Play all audios:
ABSTRACT Current intensified chemotherapy regimens have significantly increased survival rates for pediatric patients with T-cell acute lymphoblastic leukemia (T-ALL), but these treatments
can result in serious adverse effects; furthermore, patients who are resistant to chemotherapy or who relapse have inferior outcomes, together highlighting the need for improved therapeutic
strategies. Despite recent advances in stratifying T-ALL into molecular subtypes with distinct driver mutations, efforts to target the tumor-intrinsic genomic alterations critical for T-ALL
progression have yet to translate into more effective and less toxic therapies. Ample evidence now indicates that extrinsic factors in the leukemic microenvironment are critical for T-ALL
growth, infiltration, and therapeutic resistance. Considering the diversity of organs infiltrated by T-ALL cells and the unique cellular components of the microenvironment encountered at
each site, it is likely that there are both shared features of tumor-supportive niches across multiple organs and site-specific features that are key to leukemia cell survival. Therefore,
elucidating the distinct microenvironmental cues supporting T-ALL in different anatomic locations could reveal novel therapeutic targets to improve therapies. This review summarizes the
current understanding of the intricate interplay between leukemia cells and the diverse cells they encounter within their tumor microenvironments (TMEs), as well as opportunities to
therapeutically target the leukemic microenvironment. SIMILAR CONTENT BEING VIEWED BY OTHERS ACUTE LYMPHOBLASTIC LEUKAEMIA Article 13 June 2024 A MULTIOMIC ATLAS IDENTIFIES A
TREATMENT-RESISTANT, BONE MARROW PROGENITOR-LIKE CELL POPULATION IN T CELL ACUTE LYMPHOBLASTIC LEUKEMIA Article Open access 25 November 2024 IMPACT OF TUMOR MICROENVIRONMENT ON EFFICACY OF
ANTI-CD19 CAR T CELL THERAPY OR CHEMOTHERAPY AND TRANSPLANT IN LARGE B CELL LYMPHOMA Article Open access 17 January 2024 INTRODUCTION Acute lymphoblastic leukemia (ALL) is one of the four
primary types of leukemia, along with acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), and chronic myeloid leukemia (CML). While AML, CLL, and CML primarily affect adults,
approximately 80% of ALL cases occur in children1,2. ALL is classified into two main types: T-cell acute lymphoblastic leukemia (T-ALL) and B-cell acute lymphoblastic leukemia (B-ALL)2,3.
T-ALL accounts for 15% of newly diagnosed cases in children and 25% in adults4,5. T-ALL circulates in peripheral blood6 and is characterized by the aggressive clonal proliferation of
immature T-cell precursors originating in the thymus, which infiltrate secondary organs, including the spleen, the liver, bone marrow (BM), lymph nodes, and the central nervous system (CNS).
Substantial progress has been made in T-ALL treatment in recent decades, leading to long-term remission in more than 90% of children and 60% of adult patients7. However, approximately
15–20% of pediatric and 50% of adult patients relapse or do not respond to initial therapy within two years of diagnosis6. Pediatric and adult patients with relapsed T-ALL have poor
prognoses, with approximately 30–50% and 30% survival rates over 5 years, respectively8. Furthermore, current therapies cause adverse events, even in patients who are cured9. Therefore,
there is an evident clinical need to develop less toxic therapies for T-ALL patients. IMMUNOPHENOTYPIC AND MOLECULAR CHARACTERIZATION OF T-ALL T-ALL cells originate in the thymus as a
deviation from canonical T-cell development. In leukemic blasts, oncogenes are frequently activated via chromosomal translocations between T-cell receptor (TCR) gene loci and coding and
regulatory regions of genes encoding transcription factors6. T-ALL is classified into multiple subgroups on the basis of both phenotypic and genomic features. Initially, T-ALL subtype
definitions were based on the immunophenotypes of leukemic blasts and their similarity to thymocytes at distinct stages of differentiation10,11. However, advances in sequencing technologies
have revealed additional heterogeneity in the molecular subtypes of T-ALL due to coordinated dysregulation of gene expression programs, with a particular focus on classes of transcription
factors. A recent genomic and transcriptional profiling study of T-ALL samples from a large pediatric cohort identified ten molecular subgroups, _HOXA_, _TLX3_, _TLX1_, _NKX2-1_, _LMO1/2_,
_TAL1_, _TAL2_, _BCL11B_, _SPI1_, and T-other, which are distinguished by aberrant transcription factor expression, gene expression profiles, and genomic alterations12. This study
highlighted several key biological pathways altered by genomic modifications in T-ALL, including transcription factor activity, epigenetic regulation, NOTCH signaling, cell cycle regulation,
JAK-STAT signaling, and PI3K signaling. T-ALL subtypes are associated with distinct genomic alterations; for example, alterations in _SMARCA4_, encoding a transcriptional activator, are
common in the _TLX3_ T-ALL subtype, whereas alterations in the transcription factor _LEF1_ are common in the NKX2-1 subtype. Alterations in _NOTCH1_ and _CDKN2A_ were prevalent in all T-ALL
subtypes, occurring in 67% and 74% of T-ALL samples, respectively, relative to 0% and <30%, respectively, in B-ALL samples. Moreover, disruptions in genes related to RNA machinery account
for 11% of T-ALL cases, with _NKX2-1_-rearranged T-ALL being driven by the RNA helicase _DDX3X_. Genomic alterations were also associated with differential patient outcomes in some
subtypes; for example, in the TAL1 subtype, alterations in _PHF6_, encoding a transcriptional regulator, or in _PTEN_, a negative regulator of PI3K signaling, were associated with inferior
event-free and overall survival. Below, we discuss correlations between major immunophenotypic and molecular features of T-ALL subtypes. EARLY T-LINEAGE PROGENITOR (ETP)-ALL ETP-ALL, one of
the three immunophenotypic subgroups of T-ALL, is associated with a high risk of relapse or failure to achieve remission, particularly in adults. ETP-ALL accounts for 10–15% of newly
diagnosed pediatric T-ALL cases and results in a higher than typical rate of induction failure: ~6% for ETP-ALL versus ~1% for non-ETP-T-ALL13. Prior studies have identified ETP-ALL as a
distinct subtype with notably poor responsiveness to chemotherapy in both pediatric and adult patients14,15,16. Compared with non-ETP-ALL patients, ETP-ALL patients have slower treatment
responses, necessitating high-risk classification and more frequent consideration of hematopoietic stem cell transplantation17. Phenotypically, ETP-ALL blasts are characterized by flow
cytometry with a CD1a-CD4-CD5loCD8- phenotype, indicating leukemic transformation of double-negative (DN; CD4-CD8-) immature thymocyte progenitors14. Dysregulated expression of the
transcription factors LMO2/LYL1 and HOXA is common in ETP-ALL18. Moreover, ETP T-ALL exhibits distinct genomic characteristics and gene expression patterns compared to non-ETP T-ALL, often
with fewer NOTCH1 mutations18, suggesting the involvement of alternative oncogenic pathways. In a recent subgroup analysis, ETP-ALL was most frequently associated with the HOXA and T-other
molecular subgroups, which have inferior overall survival compared with most other subgroups12. Approximately 20% of pediatric ETP-ALLs have activating mutations in genes encoding
interleukin-7 receptor (IL-7R) or the downstream Janus kinases JAK1 and JAK3, both of which are frequently altered in the HOXA molecular subgroup12,19. Consistent with an important role for
aberrant IL-7R signaling in driving ETP-ALL, preclinical studies have shown that activating IL-7R mutations initiate ETP-ALL through blockade of thymocyte development20. ETP-ALL also
aberrantly expresses multiple myeloid and hematopoietic stem cell markers, including CD13, CD33, and CD117, reflecting a progenitor state that precedes T-lineage commitment14,19. The
similarity of ETP-ALL to immature progenitors is further supported by mutations in genes commonly altered in hematopoietic malignancies of other lineages, such as fms-related tyrosine kinase
(FLT3) and isocitrate dehydrogenase 1 (IDH1), which are frequently mutated in myeloid leukemias19. Additionally, mutations in genes encoding transcription factors involved in hematopoiesis,
such as runt-related transcription factor 1 (RUNX1) and ETS variant 6 (ETV 6), are characteristic of ETP-ALL19. EARLY CORTICAL T-ALL Early cortical T-ALL represents a subgroup with
relatively favorable clinical outcomes21. This subtype accounts for 30–35% of pediatric T-ALL cases22 and is characterized phenotypically by CD1a+ membrane CD3 (mCD3)-CD4+CD8+ blasts,
indicating a block at the early cortical double-positive (DP) stage of thymocyte maturation21. Common features of early cortical T-ALL include aberrant expression of the TLX1 (HOX11), TLX3,
NKX2-1, and NKX2-2 transcription factors18,23. Moreover, this subgroup frequently exhibits gain-of-function mutations in NOTCH1 and loss of the tumor suppressor CDKN2A locus18. Early
cortical T-ALL is strongly associated with elevated expression of genes involved in cell cycle progression (E2F7 and CDC2)23. LATE CORTICAL T-ALL Late cortical T-ALL is the most prevalent
subgroup of T-ALL, accounting for 35–60% of T-ALL pediatric cases, with blasts expressing a more mature cortical thymocyte immunophenotype (mCD3+CD4+CD8+)22. It is typically characterized by
aberrant expression of TAL1 with either LMO1 or LMO218. Late cortical T-ALL often presents with favorable patient outcomes24, with a particular trend toward improved event-free survival
rates in patients with TAL1 rearrangements25,26. While late cortical T-ALL exhibits fewer activating mutations in NOTCH1, deletions of the CDKN2A locus are commonly observed in this
subtype18,27. Moreover, the PI3K/AKT pathway is frequently activated through the loss of PTEN, a negative regulator of PI3K signaling, or through activating mutations in PI3K18. CURRENT
T-ALL THERAPIES Patients diagnosed with T-ALL typically undergo a 2-year course of risk-based multi-agent chemotherapy, with or without cranial radiotherapy (CRT)28,29. Treatment includes
remission induction, consolidation, and maintenance therapy phases. The most significant predictor of patient outcomes is minimal residual disease (MRD) status at the conclusion of
consolidation therapy. Risk stratification and treatment for adults and children with T-ALL vary. The Children’s Oncology Group (COG) has developed a risk classification schema for guiding
pediatric T-ALL treatment, with ongoing efforts focused on optimizing conventional chemotherapy agents. While previous treatment regimens for T-ALL frequently included CRT to prevent T-ALL
recurrence in the CNS, more recent therapies have shifted to intrathecal chemotherapy administration and/or higher doses of intravenous chemotherapy, leading to increased survival rates for
pediatric T-ALL patients1. Moreover, the dose regimens of chemotherapy have evolved, with reduced anthracycline concentrations and increased utilization of asparaginase, dexamethasone, and
high-dose methotrexate, and incorporation of nelarabine, particularly in high-risk groups9,30. Findings from the COG AALL0434 study, which was performed from 2009 to 2014 and involved 1256
children, adolescents, and young adults newly diagnosed with T-ALL, highlighted the significance of MRD monitoring for adjusting treatment intensity and improving outcomes13. The authors
emphasized the importance of recognizing ETP-T-ALL patients for early risk assessment and appropriate clinical management. Patients with ETP-T-ALL, as well as near-ETP-T-ALL, exhibited
slower responses to treatment than non-ETP-T-ALL patients, resulting in a higher rate of induction failure for the ETP-T-ALL and near-ETP-T-ALL subtypes. Participants with induction failure
were treated with high-dose methotrexate and nelarabine. In the context of this more aggressive therapy, ETP-T-ALL and near-ETP-T-ALL patients did not experience inferior outcomes relative
to non-ETP-T-ALL patients. Instead, MRD status after consolidation therapy best predicted overall outcomes irrespective of ETP subtype. In a more recent COG phase III trial for T-ALL
(AALL1231), the treatment protocol employed dexamethasone and increased asparaginase, restricted CRT to the 10–15% of patients with CNS disease or persistent MRD positivity, and evaluated
the potential benefits of incorporating bortezomib, an antineoplastic proteasome inhibitor, during induction and delayed intensification31. This study showed that limiting radiation to those
with CNS3 disease or persistent MRD positivity was successful and decreased the incidence of long-term side effects from cranial radiation13 Together, current strategies use intensified
chemotherapy regimens and optimized treatments, such as employing hematopoietic stem cell transplantation or CRT on the basis of MRD and CNS involvement, respectively, leading to significant
improvements in overall treatment outcomes13. RATIONALE FOR INVESTIGATING THE TUMOR MICROENVIRONMENT (TME) IN T-ALL Despite the aforementioned therapeutic advances, survivors often face
multiple morbidities, including secondary malignancies, cardiac issues such as decreased left ventricular ejection fraction, neurological complications, and endocrine disorders,
significantly impacting their quality of life and contributing to premature mortality32. In addition to causing morbidity, current treatments can also result in selection of leukemic clones
carrying mutations that confer chemoresistance, resulting in relapse33,34. Using xenograft models to evaluate clones present before and after chemotherapy, one study found genetic mutations
in key human oncogenes and/or tumor suppressor genes, such as _PTEN_ and _MYC_, resulting in elevated leukemia-initiating cell (LIC) activity in relapsed T-ALL35. To identify novel targets
for more effective treatment of primary and relapsed T-ALL, cell-intrinsic drivers of disease have been identified through genome-wide profiling approaches12, and ongoing clinical trials are
evaluating the potential of targeting some of these factors36. Despite promising anti-leukemic effects observed in vitro and in preclinical studies, therapeutics targeting such
cell-intrinsic drivers of disease have resulted in considerable systemic side effects. For example, gamma secretase inhibitors (GSIs) targeting hyperactivated NOTCH signaling lead to
systemic toxicity in T-ALL patients, particularly affecting the gastrointestinal tract37,38. Similarly, drugs targeting the PI3K signaling pathway cause severe adverse effects, such as
diarrhea and nausea, in patients with acute leukemias39. To date, the development of molecularly targeted therapies that significantly improve patient outcomes remains elusive. Thus, it is
important to broaden our understanding beyond genetic and epigenetic changes to other factors that promote T-ALL progression and/or relapse. Despite multiple pro-leukemic genomic lesions,
mouse and patient T-ALL cells are unable to survive well in vitro without supportive cytokines or signals from other components of the TME40,41,42,43, indicating that targeting the TME may
be a promising novel therapeutic approach. To identify components of the TME that could be targeted to inhibit leukemia progression, it is important to consider the heterogeneity of cellular
and molecular networks that leukemia cells encounter at the distinct sites they occupy, such as the BM and CNS. In this review, we discuss recent findings regarding contributions of
multiple components of the TME to T-ALL pathogenesis. Leukemia-supportive signals provided by the TMEs of distinct organs will be discussed first before turning to perspectives on clinical
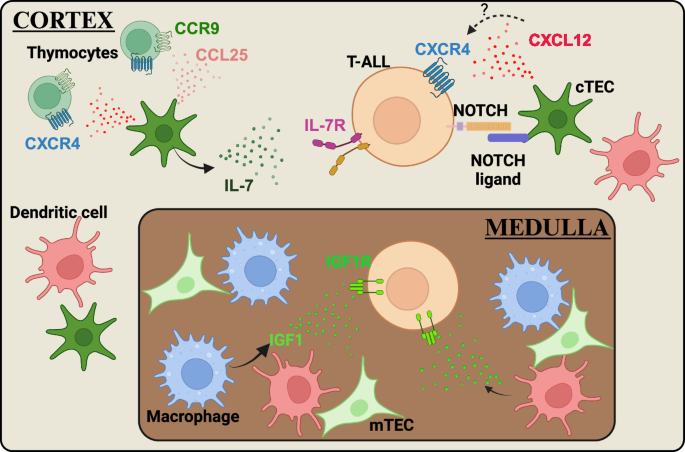
strategies to target the TME. THE ROLE OF TISSUE-SPECIFIC TMES IN SUPPORTING T-ALL THYMUS Thymocytes require bidirectional signaling with heterogeneous thymic stromal cells, including
hematopoietic cells, such as dendritic cells (DCs), and non-hematopoietic cells, such as cortical thymic epithelial cells (cTECs), to develop properly and to support differentiation of the
thymic stromal compartment44,45. Communication between thymocytes and TECs is critical for successful T-cell development. For example, cTECs express IL-7 and the NOTCH ligand DLL4, which are
essential signaling molecules that promote survival, proliferation and T-lineage commitment of immature thymocyte subsets46,47. Given that aberrant activation of NOTCH and IL-7R signaling
is common in T-ALL5, dysregulation of signaling pathways activated by cTECs could contribute to T-ALL initiation. Nascent leukemic cells in the thymus have the opportunity to exploit a wide
variety of signals from the thymic microenvironment to support their own survival and growth. Therefore, understanding the signals between T-ALL cells and the cellular elements in the thymic
TME could reveal potential targets for novel therapies (Fig. 1). IL-7, a growth factor produced in the thymus primarily by cTECs and medullary thymic epithelial cells (mTECs), plays a key
role in T-cell development48,49,50. IL-7 binds to a heterodimeric receptor composed of the IL-7Rα chain (CD127) and common cytokine-receptor γ-chain (CD132); ligand binding activates
IL-7R-associated JAK1 and JAK3 tyrosine kinases51. IL-7R signaling is essential for survival and expansion of early T-cell precursors52,53 and facilitates survival of thymocytes following
β-selection, a checkpoint that tests developing thymocytes for productive TCRβ chain gene rearrangements54. At this critical stage, thymocyte survival and proliferation are tightly
controlled: cells that fail β-selection undergo apoptosis, while those that pass proliferate extensively, expanding the pool of progenitors with productive TCRβ rearrangements. IL-7R
signaling is required to support proliferation of post β-selection thymocytes and prevent premature rearrangement of TCRα chain genes55,56. Given its essential role in thymocyte survival and
proliferation, dysregulation of IL-7R signaling could promote leukemogenesis. In fact, overexpression of wild-type IL-7Rα induces T-ALL in mice, with activation of pathways including
JAK/STAT, PI3K/AKT/mTOR, and NOTCH signaling, closely mimicking human T-ALL57. Furthermore, IL-7R is highly expressed by ~70% of adult and ~60% of pediatric T-ALL samples, regardless of
IL-7R mutational status or T-ALL immunophenotypic classification, and this elevated expression of IL-7R renders T-ALL sensitive to JAK1 inhibition58. Together, these studies indicate that
IL-7 in the leukemic TME could broadly promote progression of T-ALL. Consistent with this possibility, previous studies demonstrated that healthy thymic epithelial cells promote survival and
proliferation of primary human T-ALL cells in vitro in an IL-7-dependent manner59. Furthermore, IL-7 produced by both bone marrow and thymic stromal cells supports survival and growth of
primary patient T-ALL cells60. IL-7 also promotes leukemia expansion in mice engrafted with primary human T-ALL cells by downregulating the cyclin-dependent kinase inhibitor CDKN1B and
upregulating the anti-apoptotic protein BCL-261. Collectively, these findings implicate inhibition of IL-7 signaling as a promising thymic TME-based T-ALL therapy, which is in clinical
evaluation, as discussed below. NOTCH signaling is also essential for several stages of T-cell development, and activated NOTCH is present in the majority of T-ALL samples, where it acts as
a critical oncogene to promote T-ALL leukemogenesis62. NOTCH signaling is required for immature thymocyte progenitors to commit to the T-cell lineage and for expression of transcription
factors that regulate differentiation of CD4-CD8- DN thymocyte subsets from early progenitors through β-selection53. While multiple NOTCH receptors (NOTCH1-4) are expressed by human
thymocytes, NOTCH1 serves as the primary driver of T-cell development and commitment63,64,65. TECs express multiple NOTCH ligands, including DLL1, DLL4, JAG1 and JAG2, but only DLL4 is
essential for T-cell development46. Upon ligand binding, NOTCH1 undergoes a series of proteolytic cleavages, first in the extracellular domain, initiated by an ADAM-family protease, and then
at the transmembrane domain, mediated by the γ-secretase complex. These proteolytic events release the intracellular domain of the NOTCH receptor (ICN) from the membrane66, allowing it to
traffic to the nucleus, where it promotes expression of target genes, such as _MYC_ and _IGF1R_, that are critical for T-cell development, as well as for T-ALL initiation and
progression67,68,69,70. Initial studies identified translocations between the TCR locus and the NOTCH1 gene that resulted in expression of a truncated, constitutively activated NOTCH171.
Subsequent studies revealed numerous additional mutations that activate NOTCH1 in T-ALL, resulting in both ligand-independent and ligand-dependent gain-of-function alleles, which are
collectively present in the majority (>60%) of T-ALL patient samples72. _NOTCH1_ mutations frequently occur in the heterodimerization domain (HD) and/or the C-terminal PEST domain72.
Mutations in the HD region lead to ligand-independent activation of NOTCH1, whereas those in the PEST domain impair degradation of activated NOTCH1 but sustain the requirement for ligand
binding to induce NOTCH signaling. In addition, mutations occur in genes regulating NOTCH1 activity, such as inactivating mutations in FBW7, an E3 ubiquitin ligase that promotes NICD
degradation to terminate NOTCH signaling73,74. Although NOTCH1 functions as an oncogene critical for progression and LIC activity in T-ALL, primary patient samples with activating NOTCH1
mutations survive only when co-cultured with MS5 stromal cells expressing high levels of DLL140, indicating a continued dependence on signals from cells in their microenvironment to activate
NOTCH1 to support T-ALL cell survival. Therefore, targeting the components of the thymic TME that support activation of NOTCH1 signaling may be a promising therapeutic opportunity.
Insulin-like growth factor 1 receptor (IGF1R) represents a crucial downstream target of NOTCH signaling in T-ALL. IGF1R, a receptor tyrosine kinase, is essential for normal growth and
development75 and is particularly important for early stages of T-cell differentiation during fetal development76. IGF1R binds to three ligands: IGF1, the primary high-affinity ligand, IGF2,
and insulin77. While hepatocytes in the liver are the primary source of systemic IGF178,79, small amounts are also produced by other cell types in multiple organs. This localized production
of IGF1 has significant effects on the growth, survival, and differentiation of nearby cells79. Upon ligand binding to IGF1R, a series of downstream phosphorylation events activate both the
PI3K–AKT and MAPK pathways. Dysregulated IGF1R signaling has been implicated in development and progression of various malignancies, notably including T-ALL80,81. NOTCH1 signaling results
in elevated expression of IGF1R, and both pharmacologic and genetic perturbations of IGF1R signaling result in diminished T-ALL growth70. Additionally, T-ALL cells with reduced IGF1R
expression exhibit diminished serial transplantation in mice, indicating reduced LIC activity70. Moreover, T-ALL cells expressing lower levels of IGF1R compensate by increasing PI3K-AKT
activation, revealing the importance of this signaling pathway in T-ALL progression70. In line with these findings, in one study, pharmacologic inhibition of IGF1R decreased growth of a
subset of human T-ALL cell lines, with sensitivity to IGF1R inhibition correlating with surface IGF1R expression levels and PTEN expression82. However, combined IGF1R and PI3Kγ inhibition
did not effectively block growth of PTEN-negative T-ALL cells, suggesting the presence of additional mechanisms driving T-ALL progression82. IL-7 signaling played a distinct role in
supporting T-ALL survival in this study, suggesting that its contribution is independent of the IGF1R-PI3K axis82. Local production of IGF1 can be critical for activating IGF1R signaling in
T-ALL cells. Leukemia-associated myeloid cells, which directly support survival of mouse T-ALL cells in vitro41,42,43,83,84,85, produce IGF1. T-ALL cells show increased activation of IGF1R
relative to healthy thymocytes, and IGF1R inhibition blocks myeloid-mediated T-ALL cell survival, demonstrating that other signals provided by myeloid cells do not override the requirement
for IGF1R activation in vitro41. Subsequent studies revealed that tumor-associated myeloid cells promote initiation and progression of mouse T-ALL by activating IGF1R signaling in vivo42.
Furthermore, human M-CSF-derived macrophages support primary patient T-ALL cells through IGF1R activation in cell culture42. Taken together, these findings suggest that blocking myeloid
support could be a novel means of inhibiting IGF1R activation to specifically target survival of T-ALL cells. BONE MARROW The bone marrow (BM) microenvironment plays a critical role in
maintaining and regulating the differentiation of hematopoietic stem cells (HSCs) and supports leukemia progression86. Prior studies have highlighted the importance of signals within the BM
niche in sustaining leukemic clones and promoting therapeutic resistance in various hematologic malignancies, including T-ALL87,88. Bidirectional interactions between T-ALL cells and diverse
cellular components within the BM have been implicated in T-ALL pathogenesis; leukemia cells interact with both hematopoietic cells, such as macrophages and monocytes, and non-hematopoietic
cells, such as endothelial cells, pericytes, osteolineage cells, and mesenchymal stem cells, in the BM TME42,43,89,90,91. Therefore, a deeper understanding of the interplay between leukemia
cells and the diverse cellular components of the BM microenvironment holds promise for the identification of novel therapies for T-ALL patients (Fig. 2). The BM serves as a crucial niche
that provides factors that support HSCs92. Among these factors is CXCL12, also known as SDF1, a chemokine expressed by endothelial cells, osteoblasts, and mesenchymal stromal cells in the
BM93,94. CXCL12 is the ligand for the chemokine receptor CXCR4, which regulates HSC maintenance. The CXCR4‒CXCL12 axis also supports T-ALL progression in a calcineurin-dependent manner in
the BM95,96. Calcineurin (Cn) is a calcium-activated serine/threonine phosphatase critical for T-cell development97,98 that is also highly activated in T-ALL cells. Pharmacologic inhibition
of Cn induces apoptosis of leukemia cells and prolongs survival in a mouse model of T-ALL95. Subsequent studies using a mouse model in which Cn deletion was restricted to T-ALL cells
demonstrated that Cn was required for interactions between T-ALL cells and supportive stroma, as well as for leukemia progression and LIC activity96. Moreover, conditional deletion of Cn in
T-ALL synergized with vincristine treatment, delaying T-ALL progression96. Additional studies revealed that Cn promotes expression of CXCR4 in T-ALL cells90 and that reduced CXCR4 expression
in Cn-deficient T-ALL results in a migration defect in T-ALL cells that can be rescued by restoring CXCR4 expression90. Taken together, this study revealed that CXCR4 is essential for the
motility, survival, and proliferation of mouse and human T-ALL cells, as well as their ability to home to the BM and support disease progression90. An independent study demonstrated that
T-ALL cells closely interact in a CXCL12-dependent manner with a vascular endothelial cell niche in the BM91. CXCR4 is highly expressed on both mouse and human T-ALL cells, and genetic or
pharmacologic perturbation of CXCR4 reduced T-ALL burden, prolonged mouse survival, and decreased LIC activity91. Collectively, these studies highlight a critical role for CXCR4 signaling in
maintenance and progression of T-ALL in the BM TME, where the ligand CXCL12 is produced by endothelial cells. Given that T-ALL chemoresistance and recurrence in the BM correlate with
inferior outcomes99, targeting the CXCR4‒CXCL12 axis in the BM TME represents a promising target for emergent T-ALL therapies. The mitogen‐activated protein kinase (MAPK/MEK) signaling
pathway is frequently activated in T‐ALL cells from adult patients100. Surprisingly, a previous study revealed that pharmacologic inhibition of MEK promotes growth and proliferation of
patient T‐ALL cells when co‐cultured with stromal cells, while maintaining LIC activity101. This increased growth was found to be mediated by the secretion of IL‐18, a proinflammatory
cytokine, by BM‐derived stromal cells following MEK inhibition101. IL-18 promotes activation and proliferation of T cells and T-ALL cells102. IL-18 was found to be elevated in the peripheral
blood of both T‐ALL‐xenografted mice and T-ALL patients compared with controls, and IL-18 has been shown to support T-ALL cell survival both in vitro and in vivo101. These findings suggest
that further investigation into the role of IL-18 in T-ALL pathogenesis is warranted. The most common site of T-ALL relapse is the BM, where integrins play a key role in leukemia cell
adhesion, migration and metastasis103. Integrins promote cell adhesion by binding a variety of ligands, including the extracellular matrix (ECM) components collagen, fibronectin, and
laminin, which are present in multiple organs, including the BM104,105. Integrins also bind adhesion molecules, such as intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion
molecule 1 (VCAM-1), which are expressed by antigen-presenting cells (APCs) and endothelial cells to mediate cell‒cell contacts104. Integrin-mediated interactions of T-lineage cells with
myeloid and stromal cells are essential for thymocyte development and selection and for T-cell migration, activation, and differentiation44,106. Given the cooperative signaling between
integrins and growth factor receptors, including IGF1R107, and the fact that dysregulated integrin signaling promotes tumor growth and chemotherapeutic resistance108, integrin-mediated
interactions may represent a prime therapeutic opportunity for T-ALL. Pharmacologic blockade of either the integrin lymphocyte function-associated antigen-1 (LFA-1) or its ligand ICAM-1
significantly diminished survival of human T-ALL cell lines co-cultured with BM stromal cells109. Moreover, BM stromal cells supported the survival of patient-derived T-ALL cells in
co-cultures in an LFA-1:ICAM-1-dependent manner109. Integrin β1-mediated interactions between T-ALL cells and ECM components have also been implicated in the development of chemoresistance
and relapse in T-ALL110. In one study, interactions between human T-ALL cells and collagen or Matrigel matrices enhanced T-ALL resistance to doxorubicin in an integrin β1-dependent manner;
furthermore, blockade of integrin β1 reduced T-ALL burden in the BM, prolonged survival, and increased leukemia cell sensitivity to doxorubicin in a mouse model of T-ALL110. Chemotherapy
resistance involved doxorubicin efflux via activation of the ABCC1 drug transporter and the focal adhesion kinase (FAK)-related proline-rich tyrosine kinase 2 (PYK2) pathway110. In addition
to promoting interactions between T-ALL and stromal cells, integrins also play an important role in the interactions of T-ALL cells with myeloid cells in the BM of leukemic mice. T-ALL cells
isolated from the BM of leukemic mice required leukemia-associated myeloid cells to survive in vitro42, and pharmacologic or genetic depletion of myeloid cells from leukemic mice led to a
reduction in T-ALL burden across multiple organs, including the BM42. Subsequent studies used transcriptional profiling and in vitro transwell assays to demonstrate that integrin-mediated
contacts between T-ALL cells and myeloid cells are required for T-ALL survival in vitro43. Blocking integrin ligands or inhibiting downstream FAK/PYK2 signaling not only reduced T-ALL burden
across multiple organs, including the BM, but also prolonged survival of leukemic mice43. Additionally, inhibition of integrin-mediated adhesion or FAK/PYK2 signaling diminished survival of
primary patient T-ALL cells co-cultured with peripheral blood mononuclear cell (PBMC)-derived myeloid cells43. Together, these findings demonstrate that integrin signaling, whether
activated by interactions with the ECM, stromal cells, or myeloid cells, is a key pathway activated by the leukemic TME that promotes T-ALL survival. Thus, integrins and/or downstream
signals could serve as promising therapeutic targets. While the above studies suggest that multiple specific cellular interactions and signals in the BM TME support T-ALL progression,
intravital microscopy revealed dynamic and promiscuous cellular interactions between T-ALL cells and multiple BM elements during disease progression that did not implicate any particular
stromal element in supporting T-ALL111. Thus, further investigation into the interplay between T-ALL cells and diverse cellular components of the BM microenvironment is warranted. SPLEEN
T-ALL cells are frequently detected in the spleen at the time of diagnosis8. Previous studies have highlighted the importance of extrathymic sites, including the spleen, in supporting T-cell
development, particularly following BM transplantation112,113. These findings suggest that the splenic microenvironment may play a role in T-ALL initiation. A previous study revealed that
surgical removal of the spleen prevented the development of T-ALL in a mouse model of DLL4-driven T-ALL114. Moreover, the spleen has been identified as a site of residual disease following
chemotherapy in T-ALL patients115. Previous research emphasized the crucial role of the splenic environment in driving disease progression and inducing therapeutic resistance in multiple
hematologic malignancies, including T-ALL115,116,117. CXCL12 is expressed by splenic stromal cells, including fibroblastic reticular cells118,119, potentially promoting T-ALL survival.
Moreover, emerging evidence indicates that immune cells, notably myeloid cells in the splenic TME, play a role in driving T-ALL progression41,42,43,83,85. Therefore, exploring the cellular
and molecular interactions between T-ALL cells and the splenic TME may aid in identification of therapeutic targets for T-ALL (Fig. 3). The supportive function of myeloid cells in T-ALL
relies on activation of IGF1R, which, as discussed above, is a critical growth factor receptor for LIC activity70. In multiple mouse models, T-ALL cells isolated from the spleen required
myeloid support to survive in vitro41. Furthermore, pharmacologic or genetic ablation of myeloid cells led to a significant reduction in T-ALL burden across multiple organs, including the
spleen and liver, thereby extending mouse survival42. Notably, IGF1R activation was reduced in splenic T-ALL cells following acute myeloid depletion in vivo, suggesting that IGF1R signaling
is an important mechanism by which myeloid cells support T-ALL progression42. Together with the finding that enriched macrophage gene signatures are associated with inferior outcomes in
pediatric T-ALL patients42, the potential of IGF1R signaling as a target for novel patient therapies is evident. Despite the important role of the IGF1R pathway in myeloid-mediated T-ALL
support, exogenous IGF1 is insufficient to sustain T-ALL cell survival in vitro, indicating a role for additional myeloid-derived signals in supporting leukemia cell survival. Notably, T-ALL
cells rely on physical interactions with myeloid cells to survive in vitro43. Aberrant integrin signaling has been shown to drive disease progression and resistance to therapy across
various hematologic malignancies, supporting leukemic cell survival and tissue invasion108. For example, integrin signaling enhances survival and proliferation of AML cells through the
activation of the transcription factors STAT3 and STAT5, as well as the kinase Syk120. As discussed above, when mouse or human T-ALL cells interact with myeloid cells, integrin signaling
activates FAK and PYK243. Inhibiting ICAM-1 and VCAM-1, which respectively bind the integrins LFA-1 and VLA-4 that are expressed by T-ALL cells, or inhibiting FAK/PYK2 signaling reduced
leukemia burden across multiple organs, including the spleen and liver, and prolonged mouse survival43. In PBMCs from pediatric T-ALL patients, elevated gene signatures of the integrin and
FAK signaling pathways correlated with one another, as well as with increased myeloid gene signatures and unfavorable outcomes43. Taken together, these findings suggest that targeting
integrin activation or downstream FAK/PYK2 signaling could be a therapeutic strategy for T-ALL in multiple organs, including the spleen. CENTRAL NERVOUS SYSTEM CNS involvement occurs in
approximately 10% of pediatric and adult ALL patients at diagnosis121,122, which is relatively more common than in other leukemias, such as AML, where the CNS is involved in approximately
1–2% of patients. Notably, the true incidence of CNS involvement may exceed reported rates because leukemia cell counts are below the level of detection at diagnosis in some patients122,123.
Several studies have shown that infiltration of T-ALL cells into the CNS correlates with subsequent disease relapse and poor prognosis124,125. For example, in a study of pediatric ALL
patients experiencing isolated CNS relapse, those who maintained an initial remission duration of over 18 months had a 4-year survival rate of approximately 78%, whereas individuals with a
remission duration shorter than 18 months had a survival rate of approximately 51%124. Patients with the ETP-ALL subtype have been reported to be at an elevated risk of CNS involvement at
diagnosis (e.g., in one study, 4 of the 6 ETP-ALL patients had CNS involvement)15. Leukemic cell infiltration into the CNS can occur through multiple potential routes, including from the BM
of the skull by bridging veins and into the cerebrospinal fluid via the choroid plexus126. Therefore, gaining a better understanding of the mechanisms supporting T-ALL trafficking to and
survival within these regions, including the involvement of chemokines, is critical for developing efficacious treatment options, particularly for patients with CNS involvement who fail to
respond to conventional therapies127,128. Here, we focus on molecular signals directly implicated in the infiltration of T-ALL cells into the CNS (Fig. 4). CCR7 signaling has been identified
as a critical signal for T-ALL trafficking to the CNS129. The CCR7 ligands CCL19 and CCL21 are produced in the CNS by diverse cell types, including macrophages, DCs, microglia, endothelial
cells, and astrocytes130,131,132. In a NOTCH-induced mouse model of T-ALL, CCR7 expression by T-ALL cells was shown to be required for CNS infiltration, as was expression of CCR7 ligands by
the leukemic host129. Furthermore, enforced CCR7 expression in human T-ALL cell lines was sufficient for T-ALL cells to enter the CNS129. Notably, CCR7 expression by T-ALL cells was induced
by the activated NOTCH1 oncogene129. CARMA1, a signaling protein that plays a critical role in lymphocyte activation via the NFκB pathway133, is another key mediator of T-ALL infiltration
into the CNS in mouse models134. Knockdown of CARMA1 in human T-ALL cell lines conferred a survival advantage to mouse hosts, and T-ALL patients with CNS involvement had elevated CARMA1
levels in the BM, supporting a role for CARMA1 in promoting CNS disease in T-ALL patients134. The diminished migration of T-ALL cells after knockdown of CARMA1 in response to CCL21 in vitro
suggests that CARMA1 is linked to downstream signaling of CCR7, although CARMA1 and CCR7 could also independently regulate T-ALL migration and survival through additional signaling
pathways134. In addition to its established role in T-ALL initiation and progression within the BM, the CXCR4‒CXCL12 axis has also been implicated in the invasion of T-ALL cells into the
CNS. Pharmacologic inhibition of CXCR4 significantly diminished the colonization of T-ALL cells in the BM and reduced neuropathologic aspects of the disease126. This study highlighted the
important interplay between CXCR4-mediated BM colonization and infiltration of T-ALL cells into the CNS126. An additional study revealed that both ruxolitinib, a JAK1/2 inhibitor, and
venetoclax, a BCL2 inhibitor, were ineffective in vivo for treating T-ALL in a mouse model because of leukemia cell infiltration into the CNS135. In this study, CXCR4 was found to be
overexpressed in human T-ALL cell lines compared with healthy human T cells and was necessary for CNS infiltration135. Genetic deletion or pharmacologic inhibition of CXCR4 in T-ALL cells
prolonged mouse survival and reduced T-ALL infiltration into the CNS, thus demonstrating the potential efficacy of targeting CXCR4 in combination with conventional chemotherapies135.
Additional recent evidence has highlighted the complexity of the CNS immune microenvironment, suggesting potential novel targets for increasing T-ALL patient survival rates. For example, the
meninges have been shown to contain a diverse array of immune cells specific to CNS surveillance, including non-blood-derived monocyte and neutrophil populations originating in the cranial
BM that populate the CNS borders136. Classical CNS macrophages, specifically meningeal and perivascular macrophages, undergo integrin-mediated interactions with vascular smooth muscle cells
that are critical for their ontogeny and maturation. These studies demonstrate the potential for prominent and supportive interactions between myeloid cells and other cells in the CNS that
have not yet been formally investigated in the context of leukemias137. DISCUSSION AND CLINICAL PERSPECTIVES Although survival rates for T-ALL patients have significantly increased with
current intensified chemotherapy regimens29, significant side effects, including neurotoxicity, seizures, and stroke-like symptoms, remain a major obstacle. As a result, research exploring
therapeutic targets has broadened beyond identifying genetic alterations that impact the survival and progression of T-ALL cells, notably including elucidation of signals in the leukemia
microenvironment. The significant role of the TME in promoting tumor survival and progression across various cancer types, including solid tumors and hematologic malignancies such as T-ALL,
is widely recognized88,138. As discussed above, each leukemic organ comprises a unique TME with distinct supportive signals for T-ALL. Among the key components of the T-ALL TME,
tumor-associated myeloid cells and associated signals have emerged as potential therapeutic targets, as they stand out for their presence across diverse anatomical sites. Myeloid cells
promote T-ALL progression by activating IGF1R and integrin-mediated signaling in T-ALL cells41,42,43. Hypoxia also represents a potential therapeutic target in the TME; recent studies have
shown that hypoxia in the BM inhibits T-ALL cell growth by slowing cell cycle progression and promoting resistance to antileukemic drugs139. Targeting hypoxia-induced factor 1α (HIF-1α), a
key regulator of the cellular response to hypoxia, and activating the mTORC1 pathway may hold promise in overcoming drug resistance in T-ALL within the hypoxic BM TME139. Mesenchymal stem
cells in the BM TME may also support T-ALL by accepting damaged mitochondria from leukemia cells through ICAM-1:integrin-mediated cell adhesion140. Overall, targeting signals provided by
cellular components of the leukemic TME, along with inhibiting cell-intrinsic genetic alterations that drive T-ALL progression, has the potential to enhance the efficacy of T-ALL therapies.
Several clinical-stage inhibitors and antibodies that target signals activated by the TME have been assessed for their efficacy in treating T-ALL and other hematologic malignancies (Table
1). MK-0752, a NOTCH inhibitor, was evaluated in a phase 1 clinical trial for pediatric and adult T-ALL (NCT00100152). However, this study was terminated because of excessive toxicity
related to on-target effects in the intestine, resulting in dose-limiting diarrhea37. CB-103, another pan-NOTCH inhibitor, was also evaluated in adult T-ALL patients (NCT03422679). Despite
exhibiting favorable tolerability with fewer severe side effects, the clinical anti-tumor efficacy of this drug as monotherapy was limited141. BL-8040, a peptide-based CXCR4 antagonist, was
evaluated in combination with chemotherapy in patients with T-ALL (NCT02763384). In this trial involving patients with relapsed or refractory T-ALL, BL-8040 was tested in combination with
nelarabine. BL-8040 was well tolerated and resulted in complete remission in 5 of 9 adult patients142. These studies suggest that disruption of interactions between leukemic blasts and the
BM TME could synergize with chemotherapy. A human monoclonal antibody against IGF1R, figitumumab (also known as CP-751,871), was studied in a phase I trial (NCT01536145) in multiple myeloma
patients. Considering that this drug was well-tolerated, lowered granulocyte IGF1R expression, increased serum IGF1 levels, and reduced the tumor burden in 9 of 27 patients143, further
exploration of its efficacy may hold promise for T-ALL patients. BI-505, a human anti-ICAM-1 monoclonal antibody, was evaluated in multiple myeloma patients (NCT01025206), and it
demonstrated good tolerability, along with efficacy in 7 of 29 patients who had stable disease for at least 8 weeks. Given that BI-505’s efficacy was macrophage-dependent in preclinical
studies, the authors suggest that it is most effective in patients with less impaired immune function and a lower tumor burden144. Recently, the IL-7Rα blocking antibody lusvertikimab (LUSV;
formerly OSE-127) demonstrated suitable tolerability in a clinical trial involving healthy subjects, showing dose-dependent inhibition of IL-7 consumption without serious adverse events145.
Preclinical studies have also shown that anti-IL-7Rα monoclonal antibodies, including LUSV, delayed T-ALL growth and prolonged mouse survival146,147,148, which inspired the current trials
evaluating the inhibition of IL-7 signaling in T-ALL patients. Another approach to targeting IL-7R signaling employs the tyrosine kinase inhibitor ruxolitinib, which disrupts JAK 1 and 2
signaling downstream of IL-7R activation. Preclinical research and phase I and II clinical trials have assessed the safety and efficacy of ruxolitinib in treating various malignancies,
including ALL (NCT02723994)149. In addition, trials investigating CCR7 inhibition in patients with hematologic malignancies are ongoing. Given promising results from preclinical studies on
pro-tumor signals derived from the TME in T-ALL, further clinical trials targeting these pathways are warranted. In conclusion, directing future T-ALL therapies against leukemia-supportive
features of the TME introduces a novel opportunity, deviating from conventional genetic and epigenetic-centric approaches. Understanding the complex interplay between leukemia cells and
cells in their surrounding microenvironment holds promise for developing more effective and less toxic treatments that improve patient outcomes while minimizing adverse effects. These recent
discoveries carry substantial promise for refining the treatment of T-ALL, opening new opportunities for future therapeutic interventions (Fig. 5). REFERENCES * Hunger, S. P. &
Mullighan, C. G. Acute lymphoblastic leukemia in children. _N. Engl. J. Med._ 373, 1541–1552 (2015). Article PubMed CAS Google Scholar * Terwilliger, T. & Abdul-Hay, M. Acute
lymphoblastic leukemia: a comprehensive review and 2017 update. _Blood Cancer J._ 7, e577–e577 (2017). Article PubMed PubMed Central CAS Google Scholar * Sas, V. et al. Transient
leukemia of Down syndrome. _Crit. Rev. Clin. Lab Sci._ 56, 247–259 (2019). Article PubMed CAS Google Scholar * Pui, C.-H., Relling, M. V. & Downing, J. R. Acute lymphoblastic
leukemia. _N. Engl. J. Med._ 350, 1535–1548 (2004). Article PubMed CAS Google Scholar * Vadillo, E., Dorantes-Acosta, E., Pelayo, R. & Schnoor, M. T cell acute lymphoblastic leukemia
(T-ALL): new insights into the cellular origins and infiltration mechanisms common and unique among hematologic malignancies. _Blood Rev._ 32, 36–51 (2018). Article PubMed CAS Google
Scholar * Belver, L. & Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. _Nat. Rev. Cancer_ 16, 494–507 (2016). Article PubMed CAS Google Scholar *
Inaba, H. & Mullighan, C. G. Pediatric acute lymphoblastic leukemia. _Haematologica_ 105, 2524–2539 (2020). Article PubMed PubMed Central CAS Google Scholar * O’Dwyer, K. M. Optimal
approach to T-cell ALL. _Hematology_ 2022, 197–205 (2022). Article PubMed PubMed Central Google Scholar * Al-Mahayri, Z. N., AlAhmad, M. M. & Ali, B. R. Long-term effects of
pediatric acute lymphoblastic leukemia chemotherapy: can recent findings inform old strategies? _Front. Oncol._ 11, 710163 (2021). Article PubMed PubMed Central CAS Google Scholar *
Uckun, F. M. et al. Clinical features and treatment outcome of childhood T-lineage acute lymphoblastic leukemia according to the apparent maturational stage of T-lineage leukemic blasts: a
Children’s Cancer Group study. _J. Clin. Oncol._ 15, 2214–2221 (1997). Article PubMed CAS Google Scholar * Ferrando, A. A. et al. Gene expression signatures define novel oncogenic
pathways in T cell acute lymphoblastic leukemia. _Cancer Cell_ 1, 75–87 (2002). Article PubMed CAS Google Scholar * Brady, S. W. et al. The genomic landscape of pediatric acute
lymphoblastic leukemia. _Nat. Genet._ 54, 1376–1389 (2022). Article PubMed PubMed Central CAS Google Scholar * Wood, B. L. et al. Prognostic significance of ETP phenotype and minimal
residual disease in T-ALL: a Children’s Oncology Group study. _Blood_ 142, 2069–2078 (2023). Article PubMed CAS Google Scholar * Coustan-Smith, E. et al. Early T-cell precursor
leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. _Lancet Oncol._ 10, 147–156 (2009). Article PubMed PubMed Central CAS Google Scholar * Jain, N. et al. Early T-cell
precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: a high-risk subtype. _Blood_ 127, 1863–1869 (2016). Article PubMed PubMed Central CAS Google
Scholar * Conter, V. et al. Early T-cell precursor acute lymphoblastic leukaemia in children treated in AIEOP centres with AIEOP-BFM protocols: a retrospective analysis. _Lancet Haematol._
3, e80–e86 (2016). Article PubMed Google Scholar * Sin, C. & Man, P. M. Early T-Cell precursor acute lymphoblastic leukemia: diagnosis, updates in molecular pathogenesis, management,
and novel therapies. _Front. Oncol._ 11, 750789 (2021). Article PubMed PubMed Central CAS Google Scholar * Liu, Y. et al. The genomic landscape of pediatric and young adult T-lineage
acute lymphoblastic leukemia. _Nat. Genet._ 49, 1211–1218 (2017). Article PubMed PubMed Central CAS Google Scholar * Zhang, J. et al. The genetic basis of early T-cell precursor acute
lymphoblastic leukaemia. _Nature_ 481, 157–163 (2012). Article PubMed PubMed Central CAS Google Scholar * Treanor, L. M. et al. Interleukin-7 receptor mutants initiate early T cell
precursor leukemia in murine thymocyte progenitors with multipotent potential. _J. Exp. Med._ 211, 701–713 (2014). Article PubMed PubMed Central CAS Google Scholar * Niehues, T. et al.
A classification based on T cell selection-related phenotypes identifies a subgroup of childhood T-ALL with favorable outcome in the COALL studies. _Leukemia_ 13, 614–617 (1999). Article
PubMed CAS Google Scholar * Noronha, E. P. et al. The profile of immunophenotype and genotype aberrations in subsets of pediatric T-Cell acute lymphoblastic leukemia. _Front. Oncol._ 9,
316 (2019). Article PubMed PubMed Central Google Scholar * Homminga, I. et al. Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T Cell acute
lymphoblastic leukemia. _Cancer Cell_ 19, 484–497 (2011). Article PubMed CAS Google Scholar * Fattizzo, B., Rosa, J., Giannotta, J. A., Baldini, L. & Fracchiolla, N. S. The
physiopathology of T- Cell acute lymphoblastic leukemia: focus on molecular aspects. _Front. Oncol._ 10, 273 (2020). Article PubMed PubMed Central Google Scholar * Kikuchi, A. et al.
Clinical significance of TAL1 gene alteration in childhood T-cell acute lymphoblastic leukemia and lymphoma. _Leukemia_ 7, 933–938 (1993). PubMed CAS Google Scholar * Bash, R. O. et al.
Clinical features and outcome of T-cell acute lymphoblastic leukemia in childhood with respect to alterations at the TAL1 locus: a Pediatric Oncology Group study. _Blood_ 81, 2110–2117
(1993). Article PubMed CAS Google Scholar * Girardi, T., Vicente, C., Cools, J. & Keersmaecker, K. D. The genetics and molecular biology of T-ALL. _Blood_ 129, 1113–1123 (2017).
Article PubMed CAS Google Scholar * Raetz, E. A. & Teachey, D. T. T-cell acute lymphoblastic leukemia. _Hematology_ 2016, 580–588 (2016). Article PubMed PubMed Central Google
Scholar * Lim, S. J. S., Ford, J. B. & Hermiston, M. L. How I treat newly diagnosed and refractory T-cell acute lymphoblastic lymphoma in children and young adults. _Blood_ 141,
3019–3030 (2023). Google Scholar * Shimony, S., DeAngelo, D. J. & Luskin, M. R. Nelarabine: when and how to use in the treatment of T-cell acute lymphoblastic leukemia. _Blood Adv._ 8,
23–36 (2024). Article PubMed CAS Google Scholar * Teachey, D. T. et al. Children’s oncology group Trial AALL1231: a Phase III clinical trial testing Bortezomib in newly diagnosed T-Cell
acute lymphoblastic leukemia and lymphoma. _J. Clin. Oncol._ 40, 2106–2118 (2022). Article PubMed PubMed Central CAS Google Scholar * Malone, A. & Smith, O. P. Nelarabine toxicity
in children and adolescents with relapsed/refractory T‐ALL/T‐LBL: can we avoid throwing the baby out with the bathwater? _Br. J. Haematol._ 179, 179–181 (2017). Article PubMed CAS Google
Scholar * Tzoneva, G. et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. _Nat. Med._ 19, 368–371 (2013). Article PubMed PubMed
Central CAS Google Scholar * Dieck, C. L. & Ferrando, A. Genetics and mechanisms of NT5C2-driven chemotherapy resistance in relapsed ALL. _Blood_ 133, 2263–2268 (2019). Article
PubMed PubMed Central CAS Google Scholar * Clappier, E. et al. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. _J.
Exp. Med._ 208, 653–661 (2011). Article PubMed PubMed Central CAS Google Scholar * Cordo’, V., van der Zwet, J. C. G., Canté-Barrett, K., Pieters, R. & Meijerink, J. P. P. T-cell
acute lymphoblastic leukemia: a roadmap to targeted therapies. _Blood Cancer Discov._ 2, 19–31 (2021). Article PubMed Google Scholar * Deangelo, D. J. et al. A phase I clinical trial of
the notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. _J. Clin. Oncol._ 24, 6585 (2006). Article Google Scholar *
Papayannidis, C. et al. A Phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. _Blood
Cancer J._ 5, e350 (2015). Article PubMed PubMed Central CAS Google Scholar * Ragon, B. K. et al. Buparlisib, a PI3K inhibitor, demonstrates acceptable tolerability and preliminary
activity in a phase I trial of patients with advanced leukemias. _Am. J. Hematol._ 92, 7–11 (2017). Article PubMed CAS Google Scholar * Armstrong, F. et al. NOTCH is a key regulator of
human T-cell acute leukemia initiating cell activity. _Blood_ 113, 1730–1740 (2009). Article PubMed CAS Google Scholar * Triplett, T. A. et al. Endogenous dendritic cells from the tumor
microenvironment support T-ALL growth via IGF1R activation. _Proc. Natl Acad. Sci. USA_ 113, E1016–E1025 (2016). Article PubMed PubMed Central CAS Google Scholar * Lyu, A. et al.
Tumor-associated myeloid cells provide critical support for T-ALL. _Blood_ 136, 1837–1850 (2020). Article PubMed Google Scholar * Lyu, A. et al. Integrin signaling is critical for
myeloid-mediated support of T-cell acute lymphoblastic leukemia. _Nat. Commun._ 14, 6270 (2023). Article PubMed PubMed Central CAS Google Scholar * Lancaster, J. N., Li, Y. &
Ehrlich, L. I. R. Chemokine-mediated choreography of thymocyte development and selection. _Trends Immunol._ 39, 86–98 (2018). Article PubMed CAS Google Scholar * Ashby, K. M. &
Hogquist, K. A. A guide to thymic selection of T cells. _Nat. Rev. Immunol._ 24, 103–117 (2024). Article PubMed CAS Google Scholar * Koch, U. et al. Delta-like 4 is the essential,
nonredundant ligand for Notch1 during thymic T cell lineage commitment. _J. Exp. Med._ 205, 2515–2523 (2008). Article PubMed PubMed Central CAS Google Scholar * Shitara, S. et al. IL-7
produced by thymic epithelial cells plays a major role in the development of thymocytes and TCRγδ+ intraepithelial lymphocytes. _J. Immunol._ 190, 6173–6179 (2013). Article PubMed CAS
Google Scholar * Peschon, J. J. et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. _J. Exp. Med._ 180, 1955–1960 (1994). Article PubMed CAS
Google Scholar * Zamisch, M. et al. Ontogeny and regulation of IL-7-Expressing thymic epithelial cells. _J. Immunol._ 174, 60–67 (2005). Article PubMed CAS Google Scholar *
Mazzucchelli, R. I. et al. Visualization and identification of IL-7 producing cells in reporter mice. _PLoS ONE_ 4, e7637 (2009). Article PubMed PubMed Central Google Scholar *
Mazzucchelli, R. & Durum, S. K. Interleukin-7 receptor expression: intelligent design. _Nat. Rev. Immunol._ 7, 144–154 (2007). Article PubMed CAS Google Scholar * Freeden-Jeffry, U.,
von, Solvason, N., Howard, M. & Murray, R. The Earliest T Lineage–committed cells depend on IL-7 for Bcl-2 expression and normal cell cycle progression. _Immunity_ 7, 147–154 (1997).
Article Google Scholar * Shah, D. K. & Zúñiga-Pflücker, J. C. An overview of the intrathymic intricacies of T Cell development. _J. Immunol._ 192, 4017–4023 (2014). Article PubMed
CAS Google Scholar * Trigueros, C. et al. Pre‐TCR signaling regulates IL‐7 receptor α expression promoting thymocyte survival at the transition from the double‐negative to double‐positive
stage. _Eur. J. Immunol._ 33, 1968–1977 (2003). Article PubMed CAS Google Scholar * Hong, C., Luckey, M. A. & Park, J.-H. Intrathymic IL-7: the where, when, and why of IL-7 signaling
during T cell development. _Semin. Immunol._ 24, 151–158 (2012). Article PubMed PubMed Central CAS Google Scholar * Boudil, A. et al. IL-7 coordinates proliferation, differentiation
and Tcra recombination during thymocyte β-selection. _Nat. Immunol._ 16, 397–405 (2015). Article PubMed PubMed Central CAS Google Scholar * Silva, A. et al. Overexpression of wild-type
IL-7Rα promotes T-cell acute lymphoblastic leukemia/lymphoma. _Blood_ 138, 1040–1052 (2021). Article PubMed PubMed Central CAS Google Scholar * Courtois, L. et al. IL7-receptor
expression is frequent in T-cell acute lymphoblastic leukemia and predicts sensitivity to JAK-inhibition. _Blood_ 142, 158–171 (2023). PubMed CAS Google Scholar * Scupoli, M. T. et al.
Thymic epithelial cells promote survival of human T-cell acute lymphoblastic leukemia blasts: the role of interleukin-7. _Haematologica_ 88, 1229–1237 (2003). PubMed CAS Google Scholar *
Scupoli, M. T. et al. Interleukin 7 requirement for survival of T-cell acute lymphoblastic leukemia and human thymocytes on bone marrow stroma. _Haematologica_ 92, 264–266 (2007). Article
PubMed Google Scholar * Silva, A. et al. IL-7 contributes to the progression of human T-cell acute lymphoblastic leukemias. _Cancer Res._ 71, 4780–4789 (2011). Article PubMed CAS Google
Scholar * Li, X. & von Boehmer, H. Notch Signaling in T-Cell Development and T-ALL. _ISRN Hematol._ 2011, 921706 (2011). Article PubMed PubMed Central Google Scholar * Radtke, F.
et al. Deficient T cell fate specification in mice with an induced inactivation of Notch1. _Immunity_ 10, 547–558 (1999). Article PubMed CAS Google Scholar * Pui, J. C. et al. Notch1
expression in early lymphopoiesis influences B versus T lineage determination. _Immunity_ 11, 299–308 (1999). Article PubMed CAS Google Scholar * Shi, J., Fallahi, M., Luo, J.-L. &
Petrie, H. T. Nonoverlapping functions for Notch1 and Notch3 during murine steady-state thymic lymphopoiesis. _Blood_ 118, 2511–2519 (2011). Article PubMed PubMed Central CAS Google
Scholar * Struhl, G. & Greenwald, I. Presenilin-mediated transmembrane cleavage is required for Notch signal transduction in Drosophila. _Proc. Natl Acad. Sci. USA_ 98, 229–234 (2001).
Article PubMed CAS Google Scholar * Weng, A. P. et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. _Genes Dev._ 20, 2096–2109 (2006).
Article PubMed PubMed Central CAS Google Scholar * Palomero, T. et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell
growth. _Proc. Natl Acad. Sci. USA_ 103, 18261–18266 (2006). Article PubMed PubMed Central CAS Google Scholar * Sharma, V. M. et al. Notch1 contributes to mouse T-Cell leukemia by
directly inducing the expression of c-myc. _Mol. Cell Biol._ 26, 8022–8031 (2006). Article PubMed PubMed Central CAS Google Scholar * Medyouf, H. et al. High-level IGF1R expression is
required for leukemia-initiating cell activity in T-ALL and is supported by Notch signaling. _J. Exp. Med._ 208, 1809–1822 (2011). Article PubMed PubMed Central CAS Google Scholar *
Ellisen, L. W. et al. TAN-1, the human homolog of the Drosophila Notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. _Cell_ 66, 649–661 (1991). Article PubMed
CAS Google Scholar * Weng, A. P. et al. Activating Mutations of NOTCH1 in Human T Cell Acute Lymphoblastic Leukemia. _Science_ 306, 269–271 (2004). Article PubMed CAS Google Scholar
* Thompson, B. J. et al. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. _J. Exp. Med._ 204, 1825–1835 (2007). Article PubMed PubMed Central CAS Google
Scholar * Lobry, C., Oh, P. & Aifantis, I. Oncogenic and tumor suppressor functions of Notch in cancer: it’s NOTCH what you think. _J. Exp. Med._ 208, 1931–1935 (2011). Article PubMed
PubMed Central CAS Google Scholar * Ullrich, A. et al. Insulin‐like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that
define functional specificity. _EMBO J._ 5, 2503–2512 (1986). Article PubMed PubMed Central CAS Google Scholar * Kecha, O. et al. Involvement of insulin-like growth factors in early T
cell development: a study using fetal thymic organ cultures 1. _Endocrinology_ 141, 1209–1217 (2000). Article PubMed CAS Google Scholar * Adams, T. E., Epa, V. C., Garrett, T. P. J.
& Ward, C. W. Structure and function of the type 1 insulin-like growth factor receptor. _Cell Mol. Life Sci._ 57, 1050–1093 (2000). Article PubMed PubMed Central CAS Google Scholar
* Sjögren, K. et al. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. _Proc. Natl Acad.
Sci. USA_ 96, 7088–7092 (1999). Article PubMed PubMed Central Google Scholar * Laron, Z. Insulin-like growth factor 1 (IGF-1): a growth hormone. _Mol. Pathol._ 54, 311–316 (2001).
Article PubMed PubMed Central CAS Google Scholar * Pollak, M. N., Schernhammer, E. S. & Hankinson, S. E. Insulin-like growth factors and neoplasia. _Nat. Rev. Cancer_ 4, 505–518
(2004). Article PubMed CAS Google Scholar * Gusscott, S., Tamiro, F., Giambra, V. & Weng, A. P. Insulin-like growth factor (IGF) signaling in T-cell acute lymphoblastic leukemia.
_Adv. Biol. Regul._ 74, 100652 (2019). Article PubMed CAS Google Scholar * Gusscott, S. et al. IGF1R Derived PI3K/AKT Signaling Maintains Growth in a Subset of Human T-Cell Acute
Lymphoblastic Leukemias. _PLoS ONE_ 11, e0161158 (2016). Article PubMed PubMed Central Google Scholar * Chen, S.-Y. et al. Organ-specific microenvironment modifies diverse functional and
phenotypic characteristics of leukemia-associated macrophages in mouse T cell acute lymphoblastic leukemia. _J. Immunol._ 194, 2919–2929 (2015). Article PubMed CAS Google Scholar *
Yang, X. et al. Hepatic leukemia-associated macrophages exhibit a pro-inflammatory phenotype in Notch1-induced acute T cell leukemia. _Immunobiology_ 223, 73–80 (2018). Article PubMed CAS
Google Scholar * Yang, F. et al. Monocyte-derived leukemia-associated macrophages facilitate extramedullary distribution of T-cell acute lymphoblastic leukemia cells. _Cancer Res._ 80,
3677–3691 (2020). Article PubMed CAS Google Scholar * Reagan, M. R. & Rosen, C. J. Navigating the bone marrow niche: translational insights and cancer-driven dysfunction. _Nat. Rev.
Rheumatol._ 12, 154–168 (2016). Article PubMed CAS Google Scholar * Passaro, D., Quang, C. T. & Ghysdael, J. Microenvironmental cues for T‐cell acute lymphoblastic leukemia
development. _Immunol. Rev._ 271, 156–172 (2016). Article PubMed CAS Google Scholar * Witkowski, M. T., Kousteni, S. & Aifantis, I. Mapping and targeting of the leukemic
microenvironment. _J. Exp. Med._ 217, e20190589 (2019). Article PubMed Central Google Scholar * Boyerinas, B. et al. Adhesion to osteopontin in the bone marrow niche regulates
lymphoblastic leukemia cell dormancy. _Blood_ 121, 4821–4831 (2013). Article PubMed PubMed Central CAS Google Scholar * Passaro, D. et al. CXCR4 is required for leukemia-initiating cell
activity in T cell acute lymphoblastic leukemia. _Cancer Cell_ 27, 769–779 (2015). Article PubMed CAS Google Scholar * Pitt, L. A. et al. CXCL12-producing vascular endothelial niches
control acute T cell leukemia maintenance. _Cancer Cell_ 27, 755–768 (2015). Article PubMed PubMed Central CAS Google Scholar * Morrison, S. J. & Scadden, D. T. The bone marrow
niche for haematopoietic stem cells. _Nature_ 505, 327–334 (2014). Article PubMed PubMed Central CAS Google Scholar * Greenbaum, A. et al. CXCL12 in early mesenchymal progenitors is
required for haematopoietic stem-cell maintenance. _Nature_ 495, 227–230 (2013). Article PubMed PubMed Central CAS Google Scholar * Ding, L. & Morrison, S. J. Haematopoietic stem
cells and early lymphoid progenitors occupy distinct bone marrow niches. _Nature_ 495, 231–235 (2013). Article PubMed PubMed Central CAS Google Scholar * Medyouf et al. Targeting
calcineurin activation as a therapeutic strategy for T-cell acute lymphoblastic leukemia. _Nat. Med._ 13, 736–741 (2007). Article PubMed CAS Google Scholar * Gachet, S. et al.
Leukemia-initiating cell activity requires calcineurin in T-cell acute lymphoblastic leukemia. _Leukemia_ 27, 2289–2300 (2013). Article PubMed CAS Google Scholar * Bueno, O. F., Brandt,
E. B., Rothenberg, M. E. & Molkentin, J. D. Defective T cell development and function in calcineurin Aβ-deficient mice. _Proc. Natl Acad. Sci. USA_ 99, 9398–9403 (2002). Article PubMed
PubMed Central CAS Google Scholar * Neilson, J. R., Winslow, M. M., Hur, E. M. & Crabtree, G. R. Calcineurin B1 is essential for positive but not negative selection during thymocyte
development. _Immunity_ 20, 255–266 (2004). Article PubMed CAS Google Scholar * Bhojwani, D. & Pui, C.-H. Relapsed childhood acute lymphoblastic leukaemia. _Lancet Oncol._ 14,
e205–e217 (2013). Article PubMed Google Scholar * Gregorj, C. et al. ERK1/2 phosphorylation is an independent predictor of complete remission in newly diagnosed adult acute lymphoblastic
leukemia. _Blood_ 109, 5473–5476 (2007). Article PubMed CAS Google Scholar * Uzan, B. et al. Interleukin‐18 produced by bone marrow‐derived stromal cells supports T‐cell acute leukaemia
progression. _EMBO Mol. Med._ 6, 821–834 (2014). Article PubMed PubMed Central CAS Google Scholar * Nakanishi, K. Unique action of Interleukin-18 on T cells and other immune cells.
_Front. Immunol._ 9, 763 (2018). Article PubMed PubMed Central Google Scholar * Duarte, D., Hawkins, E. D. & Celso, C. L. The interplay of leukemia cells and the bone marrow
microenvironment. _Blood_ 131, 1507–1511 (2018). Article PubMed CAS Google Scholar * Humphries, J. D., Byron, A. & Humphries, M. J. Integrin ligands at a glance. _J. Cell Sci._ 119,
3901–3903 (2006). Article PubMed CAS Google Scholar * Gattazzo, F., Urciuolo, A. & Bonaldo, P. Extracellular matrix: a dynamic microenvironment for stem cell niche. _Biochim.
Biophys. Acta_ 1840, 2506–2519 (2014). Article PubMed PubMed Central CAS Google Scholar * Chapman, N. M. & Houtman, J. C. D. Functions of the FAK family kinases in T cells: beyond
actin cytoskeletal rearrangement. _Immunol. Res._ 59, 23–34 (2014). Article PubMed PubMed Central CAS Google Scholar * Cox, O. T. et al. IGF-1 receptor and adhesion signaling: an
important axis in determining cancer cell phenotype and therapy resistance. _Front. Endocrinol._ 6, 106 (2015). Article Google Scholar * Cooper, J. & Giancotti, F. G. Integrin
signaling in cancer: mechanotransduction, stemness, epithelial plasticity, and therapeutic resistance. _Cancer Cell_ 35, 347–367 (2019). Article PubMed PubMed Central CAS Google Scholar
* Winter, S. S. et al. Enhanced T‐lineage acute lymphoblastic leukaemia cell survival on bone marrow stroma requires involvement of LFA‐1 and ICAM‐1. _Br. J. Haematol._ 115, 862–871
(2001). Article PubMed CAS Google Scholar * Berrazouane, S. et al. Beta1 integrin blockade overcomes doxorubicin resistance in human T-cell acute lymphoblastic leukemia. _Cell Death
Dis._ 10, 357 (2019). Article PubMed PubMed Central Google Scholar * Hawkins, E. D. et al. T-cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments.
_Nature_ 538, 518–522 (2016). Article PubMed PubMed Central Google Scholar * Lancrin, C. et al. Major T cell progenitor activity in bone marrow–derived spleen colonies. _J. Exp. Med._
195, 919–929 (2002). Article PubMed PubMed Central CAS Google Scholar * Maillard, I. et al. Notch-dependent T-lineage commitment occurs at extrathymic sites following bone marrow
transplantation. _Blood_ 107, 3511–3519 (2006). Article PubMed PubMed Central CAS Google Scholar * Xiong, H. et al. Spleen plays a major role in DLL4-driven acute T-cell lymphoblastic
leukemia. _Theranostics_ 11, 1594–1608 (2021). Article PubMed PubMed Central CAS Google Scholar * Grande, A. D. et al. The spleen as a sanctuary site for residual leukemic cells
following ABT-199 monotherapy in ETP-ALL. _Blood Adv._ 5, 1963–1976 (2021). Article PubMed PubMed Central Google Scholar * Pelusi, N. et al. The spleen microenvironment influences
disease transformation in a mouse model of KITD816V-dependent myeloproliferative neoplasm. _Sci. Rep._ 7, 41427 (2017). Article PubMed PubMed Central CAS Google Scholar * Bührer, E. D.
et al. Splenic red pulp macrophages provide a niche for CML stem cells and induce therapy resistance. _Leukemia_ 36, 2634–2646 (2022). Article PubMed PubMed Central Google Scholar *
Ellyard, J. I., Avery, D. T., Mackay, C. R. & Tangye, S. G. Contribution of stromal cells to the migration, function and retention of plasma cells in human spleen: potential roles of
CXCL12, IL‐6 and CD54. _Eur. J. Immunol._ 35, 699–708 (2005). Article PubMed CAS Google Scholar * Umemoto, E. et al. Constitutive plasmacytoid dendritic cell migration to the splenic
white pulp is cooperatively regulated by CCR7- and CXCR4-Mediated signaling. _J. Immunol._ 189, 191–199 (2012). Article PubMed CAS Google Scholar * Oellerich, T. et al. β2
integrin–derived signals induce cell survival and proliferation of AML blasts by activating a Syk/STAT signaling axis. _Blood_ 121, 3889–3899 (2013). Article PubMed CAS Google Scholar *
Thastrup, M., Duguid, A., Mirian, C., Schmiegelow, K. & Halsey, C. Central nervous system involvement in childhood acute lymphoblastic leukemia: challenges and solutions. _Leukemia_ 36,
2751–2768 (2022). Article PubMed PubMed Central Google Scholar * Kopmar, N. E. & Cassaday, R. D. How I prevent and treat central nervous system disease in adults with acute
lymphoblastic leukemia. _Blood_ 141, 1379–1388 (2023). Article PubMed CAS Google Scholar * Ganzel, C. et al. CNS involvement in AML at diagnosis is rare and does not affect response or
survival: data from 11 ECOG-ACRIN trials. _Blood Adv._ 5, 4560–4568 (2021). Article PubMed PubMed Central CAS Google Scholar * Barredo, J. C. et al. Isolated CNS relapse of acute
lymphoblastic leukemia treated with intensive systemic chemotherapy and delayed CNS radiation: a pediatric oncology group study. _J. Clin. Oncol._ 24, 3142–3149 (2006). Article PubMed CAS
Google Scholar * Fielding, A. K. et al. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. _Blood_ 109, 944–950 (2007). Article
PubMed CAS Google Scholar * Jost, T. R. et al. Role of CXCR4‐mediated bone marrow colonization in CNS infiltration by T cell acute lymphoblastic leukemia. _J. Leukoc. Biol._ 99, 1077–1087
(2016). Article PubMed CAS Google Scholar * März, M. et al. Pediatric acute lymphoblastic leukemia—Conquering the CNS across the choroid plexus. _Leuk. Res._ 71, 47–54 (2018). Article
PubMed Google Scholar * Ribera, J.-M. T-ALL in CNS-3 status needs improvement. _Blood_ 141, 1779–1780 (2023). Article PubMed CAS Google Scholar * Buonamici, S. et al. CCR7 signalling
as an essential regulator of CNS infiltration in T-cell leukaemia. _Nature_ 459, 1000–1004 (2009). Article PubMed PubMed Central CAS Google Scholar * Columba-Cabezas, S., Serafini, B.,
Ambrosini, E. & Aloisi, F. Lymphoid Chemokines CCL19 and CCL21 are Expressed in the central nervous system during experimental autoimmune encephalomyelitis: implications for the
maintenance of chronic neuroinflammation. _Brain Pathol._ 13, 38–51 (2003). Article PubMed Google Scholar * Cupovic, J. et al. Central nervous system stromal cells control Local CD8+ T
cell responses during virus-induced neuroinflammation. _Immunity_ 44, 622–633 (2016). Article PubMed PubMed Central CAS Google Scholar * O’Connor, T. et al. Age-related gliosis promotes
central nervous system lymphoma through CCL19-mediated tumor cell retention. _Cancer Cell_ 36, 250–267.e9 (2019). Article PubMed Google Scholar * Thome, M. CARMA1, BCL-10 and MALT1 in
lymphocyte development and activation. _Nat. Rev. Immunol._ 4, 348–359 (2004). Article PubMed CAS Google Scholar * Oruganti, S. R. et al. CARMA1 is a novel regulator of T-ALL disease and
leukemic cell migration to the CNS. _Leukemia_ 31, 255–258 (2017). Article PubMed CAS Google Scholar * Walker, K. L. et al. CXCR4 allows T cell acute lymphoblastic leukemia to escape
from JAK1/2 and BCL2 inhibition through CNS infiltration. _Leuk. Lymphoma_ 62, 1167–1177 (2021). Article PubMed PubMed Central CAS Google Scholar * Cugurra, A. et al. Skull and
vertebral bone marrow are myeloid cell reservoirs for the meninges and CNS parenchyma. _Science_ 373, eabf7844 (2021). Article PubMed PubMed Central CAS Google Scholar * Masuda, T. et
al. Specification of CNS macrophage subsets occurs postnatally in defined niches. _Nature_ 604, 740–748 (2022). Article PubMed CAS Google Scholar * Binnewies, M. et al. Understanding the
tumor immune microenvironment (TIME) for effective therapy. _Nat. Med._ 24, 541–550 (2017). Article Google Scholar * Fahy, L. et al. Hypoxia favors chemoresistance in T-ALL through an
HIF1α-mediated mTORC1 inhibition loop. _Blood Adv._ 5, 513–526 (2021). Article PubMed PubMed Central CAS Google Scholar * Wang, J. et al. Cell adhesion-mediated mitochondria transfer
contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. _J. Hematol. Oncol._ 11, 11 (2018). Article PubMed PubMed Central Google Scholar
* Hanna, G. J. et al. A Phase I study of the pan-notch inhibitor CB-103 for patients with advanced adenoid cystic carcinoma and other tumors. _Cancer Res. Commun._ 3, 1853–1861 (2023).
Article PubMed PubMed Central CAS Google Scholar * Uy, G. L. et al. CXCR4 Inhibition with BL-8040 in Combination with Nelarabine in Patients with Relapsed or Refractory T-Cell Acute
Lymphoblastic Leukemia / Lymphoblastic Lymphoma. _Blood_ 134, 2630 (2019). Article Google Scholar * Lacy, M. Q. et al. Phase I, pharmacokinetic and pharmacodynamic study of the
anti–insulinlike growth factor Type 1 receptor monoclonal antibody CP-751,871 in patients with multiple myeloma. _J. Clin. Oncol._ 26, 3196–3203 (2008). Article PubMed CAS Google Scholar
* Hansson, M. et al. A Phase I dose-escalation study of antibody BI-505 in relapsed/refractory multiple myeloma. _Clin. Cancer Res._ 21, 2730–2736 (2015). Article PubMed CAS Google
Scholar * Poirier, N. et al. First-in-human study in healthy subjects with the noncytotoxic monoclonal antibody OSE-127, a strict antagonist of IL-7Rα. _J. Immunol._ 210, 753–763 (2023).
Article PubMed CAS Google Scholar * Akkapeddi, P. et al. A fully human anti-IL-7Rα antibody promotes antitumor activity against T-cell acute lymphoblastic leukemia. _Leukemia_ 33,
2155–2168 (2019). Article PubMed PubMed Central CAS Google Scholar * Hixon, J. A. et al. New anti-IL-7Rα monoclonal antibodies show efficacy against T cell acute lymphoblastic leukemia
in pre-clinical models. _Leukemia_ 34, 35–49 (2020). Article PubMed CAS Google Scholar * Lenk, L. et al. The IL-7R antagonist lusvertikimab reduces leukemic burden in xenograft ALL via
antibody-dependent cellular phagocytosis. _Blood_ 143, 2735–2748 (2024). Article PubMed PubMed Central CAS Google Scholar * Maude, S. L. et al. Targeting JAK1/2 and mTOR in murine
xenograft models of Ph-like acute lymphoblastic leukemia. _Blood_ 120, 3510–3518 (2012). Article PubMed PubMed Central CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank
all members of the Ehrlich laboratory for helpful discussion and advice. This work was supported by RP240054 from the Cancer Prevention and Research Institute of Texas (to L.I.R.E.). AUTHOR
INFORMATION AUTHORS AND AFFILIATIONS * Division of Hematology/Oncology, Department of Medicine, University of California, San Francisco, CA, USA Aram Lyu * Department of Molecular
Biosciences, The University of Texas at Austin, Austin, TX, USA Seo Hee Nam, Ryan S. Humphrey & Lauren I. R. Ehrlich * Department of Pediatrics, Baylor College of Medicine/Dan L. Duncan
Cancer Center and Texas Children’s Cancer Center, Houston, TX, USA Terzah M. Horton * Department of Oncology, Livestrong Cancer Institutes, The University of Texas at Austin Dell Medical
School, Austin, TX, USA Lauren I. R. Ehrlich Authors * Aram Lyu View author publications You can also search for this author inPubMed Google Scholar * Seo Hee Nam View author publications
You can also search for this author inPubMed Google Scholar * Ryan S. Humphrey View author publications You can also search for this author inPubMed Google Scholar * Terzah M. Horton View
author publications You can also search for this author inPubMed Google Scholar * Lauren I. R. Ehrlich View author publications You can also search for this author inPubMed Google Scholar
CORRESPONDING AUTHOR Correspondence to Lauren I. R. Ehrlich. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a
Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit
to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are
included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and
your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this
licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Lyu, A., Nam, S.H., Humphrey, R.S. _et al._ Cells and signals of the
leukemic microenvironment that support progression of T-cell acute lymphoblastic leukemia (T-ALL). _Exp Mol Med_ 56, 2337–2347 (2024). https://doi.org/10.1038/s12276-024-01335-7 Download
citation * Received: 06 June 2024 * Revised: 30 July 2024 * Accepted: 11 August 2024 * Published: 01 November 2024 * Issue Date: November 2024 * DOI:
https://doi.org/10.1038/s12276-024-01335-7 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative