa or Ryuji Hamamoto. ETHICS
DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in
published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION SUPPLEMENTARY TABLE 1 SUPPLEMENTARY TABLE 2 SUPPLEMENTARY TABLE 3 RIGHTS AND PERMISSIONS
OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or
format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or
other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in
the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the
copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Takasawa, K., Asada, K.,
Kaneko, S. _et al._ Advances in cancer DNA methylation analysis with methPLIER: use of non-negative matrix factorization and knowledge-based constraints to enhance biological
interpretability. _Exp Mol Med_ 56, 646–655 (2024). https://doi.org/10.1038/s12276-024-01173-7 Download citation * Received: 11 May 2023 * Revised: 27 November 2023 * Accepted: 13 December
2023 * Published: 04 March 2024 * Issue Date: March 2024 * DOI: https://doi.org/10.1038/s12276-024-01173-7 SHARE THIS ARTICLE Anyone you share the following link with will be able to read
this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative