Play all audios:
ABSTRACT Respiratory syncytial virus (RSV) is a leading cause of severe lower respiratory tract infections, especially in infants. Lung neutrophilia is a hallmark of RSV disease but the
mechanism by which neutrophils are recruited and activated is unclear. Here, we investigate the innate immune signaling pathways underlying neutrophil recruitment and activation in
RSV-infected mice. We show that MyD88/TRIF signaling is essential for lung neutrophil recruitment while MAVS signaling, leading to type I IFN production, is necessary for neutrophil
activation. Consistent with that notion, administration of type I IFNs to the lungs of RSV-infected _Mavs__−/−_ mice partially activates lung neutrophils recruited via the MyD88/TRIF
pathway. Conversely, lack of neutrophil recruitment to the lungs of RSV-infected _Myd88/Trif__−/−_ mice can be corrected by administration of chemoattractants and those neutrophils become
fully activated. Interestingly, _Myd88/Trif__−/−_ mice did not have increased lung viral loads during RSV infection, suggesting that neutrophils are dispensable for viral control. Thus, two
distinct pathogen sensing pathways collaborate for neutrophil recruitment and full activation during RSV infection. SIMILAR CONTENT BEING VIEWED BY OTHERS ATG5 SUPPRESSES TYPE I
IFN-DEPENDENT NEUTROPHIL EFFECTOR FUNCTIONS DURING _MYCOBACTERIUM TUBERCULOSIS_ INFECTION IN MICE Article 15 May 2025 NEUTROPHILS IN RESPIRATORY VIRAL INFECTIONS Article Open access 23 March
2021 CONTROL OF IFN-I RESPONSES BY THE AMINOPEPTIDASE IRAP IN NEONATAL C57BL/6 ALVEOLAR MACROPHAGES DURING RSV INFECTION Article Open access 12 April 2021 INTRODUCTION Neutrophils are
classically considered as among the first immune cells to respond to both infection and injury. Historically, neutrophils were considered as non-specific effector cells but it is becoming
increasingly clear that they can respond differentially to harmful stimuli.1 Pattern recognition receptor (PRR) signaling induces innate immune responses including the generation of
chemotactic gradients along which neutrophils are recruited into affected tissues.2,3 Following recruitment, additional signals are required to trigger neutrophil activation and
degranulation. During activation, neutrophils can secrete hundreds of pre-stored effector proteins and proteolytic enzymes including matrix metalloproteinases (MMPs), myeloperoxidase (MPO),
and neutrophil elastase (NE).4 Neutrophils can also drive inflammation by generating reactive oxygen species (ROS), as well as by undergoing a unique form of cell death whereby they secrete
nuclear DNA in the form of neutrophil extracellular traps (NETs).4,5,6 Whilst neutrophils are important as immediate antimicrobial responders, their recruitment and activation must be
tightly regulated to minimalize pathology caused by bystander effects on host tissues.7,8 This is particularly relevant in the lung where the disruption of delicate alveolar structures can
rapidly have life-threatening consequences if gas exchange is compromised. The molecular mechanisms regulating neutrophil recruitment and activation in response to bacterial and fungal
respiratory infections have been well characterized.9 However, considerably less is known about the regulation of neutrophil action during respiratory viral infections.10,11,12 Respiratory
syncytial virus (RSV) is the greatest cause of infant hospitalizations in the developed world.13,14 More than 90% of children encounter RSV within their first 24 months and a minority
develops severe disease; 1–3% will require hospitalization.15 In addition to acute disease, RSV-induced bronchiolitis and viral pneumonia in infancy are associated with debilitating,
long-term respiratory disorders, such as recurrent wheeze16 and asthma.17 Although it is not well understood why some infants are asymptomatic while other infants develop severe disease,
host immune factors have been implicated in disease severity.18,19 During RSV infection, neutrophils are recruited to the lungs of both mouse and man.20,21,22 Clinical studies suggest that
neutrophils contribute to immune pathology during disease20,22,23 and they are the most abundant cell type in the bronchoalveolar lavage (BAL) of infants with RSV-induced bronchiolitis.20,22
Neutrophil activation during RSV infection has been more difficult to assess clinically than neutrophil recruitment. However, elevated NE concentrations have been observed in both the serum
and nasal lavage of children with confirmed RSV infection.23 Together, these studies suggest that a hyperactive or dysregulated neutrophil response to RSV may underlie disease severity and
indicate the importance of studying the signaling pathways that regulate neutrophil recruitment and activation in the infected lung. Innate immune receptors on epithelial cells and alveolar
macrophages (AMs) initiate the immune response to RSV within the respiratory tract.24,25 The plasma membrane expressed Toll-like receptors (TLRs), TLR2/TLR6 and TLR4, and the
endosomally-expressed TLR3 and TLR7, have all been implicated in RSV detection.25,26 RSV is also detected in the cytosol by retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs):
RIG-I and melanoma differentiation–associated protein 5 (MDA5).27,28,29 RLR signaling occurs through the adaptor protein mitochondrial antiviral signaling protein (MAVS)30,31 while TLRs
signal via the adaptor proteins MyD88 and/or TRIF.32 Signaling via MAVS, MyD88 and TRIF induces the expression of type I and type III interferons (IFNs), as well as other pro-inflammatory
cytokines and chemokines.33 Type I IFNs bind to the interferon-α/β receptor (IFNAR) expressed on all nucleated cells and induce the expression of hundreds of IFN-stimulated genes (ISGs).34
ISG protein products have direct antiviral properties and can also promote the recruitment and activation of other components of the immune response, including antiviral monocytes.21,27 In
this study, we investigated the necessity of MAVS and MyD88/TRIF dependent signaling pathways to lung neutrophil recruitment and activation during RSV infection in vivo. _Mavs__−/−_ mice,
unable to signal via MDA-5 or RIG-I, and _Myd88/Trif__−/−_ mice, unable to signal via TLRs (and the IL-1 receptor (IL-1R) and IL-18 receptor (IL-18R)) were infected with RSV. We found that
neutrophil recruitment to the lung was dependent on MyD88/TRIF signaling but mostly independent of MAVS signaling. The neutrophil chemoattractant CXCL1 was shown to be mainly produced by
non-epithelial, non-endothelial (CD45−, Epcam−, CD31−) lung cells. However, neutrophil activation in the lung was dependent on the type I IFN-driven, pro-inflammatory lung environment
induced by MAVS signaling. Restoring the pro-inflammatory environment in the lungs of _Mavs__−/−_ mice using recombinant IFN-α (rIFN-α) was sufficient to partially activate neutrophils
during RSV infection. Conversely, neutrophils recruited into the lungs of _Myd88/Trif__−/−_ mice using recombinant CXCL1 (rCXCL1) became fully activated during RSV infection. This study
sheds light on the molecular mechanisms that drive neutrophil recruitment and activation in the lungs during RSV infection and demonstrates how two distinct PRR signaling pathways
collaborate to drive neutrophil responses to virus infection. RESULTS RSV INFECTION IS CHARACTERIZED BY EARLY NEUTROPHIL RECRUITMENT AND ACTIVATION IN THE LUNG Neutrophils are recruited to
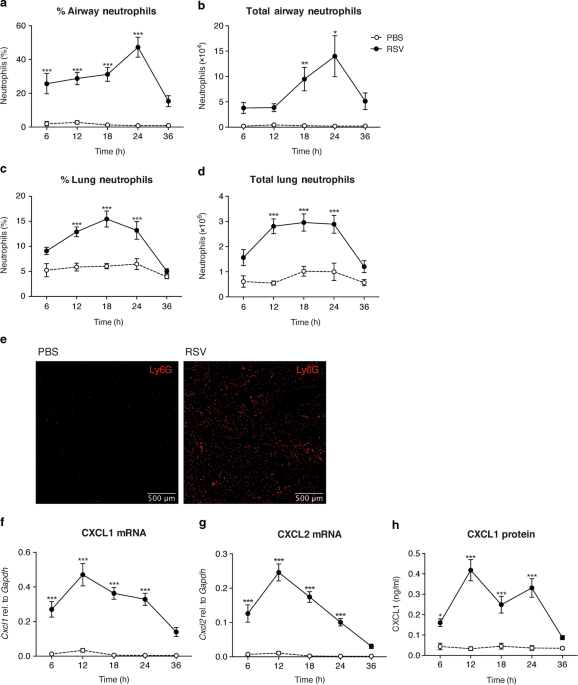
the lungs during RSV infection in both mice and man.20,23,27 To establish the exact timing of neutrophil recruitment to the airways and lungs in the C57BL/6 mouse model, mice were
intranasally (i.n.) infected with RSV. The frequencies and total numbers of neutrophils were quantified over time in the airways, by sampling the BAL, and in the lungs using flow cytometry
(Fig. 1a–d). Neutrophils were defined as CD45+, CD3−, CD19−, MHC II−, Ly6G+ cells (gating strategy shown in Supplementary Fig. 1). Neutrophil recruitment to the airways and to the lung
peaked at 18–24 h post infection (p.i.) and was not induced by mock (PBS) infection (Fig. 1a–d). Fluorescence microscopy of lung sections demonstrated that Ly6G+ cells were evenly
distributed throughout the lungs during RSV infection at 24 h p.i. (Fig. 1e). CD64+ inflammatory monocyte recruitment to the lung also occurred early following RSV infection (Supplementary
Fig. 2a, b), with their recruitment peaking after lung neutrophilia has resolved at 48 h p.i.,27 but, in contrast, RSV infection did not have much of an impact on the number of AMs in the
airways at any of the early timepoints (Supplementary Fig. 2c). Expression of the neutrophil chemoattractants _Cxcl1_ and _Cxcl2_ peaked prior to neutrophil influx at 12 h p.i. and this was
confirmed at the protein level for CXCL1 (Fig. 1f–h). Next, we assessed the activation status of neutrophils recruited to the airways and lung during RSV infection (Fig. 2). Neutrophil
activation status was initially determined by the detection of the proteolytic enzymes MMP-9, MPO and NE, all of which are pre-stored in cytoplasmic granules and released during neutrophil
degranulation.35 Whilst neutrophil recruitment peaked at 18–24 h p.i., the secretion of MMP-9, MPO and NE into the airways, as detected in the BAL fluid, peaked at 24 h p.i. (Fig. 2a).
Neutrophils were confirmed to be the major cellular source of MMP-9 and NE in BAL as these mediators were not detected after anti-Ly6G depletion (Fig. 2b, c). In contrast, neutrophil
depletion with anti-Ly6G reduced but did not entirely abrogate MPO levels in BAL suggesting that there could be additional cellular sources of MPO in the airways after RSV infection (Fig.
2c). To further assess neutrophil activation status during RSV infection, we quantified the cell surface expression of a variety of markers associated with neutrophil activation in other
inflammatory contexts on lung, airway and peripheral blood neutrophils (Supplementary Fig. 3). Upregulation of CD64, CD69 and CD11b on the cell surface of neutrophils has previously been
associated with neutrophil activation,36,37,38 as has the downregulation of CD62L and CD182.39,40 During RSV infection, we found that CD69 expression remained low on neutrophils in all
tissues, regardless of infection status. CD11b was upregulated upon migration into the airways and lungs, but this was also seen in the absence of an active RSV infection. Likewise, CD62L
and CD182 were downregulated on airway and lung neutrophils relative to blood neutrophils, but not specifically in response to RSV infection (Supplementary Fig. 3). Of the activation markers
tested, CD64 was the most specific marker of neutrophil activation in the lungs (Fig. 2d, e and Supplementary Fig. 3). CD64 expression levels were low on blood neutrophils and high on lung
and airway neutrophil during RSV infection (Supplementary Fig. 3), peaking at 18–24 h p.i. (Fig. 2e). We confirmed that mock infection did not induce the secretion of MMP-9, MPO or NE into
the airways or the upregulation of cell surface activation marker CD64 at any time point (Fig. 2). We also confirmed that live virus, and not any mediator in the virus preparation, was
necessary for neutrophil recruitment and activation as ultra-violet inactivated RSV (UV RSV) did not recruit neutrophils, induce MMP-9, MPO or NE secretion, or drive CD64 cell surface
upregulation on lung neutrophils (Supplementary Fig. 4). Also, HEp-2 cell supernatant did not induce neutrophil recruitment (data not shown). Together, these data confirm that neutrophils
are a major cell type recruited to the lungs early during RSV infection in the murine model and demonstrate that RSV infection induces neutrophil activation in the lung. NEUTROPHIL
RECRUITMENT IS DEPENDENT ON SIGNALING VIA MYD88/TRIF RSV is recognized by both RLRs and TLRs, which signal via the cytoplasmic adaptor proteins MAVS and MyD88/TRIF, respectively.25,26 To
investigate the lack of redundancy of each of these two distinct signaling pathways in neutrophil recruitment, _Mavs__−/−_ and _Myd88/Trif__−/−_ mice were infected with RSV and airway
neutrophils were quantified at 18 h p.i. (Fig. 3a). After RSV infection, neutrophils composed 40% of the airway cells in wild-type (wt) mice and 25% of the airway cells in _Mavs__−/−_ mice.
No neutrophils were recruited during RSV infection in _Myd88/Trif__−/−_ mice (Fig. 3a). These data indicate that MyD88/TRIF signaling is essential for neutrophil recruitment to the lungs
during RSV infection as has previously been implied in _Myd88__−/−_ mice.41 As reported,27,28,29 _Mavs__−/−_ mice had a higher viral load than wt mice at 18 h p.i. and at day 4 p.i. (the
peak of viral load in the mouse model21). In contrast, _Myd88/Trif__−/−_ mice had a similar viral load to wt mice at both timepoints measured (Supplementary Fig. 5). To further investigate
the mechanism driving neutrophil recruitment to the lung during RSV infection, we assessed the source of one of the neutrophil chemoattractants, CXCL1. Wt and _Myd88/Trif__−/−_ mice were
mock (PBS) or RSV infected and resident AMs, endothelial cells, ATII epithelial cells and CD45−, EpCAM− CD31− cells (other stromal cells) were sorted at 12 h p.i. (Supp. Fig. 1b for FACS
gating strategy) and compared to a presorted sample (total lung). RSV L gene transcripts were detected in all cell populations in RSV-infected mice (Fig. 3b). _Ifna5_ expression was detected
in the total lung sample (presort) and in sorted AMs (Fig. 3c) from both wt and _Myd88/Trif__−/−_ mice, confirming previous findings.24,27 Interestingly, _Cxcl1_ mRNA was significantly
enriched in the CD45-, EpCAM-, CD31- cell population (other stromal cells) from wt mice, suggesting that non-epithelial, non-endothelial cells are the major source of CXCL1 during RSV
infection (Fig. 3d). _Cxcl1_ mRNA was not detected in any lung cell populations from RSV-infected _Myd88/Trif__−/−_ mice (Fig. 3d). These data suggest that MyD88/TRIF signaling is crucial
for recruitment of neutrophils into the lungs during RSV infection and that signaling via MyD88/TRIF is necessary for inducing _Cxcl1_ and, possibly, other neutrophil chemoattractants in the
lung stromal cell compartment. NEUTROPHIL ACTIVATION IS DEPENDENT ON SIGNALING VIA MAVS We next investigated how the MAVS and MyD88/TRIF dependent PRR signaling pathways regulate neutrophil
activation in the lung during RSV infection (Fig. 4a–c). MMP-9, MPO and NE were measured in the airways of wt, _Mavs__−/−_ and _Myd88/Trif__−/−_ mice at 18 h p.i. (Fig. 4a). Consistent with
their inability to recruit neutrophils to the lung, _Myd88/Trif__−/−_ mice did not display increased levels of MMP-9, MPO or NE in the airways after RSV infection (Fig. 4a). Notably
_Mavs__−/−_ mice, which did recruit neutrophils during RSV infection, also did not show elevated MMP-9, MPO or NE in BAL during RSV infection (Fig. 4a). Furthermore, lung neutrophils from
RSV-infected _Mavs__−/−_ mice lacked cell surface CD64 expression (Fig. 4b, c). In contrast, the very few lung neutrophils found in RSV infected _Myd88/Trif__−/−_ mice (that have functional
MAVS signaling) did upregulate CD64 normally (Supplementary Fig. 6). These data suggest that MAVS signaling is required for neutrophil activation in the lung during RSV infection, at least
as measured by CD64 upregulation and the secretion of some granule contents. Signaling via MAVS is essential for the production of type I IFNs and lung inflammation during RSV infection.27
We confirmed that IFN-α and IL-6 were not present in the airways of _Mavs__−/−_ mice at 18 h p.i. (Fig. 4d). However, _Myd88/Trif__−/−_ mice had comparable levels of IFN-α and IL-6 in the
BAL to wt mice (Fig. 4d). Therefore, although signaling via MyD88/TRIF is required for neutrophil recruitment during RSV infection, it is not required for the overall establishment of a
cytokine-driven, pro-inflammatory lung environment as assessed by IFN-α and IL-6 levels. These observations lead to the hypothesis that, during RSV infection, pro-inflammatory factors
induced downstream of MAVS signaling activate lung neutrophils after they are recruited into the lungs as a downstream consequence of MyD88/TRIF signaling. RECOMBINANT IFN-Α RESTORES THE
PRO-INFLAMMATORY LUNG ENVIRONMENT IN THE ABSENCE OF MAVS SIGNALING AND IS SUFFICIENT TO ACTIVATE LUNG NEUTROPHILS DURING RSV INFECTION We tested this hypothesis firstly by restoring the
pro-inflammatory environment in the lungs of _Mavs__−/−_ mice during RSV infection by intranasal administration of rIFN-α, which induces pro-inflammatory cytokines and monocyte recruitment
in the lungs of wt mice.21 We confirmed that this also works in _Mavs__−/−_ mice (Fig. 5a, b) and in mock infected wt mice (Supplementary Fig. 7a) without impacting neutrophil or AM numbers
(Fig. 5c, d and Supplementary Fig. 7b). Consistent with induction of pro-inflammatory mediators, rIFN-α but not BSA (a protein control for rIFN-α) was sufficient to cause upregulation of
CD64 on lung neutrophils present at baseline in wt mice (Supplementary Fig. 6c). Likewise, rIFN-α was sufficient to drive upregulation of CD64 in RSV-infected _Mavs__−/−_ mice to levels
comparable to those in RSV-infected wt mice (Fig. 5e, f). This was accompanied by higher levels of MPO in the BAL (Fig. 5g) and a trend for increased levels of airway MMP-9 and NE, which did
not reach statistical significance (Fig. 5g). Thus, rIFN-α is sufficient to drive an inflammatory environment in the lung that results, at least partially, in neutrophil activation in
RSV-infected _Mavs__−/−_ mice. Secondly, we used _Myd88/Trif__−/−_ mice to test our hypothesis that factors induced downstream of MAVS signaling activate lung neutrophils during RSV
infection. _Myd88/Trif__−/−_ mice produce type I IFNs in response to RSV as they have intact MAVS signaling and therefore have a highly pro-inflammatory lung environment early during RSV
infection (Fig. 4). We postulated that _Myd88/Trif__−/−_ mice do produce the neutrophil activating factors in the lung in response to RSV, despite neutrophil recruitment being abrogated.
Indeed, as mentioned, the few neutrophils that can be found in RSV infected _Myd88/Trif__−/−_ mice upregulated the activation marker CD64 (Supplementary Fig. 6). However, to further test
this hypothesis, _Myd88/Trif__−/−_ mice were treated with rCXCL1 i.n. 6 h p.i. to recruit neutrophils into the lung after RSV infection (Fig. 6). Neutrophil responses were evaluated at 18 h
p.i. Treatment of _Myd88/Trif__−/−_ mice with rCXCL1 during RSV infection did not influence the number of airway AMs or lung monocytes as compared to mock treatment (Fig. 6a, b). As
previously observed, _Myd88/Trif__−/−_ mice did not recruit neutrophils to the airways or lungs 18 h p.i. (Fig. 6c) but treatment with 10 μg rCXCL1 was sufficient to recruit neutrophils
(Fig. 6c). There was no difference in neutrophil recruitment between mock infected or RSV infected _Myd88/Trif__−/−_ mice treated with rCXCL1 (Fig. 6c). Lung neutrophils recruited into
RSV-infected _Myd88/Trif__−/−_ mice displayed an activated phenotype as measured by the upregulation of cell surface CD64, which was comparable to that observed on neutrophils during RSV
infection of wt mice (Fig. 6d). CXCL1 treatment in the absence of RSV did not cause upregulation of CD64 on lung neutrophils in _Myd88/Trif__−/−_ mice (Fig. 6d), confirming that the
MAVS-dependent inflammatory environment is required for full neutrophil activation. On its own, rCXCL1 did induce the accumulation of some MMP-9, MPO and NE in the BAL of _Myd88/Trif__−/−_
mice (Fig. 6e), but this was significantly increased during RSV infection. Together, these data demonstrate that the factors which activate lung neutrophils during RSV infection are present
in the lungs of _Myd88/Trif__−/−_ mice despite the fact that these mice cannot recruit neutrophils to the lung during the infection. DISCUSSION Neutrophils are one of the first cell types to
be recruited to the lung during RSV infection21 and lung neutrophilia is a hallmark of severe RSV infection in infants.20,22,23 Here, innate immune signaling deficient _Mavs__−/−_ and
_Myd88/Trif__−/−_ mice were used to understand which RSV sensing pathways are required for the early recruitment and activation of lung neutrophils during RSV infection. Interestingly, we
found that both MyD88/TRIF and MAVS signaling are required but that they control distinct aspects of the neutrophilic response (Fig. 7). Indeed, while MyD88/TRIF signaling was necessary for
neutrophils to infiltrate the lung, it was dispensable for their activation whereas the opposite was true for the MAVS pathway. To our knowledge, this is the first study to demonstrate that
distinct innate immune signaling pathways synergize to drive lung neutrophilia and neutrophil activation during a respiratory virus infection. Neutrophils are classically considered rapid
responders and, consistent with that, early neutrophil recruitment was detected in the lung post RSV infection. To assess the activation status of lung neutrophils, the cell surface
expression of known activation markers was investigated by flow cytometry. This provides a sensitive readout of neutrophil activation as the expression level on each cell can be assessed
individually. For a more global and functional readout of neutrophil activation in the lung the accumulation of MMP-9, MPO and NE in BAL fluid was assessed. NE is exclusively produced by
neutrophils and our data in RSV-infected mice support clinical observations of increased NE in the nasal lavage of children with RSV-induced bronchiolitis.23 Neutrophil depletion experiments
confirmed that neutrophils are the major source of secreted MMP-9 and NE in response to RSV. However, neutrophil depletion did not completely abolish airway MPO during the infection,
suggesting there may be an alternative cellular source of this mediator. It is possible that monocytes also secrete MPO in the lung in response to RSV as human monocytes have been shown to
contain moderate levels of intracellular MPO.42 Neutrophil granules contain >1200 unique proteins43 and it will be of interest to further investigate which additional factors are secreted
during RSV infection. Of the cell surface activation markers tested, only CD64 (high affinity FcγRI) upregulation occurred on lung neutrophils specifically during RSV infection and not mock
infection CD64 upregulation is associated with neutrophil activation in bacterial infections,36 however the functional relevance of CD64 cell surface expression on neutrophils in viral
infections is not known. Interestingly, CD69 was upregulated on airway neutrophils during influenza infection37 but remained low during RSV infection, suggesting that neutrophils may respond
differently to these two respiratory viruses. Signaling via MyD88/TRIF was essential for neutrophil recruitment to the RSV infected lung (shown here and in _Myd88__−/−_ mice41). MyD88/TRIF
signaling has also been shown to be required for neutrophil recruitment during infection with influenza virus, _Staphylococcus aureus_ and _Streptococcus pneumoniae_, reflecting
TLR-dependent signaling or signals from IL-1 receptor family members, which also employ MyD88.8,44,45 However, it is not known whether the lung cell types involved, and the receptors used
for initiating the neutrophil recruitment, are the same for different infections. For example, during _S. pneumoniae_ infection, both hematopoietic and non-hematopoietic lung cells
contribute to the production of neutrophil chemoattractants via MyD88 signaling.44 Furthermore, TLR signaling in non-hematopoietic cells drives neutrophil recruitment during influenza virus
infection.8 During RSV infection, we found that non-epithelial, non-endothelial stromal cells (CD45-, EpCAM-, CD31- cells) are the major producers of CXCL1 and that this production is
dependent on signaling via MyD88/TRIF. The cell type in this mix of “other stromal cells” which is required for neutrophil recruitment, the receptors involved, the signaling which occurs
downstream of MyD88/TRIF and the production of other neutrophil chemoattractants remain to be elucidated. While neutrophil recruitment to the lung was not dependent on signaling via MAVS,
concordant with previous data,27 activation was dependent on a functional MAVS signaling pathway. Neutrophils in the _Mavs__−/−_ mice did not become activated during RSV infection, despite
these mice having a higher viral load than wt mice. _Myd88/Trif__−/−_ mice did not recruit neutrophils during RSV infection, yet this did not impair the ability of the host to control viral
replication. We have previously demonstrated that impaired type I IFN production, via loss of MAVS signaling or type I IFN receptor signaling, results in increased viral loads,21,27 and that
viral control is reestablished when mice are treated with CCL2 to restore the recruitment of anti-viral monocytes.27 Together, these studies suggest that type I IFNs, acting in part via
monocytes, are the main drivers of early protection during RSV infection and that neutrophil recruitment and activation in the lung are dispensable for viral control. While we cannot
completely exclude that neutrophil activation requires MAVS signaling in neutrophils themselves, it is not thought that neutrophils can be activated directly by RSV particles.46 During RSV
infection, MAVS signaling initiates type I IFN production and the amplification of type I IFN signaling via the IFNAR receptor drives a downstream signaling cascade that leads to the
production of many anti-viral ISGs and pro-inflammatory mediators.21,27 Cytokines such as IFN-γ and GM-CSF, which were induced when RSV-infected _Mavs__−/−_ mice were treated with rIFN-α,
can activate neutrophils.47 When the pro-inflammatory environment was restored in RSV-infected _Mavs__−/−_ mice by treatment with rIFN-α, neutrophils displayed an activated phenotype as
measured by the upregulation of CD64 and by the secretion of MPO in the airways. However, these mice did not display significantly more MMP-9 and NE levels in BAL than mock-treated
_Mavs__−/−_ mice. It is possible that this may be a quantitative defect due to the fact that a single administration of IFN-α at 6 h p.i. may not sufficiently replicate the kinetics of IFN-α
production in wt mice. Alternatively, we cannot exclude the possibility that factors in addition to IFN-α are required to drive full neutrophil activation. Therefore, our data suggest that
_Mavs__−/−_ deficient neutrophils become at least partially activated when they are in an inflammatory environment induced downstream of IFN-α. This likely involves ISG protein products
upregulated downstream of type I IFN receptor signaling.27,28,29 It is interesting to consider that while type I IFNs have a beneficial inhibitory effect on viral load they also drive lung
inflammation21 and, as we show in this work, neutrophil activation during RSV infection. Therefore, the potential use of type I IFNs as a therapeutic in RSV infection must be carefully
considered to avoid immunopathology. As RSV infected _Myd88/Trif__−/−_ mice have intact MAVS signaling, we hypothesized that RSV infection would induce the upregulation of the factor/s which
are required for neutrophil activation. Indeed, the very few lung neutrophils found in _Myd88/Trif__−/−_ mice upregulated CD64. We suspect that the reason MMP-9, MPO and NE were not
detected in the airways of these mice was because there were too few neutrophils to release detectable quantities of these mediators. Consistent with that possibility, when neutrophils were
artificially recruited into the lungs of RSV-infected _Myd88/Trif__−/−_ mice by administration of rCXCL1, the lung neutrophils upregulated CD64 and MMP-9, MPO and NE were detected in the
airways. It is known that CXCL1 has a role in both the recruitment and activation of neutrophils35 and treatment with rCXCL1 on its own did induce some production of MMP-9, MPO and NE in the
lungs of _Myd88/Trif__−/−_ mice. However, rCXCL1 treatment in the absence of RSV did not induce the upregulation of CD64 on lung neutrophils. Furthermore, neutrophil mediator production was
amplified and CD64 upregulated on neutrophils by RSV infection during rCXCL1 treatment of _Myd88/Trif__−/−_ mice, demonstrating that the inflammatory environment induced by RSV infection
was required for full neutrophil activation. These data support our finding that an intact MAVS signaling pathway is sufficient to induce an inflammatory environment that at least partially
activates neutrophils during RSV infection. To summarize, this paper demonstrates for the first time that two distinct innate immune signaling pathways collaborate to ensure that both
neutrophil recruitment and activation occur during RSV infection. MyD88/TRIF signaling is necessary to attract and recruit neutrophils and MAVS signaling is crucial to create an inflammatory
environment in the lung that drives at least partial neutrophil activation. Excessive neutrophilia can be detrimental to the health of lung tissue and compromise efficient gas exchange. As
such, a deeper understanding of the molecular pathways that regulate neutrophil recruitment and activation may help pave the way towards development of potential therapies. METHODS MICE Wt
C57BL/6 mice were purchased from Charles River. _Ifna6gfp_+/−, _Ifna6gfp_+/− _Myd88Trif__−/−_ and _Ifna6gfp_+/− _Mavs__−/−_ mice were bred in-house (obtained from S. Akira, Japan). The GFP
signal has not been quantified in this work so the mice will be denoted as wt, _MyD88/Trif__−/−_ and _Mavs__−/−_ mice, respectively. All mice were bred and housed in specific pathogen-free
conditions and were gender and age-matched (7–12 weeks). Different strains of mice were not co-housed but kept in the same breeding room. The breeders of _MyD88/Trif__−/−_ mice were kept on
Septrin in the drinking water because of their immunocompromised status. All animal experiments were reviewed and approved by the Animal Welfare and Ethical Review Board (AWERB) within
Imperial College London and approved by the UK Home Office in accordance with the Animals (Scientific Procedures) Act 1986 and the ARRIVE guidelines. VIRUS AND INFECTIONS Plaque-purified
human RSV (originally A2 strain from ATCC, US) was grown in Hep2 cells.48 RSV was inactivated by exposing virus to UV light for 2 min (UV RSV) in a CX-2000 UV cross-linker (UVP). Mice were
lightly anaesthetized before administration intranasally (i.n.) of 100 µl containing 106 FFU RSV or PBS control. In some instances, this was followed by instillation of recombinant proteins
(1 µg IFN-α (Miltenyi Biotec), 10 µg CXCL1 (Biolegend)) or a protein control, bovine serum albumin (BSA; Sigma Aldrich). Mice were sacrificed post-infection (p.i.) by a fatal dose of
pentobarbital injected intraperitonially (i.p.). ANTIBODY MEDIATED NEUTROPHIL DEPLETION Mice were given 200 µg i.n. and 500 µg i.p. anti-Ly6G MAb or isotype rat Ig2A MAb (Bio X Cell) 1 day
pre-infection.49,50 AIRWAY CELL PROCESSING To recover the airway cells and immune mediators, BAL was performed. One ml PBS supplemented with 0.5 mM EDTA was used to flush the lungs three
times. The BAL fluid was centrifuged and supernatant stored at −80 °C. Red blood cells (RBCs) were removed by lysis with ACK (0.15 M NH4Cl, 1.0 mM KHCO3, 0.1 mM Na2EDTA). The cell number was
determined by Trypan Blue (Thermo Fisher Scientific) exclusion of dead cells. BAL cells were termed airway cells throughout. Airway cells were either stained for flow cytometry or the
cellular composition was determined by spinning 1–2 × 105 cells onto a microscope slide (Thermo Scientific) at 450 rpm for 5 min using Cytospin 4 Cytocentrifuge (Thermo Fisher Scientific).
Slides were H&E stained using Reastain Quick-Diff kit (Gentaur), according to the manufacturer’s instructions. Cells were classified as neutrophils using a microscope and ≥300 total
cells were counted. ISOLATION OF LUNG CELLS AND PERIPHERAL MONONUCLEAR BLOOD CELLS For RNA extractions, lung lobes were snap-frozen in liquid nitrogen and stored at −80 °C. For flow
cytometry, 1–3 lung lobes were collected in complete DMEM (cDMEM; supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin). Collagenase D
(1 mg/ml; Roche) and DNase I (30 µg/ml; Invitrogen) was added and samples processed with a gentle MACS dissociator (Miltenyi Biotech). Lungs were incubated shaking at 37 °C for 1 h and then
processed again with a gentle MACS dissociator. RBCs were removed by ACK lysis. Cells were re-suspended in PBS for flow cytometry and filtered through a 100 µM cell strainer (Greiner
BioOne). The lung cell count was quantified by Trypan Blue (Thermo Fisher Scientific) exclusion of dead cells. At least 75 µl blood was collected in 1 ml PBS supplemented with 5 mM EDTA.
RBCs were removed by lysis with ACK. Cells were re-suspended in FACS buffer (PBS supplemented with 1% BSA, 0.05 mM EDTA) before flow cytometry staining. FLOW CYTOMETRY For flow cytometry
staining, 2.5 × 106 lung cells were incubated for 20 min at 4 °C with a purified rat IgG2b anti–mouse CD16/CD32 receptor antibody (BD) to block Fc binding in FACS buffer. For surface
staining, cells were stained with fluorochrome-conjugated antibodies against CD45 (30-F11, BV605), CD11b (M1/70, AF700), CD64 (X54-5/7.1, APC), Ly6G (1A8, BV570/BV785), CD3ε (145-2C11,
FITC), Ly6C (HK1.4, BV421), CD19 (6D5, FITC), CD69 (H1.2F3, Per CP Cy5.5), MHC-II (M5/114.15.2, APC eF870), CD62L (MEL-14, BV421), CD182 (SA044G4, PE), CD11c (HL3, PE-CF594) for 25 min at 4
°C. Cells were washed with PBS and stained with fixable live-dead Aqua dye (Invitrogen) for 30 min. Cells were fixed by incubation with 100 µl 1% paraformaldehyde or Cytofix™ Fixation Buffer
(BD) for 20 mins at 4 °C and stored in FACS buffer. Analysis was performed on a BD LSR Fortessa. Acquisition was set to 250,000 single, live, CD45+ cells. All antibodies were purchased from
BD, BioLegend, or eBioscience. Data were analyzed with FlowJo software (Tree Star). Total cell populations were quantified as the whole lung count × (%population of CD45+ cells) × (%CD45+
cell of live cells) × (proportion of lung tissue sampled). FACS For cell sorting of lung cell populations, single cell suspensions were obtained by dispase digestion (5 mg/ml; Roche) and
DNase I (250 μg/ml; Sigma), as reported.27 Cells were incubated with a purified rat IgG2b anti–mouse CD16/CD32 receptor antibody (BD) and stained with fluorochrome-conjugated antibodies
against CD11c (HL3, PE-CF594), CD31 (390, PE), CD45 (30-F11, APC-Cy7), and EpCAM (G8.8, PerCP/Cy5.5) as described above. Sytox Blue Dead Cell Stain (1:8000; Life Technologies, UK) was added
to cells before running on a BD FACSAria III using FACSDiVa software (BD Bioscience). Sorted cells were stored in Trizol™ Reagent (Invitrogen, UK) until RNA extraction was performed.
FLUORESCENT MICROSCOPY Lungs were inflated with 1 ml 50% OCT (VWR) diluted in PBS and frozen on dry ice. Ten micrometers lung cryosections were cut using a cryostat (Bright OTF5000 LS) and
sections were fixed in acetone for 10 min at room temperature. Sections were rehydrated twice in PBS before blocking with purified rat IgG2b anti–mouse CD16/CD32 receptor antibody (BD) to
block Fc binding in FACS buffer for 30 min at room temperature. To detect Ly6G+ neutrophils, sections were stained with α-Ly6G 1A8 (1:800, Abcam) overnight at 4 °C. Sections were then washed
twice in PBS and stained with a species-specific secondary antibody conjugated to Alexa Fluor 647 (1:200, Abcam) for 2 h in the dark at 4 °C. Sections were washed twice in PBS and
coverslips were mounted into glass slides with ProLong® Gold Anti-fade Mountant with DAPI (ThermoFisher Scientific). The Zeiss Inverted Widefield Microscope on a 20× dry lens was used for
obtaining images of the cells. Analysis of the images was performed on Fiji, an open-source imaging processing software (ImageJ). RNA EXTRACTION AND QUANTITATIVE RT-PCR Lungs were
homogenized using a TissueLyser LT (Qiagen) and total RNA was extracted from the lung tissue supernatant using RNeasy Mini kit (Qiagen). RNA extraction from sorted lung cells was performed
using Trizol (Invitrogen). Following the chloroform step, the aqueous phase containing RNA was further processed using the RNeasy Mini or Micro Kit according to manufacturer’s instructions
(Qiagen). RNA concentration was determined by NanoDrop (Thermo Scientific). In all, 2 µg or 9 µl (sorted cells) of RNA was converted to cDNA using High Capacity RNA-to-cDNA kit (Applied
Biosystems). RT-qPCR was performed with Quantitect Probe PCR Master Mix (Qiagen). For mRNA analysis of _Gapdh, Cxcl1, Cxcl2, Il6_, _Csf2_, and _Ifna5_ gene specific primers and probes were
used (all Applied Biosystems). For absolute quantification of RSV L gene, TNF-α and IFN-γ mRNA, the exact number of copies of the gene of interest was calculated using a plasmid DNA standard
curve for each gene and normalized to _Gapdh_.21 RT-qPCR was performed using the 7500 Fast Real-Time PCR System (Applied Biosystems). To quantify relative mRNA expression the mean ΔCT was
calculated for each target gene relative to _Gapdh_ (encoding glyceraldehyde-3-phosphate dehydrogenase) and expressed as 2−ΔCT. Analysis was performed using 7500 Fast System SDS Software
(Applied Biosystems). IMMUNE MEDIATOR DETECTION ELISA was used to measure the concentration of CXCL1, MMP-9, MPO, NE (all DuoSet ELISA kits from R&D systems), IL-621 and IFN-α.27
Absorbance was determined at 450 nm, on SpectraMax Plus (Molecular Devices) or FLUOstar Omega (BMG Labtech) plate readers and analyzed using SoftMax (Molecular Devices) or Mars (BMG Labtech)
software. STATISTICAL ANALYSIS Statistical analysis was performed using Prism (GraphPad software) version 6. Data are presented as the mean ± SEM. As indicated, one-way ANOVA followed by
Tukey’s post hoc was used to compare multiple groups. To compare genotypes during mock and RSV infection, a two-way ANOVA, followed by either Bonferroni’s or Tukey’s post hoc test, was used
as indicated. _P_ values < 0.05 were considered statistically significant for all tests. *_P_ < 0.05; **_P_ < 0.01; ***_P_ < 0.001. REFERENCES * Papayannopoulos, V. Neutrophil
extracellular traps in immunity and disease. _Nat. Rev. Immunol._ 18, 134–147 (2018). Article CAS Google Scholar * Hayashi, F., Means, T. K. & Luster, A. D. Toll-like receptors
stimulate human neutrophil function. _Blood_ 102, 2660–2669 (2003). Article CAS Google Scholar * Sabroe, I., Dower, S. K. & Whyte, M. K. B. The role of Toll-like receptors in the
regulation of neutrophil migration, activation, and apoptosis. _Clin. Infect. Dis._ 41, S421–S426 (2005). Article CAS Google Scholar * Sørensen, O. E. & Borregaard, N. Neutrophil
extracellular traps - the dark side of neutrophils. _J. Clin. Invest_ 126, 1612–1620 (2016). Article Google Scholar * Brinkmann, V. et al. Neutrophil extracellular traps kill bacteria.
_Science_ 303, 1532–1535 (2004). Article CAS Google Scholar * Kaplan, M. J. & Radic, M. Neutrophil extracellular traps: double-edged swords of innate immunity. _J. Immunol._ 189,
2689–2695 (2012). Article CAS Google Scholar * Bardoel, B. W., Kenny, E. F., Sollberger, G. & Zychlinsky, A. The balancing act of neutrophils. _Cell Host Microbe_ 15, 526–536 (2014).
Article CAS Google Scholar * Bradley, L. M., Douglass, M. F., Chatterjee, D., Akira, S. & Baaten, B. J. G. Matrix metalloprotease 9 mediates neutrophil migration into the airways in
response to influenza virus-induced toll-like receptor signaling. _PLoS Pathog._ 8, e1002641 (2012). Article CAS Google Scholar * Kruger, P. et al. Neutrophils: between host defence,
immune modulation, and tissue injury. _PLoS Pathog._ 11, e1004651 (2015). Article Google Scholar * Toussaint, M. et al. Host DNA released by NETosis promotes rhinovirus-induced type-2
allergic asthma exacerbation. _Nat. Med._ 23, 681–691 (2017). Article CAS Google Scholar * Geerdink, R. J., Pillay, J., Meyaard, L. & Bont, L. Neutrophils in respiratory syncytial
virus infection: a target for asthma prevention. _J. Allergy Clin. Immunol._ 136, 838–847 (2015). Article CAS Google Scholar * Tate, M. D. et al. The role of neutrophils during mild and
severe influenza virus infections of mice. _PLoS ONE_ 6, e17618 (2011). Article CAS Google Scholar * Smyth, R. L. & Openshaw, P. J. M. Bronchiolitis. _Lancet_ 368, 312–322 (2006).
Article Google Scholar * Shi, T. et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children
in 2015: a Systematic Review and Modelling Study. _Lancet_ 390, 946–958 (2017). Article Google Scholar * Hall, C. B. et al. The burden of respiratory syncytial virus infection in young
children. _N. Engl. J. Med._ 360, 588–598 (2009). Article CAS Google Scholar * Henderson, J. et al. Hospitalization for RSV bronchiolitis before 12 months of age and subsequent asthma,
atopy and wheeze: a Longitudinal Birth Cohort Study. _Pedia. Allergy Immunol._ 16, 386–392 (2005). Article Google Scholar * Stern, D. A., Morgan, W. J., Halonen, M., Wright, A. L. &
Martinez, F. D. Wheezing and bronchial hyper-responsiveness in early childhood as predictors of newly diagnosed asthma in early adulthood: a longitudinal birth-cohort study. _Lancet_ 372,
1058–1064 (2008). Article Google Scholar * Collins, P. L. & Graham, B. S. Viral and host factors in human respiratory syncytial virus pathogenesis. _J. Virol._ 82, 2040–2055 (2008).
Article CAS Google Scholar * Openshaw, P. J. M., Chiu, C., Culley, F. J. & Johansson, C. Protective and harmful immunity to RSV infection. _Annu. Rev. Immunol._ 35, 501–532 (2017).
Article CAS Google Scholar * McNamara, P. S. Bronchoalveolar lavage cellularity in infants with severe respiratory syncytial virus bronchiolitis. _Arch. Dis. Child._ 88, 922–926 (2003).
Article CAS Google Scholar * Goritzka, M. et al. Alpha/beta interferon receptor signaling amplifies early proinflammatory cytokine production in the lung during respiratory syncytial
virus infection. _J. Virol._ 88, 6128–6136 (2014). Article CAS Google Scholar * Everard, M. L. et al. Analysis of cells obtained by bronchial lavage of infants with respiratory syncytial
virus infection. _Arch. Dis. Child._ 71, 428–432 (1994). Article CAS Google Scholar * Emboriadou, M. et al. Human neutrophil elastase in RSV bronchiolitis. _Ann. Clin. Lab. Sci._ 37,
79–84 (2007). CAS PubMed Google Scholar * Makris, S., Bajorek, M., Culley, F. J., Goritzka, M. & Johansson, C. Alveolar macrophages can control respiratory syncytial virus infection
in the absence of type I interferons. _J. Innate Immun._ 8, 452–463 (2016). Article CAS Google Scholar * Johansson, C. Respiratory syncytial virus infection: an innate perspective.
_F1000Research_ 5, 2898 (2016). Article Google Scholar * Marr, N., Turvey, S. E. & Grandvaux, N. Pathogen recognition receptor crosstalk in respiratory syncytial virus sensing: a host
and cell type perspective. _Trends Microbiol._ 21, 568–574 (2013). Article CAS Google Scholar * Goritzka, M. et al. Alveolar macrophage-derived type I interferons orchestrate innate
immunity to RSV through recruitment of antiviral monocytes. _J. Exp. Med_ 212, 699–714 (2015). Article CAS Google Scholar * Demoor, T. et al. IPS-1 signaling has a nonredundant role in
mediating antiviral responses and the clearance of respiratory syncytial virus. _J. Immunol._ 189, 5942–5953 (2012). Article CAS Google Scholar * Bhoj, V. G. et al. MAVS and MyD88 are
essential for innate immunity but not cytotoxic T lymphocyte response against respiratory syncytial virus. _Proc. Natl Acad. Sci. USA_ 105, 14046–14051 (2008). Article CAS Google Scholar
* Wu, B. & Hur, S. How RIG-I like receptors activate MAVS. _Curr. Opin. Virol._ 12, 91–98 (2015). Article CAS Google Scholar * Seth, R. B., Sun, L., Ea, C.-K. & Chen, Z. J.
Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. _Cell_ 122, 669–682 (2005). Article CAS Google Scholar *
Kawasaki, T. & Kawai, T. Toll-like receptor signaling pathways. _Front Immunol._ 5, 461 (2014). Article Google Scholar * Kawai, T. & Akira, S. The role of pattern-recognition
receptors in innate immunity: update on Toll-like receptors. _Nat. Immunol._ 11, 373–384 (2010). Article CAS Google Scholar * Ivashkiv, L. B. & Donlin, L. T. Regulation of type I
interferon responses. _Nat. Rev. Immunol._ 14, 36–49 (2014). Article CAS Google Scholar * Kolaczkowska, E. & Kubes, P. Neutrophil recruitment and function in health and inflammation.
_Nat. Rev. Immunol._ 13, 159–175 (2013). Article CAS Google Scholar * Cid, J., Aguinaco, R., Sánchez, R., García-Pardo, G. & Llorente, A. Neutrophil CD64 expression as marker of
bacterial infection: a systematic review and meta-analysis. _J. Infect._ 60, 313–319 (2010). Article Google Scholar * Tate, M. D., Brooks, A. G. & Reading, P. C. The role of
neutrophils in the upper and lower respiratory tract during influenza virus infection of mice. _Respir. Res_ 9, 932–13 (2008). Article Google Scholar * Costantini, C. et al. Neutrophil
activation and survival are modulated by interaction with NK cells. _Int. Immunol._ 22, 827–838 (2010). Article CAS Google Scholar * Fortunati, E., Kazemier, K. M., Grutters, J. C.,
Koenderman, L.& Van den Bosch, vJ. Human neutrophils switch to an activated phenotype after homing to the lung irrespective of inflammatory disease. _Clin. Exp. Immunol._ 155, 559–566
(2009). Article CAS Google Scholar * Pignatti, P. et al. Downmodulation of CXCL8/IL-8 receptors on neutrophils after recruitment in the airways. _J. Allergy Clin. Immunol._ 115, 88–94
(2005). Article CAS Google Scholar * Murawski, M. R. et al. Respiratory syncytial virus activates innate immunity through Toll-like receptor 2. _J. Virol._ 83, 1492–1500 (2009). Article
CAS Google Scholar * Tay, S. P., Cheong, S. K., Hamidah, N. H. & Ainoon, O. Flow cytometric analysis of intracellular myeloperoxidase distinguishes lymphocytes, monocytes and
granulocytes. _Malays. J. Pathol._ 20, 91–94 (1998). CAS PubMed Google Scholar * Rørvig, S., Østergaard, O., Heegaard, N. H. H. & Borregaard, N. Proteome profiling of human neutrophil
granule subsets, secretory vesicles, and cell membrane: correlation with transcriptome profiling of neutrophil precursors. _J. Leukoc. Biol._ 94, 711–721 (2013). Article Google Scholar *
Dudek, M. et al. Lung epithelium and myeloid cells cooperate to clear acute pneumococcal infection. _Mucosal Immunol._ 9, 1288–1302 (2016). Article CAS Google Scholar * Miller, L. S. et
al. MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against Staphylococcus aureus. _Immunity_ 24, 79–91 (2006). Article CAS Google Scholar *
Bataki, E. L., Evans, G. S. & Everard, M. L. Respiratory syncytial virus and neutrophil activation. _Clin. Exp. Immunol._ 140, 470–477 (2005). Article CAS Google Scholar * Buckle, A.
M. & Hogg, N. The effect of IFN-gamma and colony-stimulating factors on the expression of neutrophil cell membrane receptors. _J. Immunol._ 143, 2295–2301 (1989). CAS PubMed Google
Scholar * Lee, D. C. P. et al. CD25+natural regulatory T cells are critical in limiting innate and adaptive immunity and resolving disease following respiratory syncytial virus infection.
_J. Virol._ 84, 8790–8798 (2010). Article CAS Google Scholar * Akthar, S. et al. Matrikines are key regulators in modulating the amplitude of lung inflammation in acute pulmonary
infection. _Nat. Commun._ 6, 8423 (2015). Article CAS Google Scholar * Lim, K. et al. Neutrophil trails guide influenza-specific CD8+ T cells in the airways. _Science_ 349, aaa4352
(2015). Article Google Scholar Download references ACKNOWLEDGEMENTS We thank S. Akira and Y. Kumagai (World Premier International Immunology Frontier Research Center, Osaka University,
Osaka, Japan) for providing _Mavs__−/−_ and _Myd88/Trif__−/−_ mice. We thank Caetano Reis e Sousa, Robert Snelgrove and Ryan Thwaites for critically reading the manuscript. We also thank the
other members of the Respiratory Infections section for suggestions and critique. Finally, we thank Yanping Guo of the St Mary’s flow cytometry facility for her help and the staff of the St
Mary’s animal facility for their assistance. F.C.M.K. was supported by a PhD Fellowship from the Wellcome Trust (109058/Z/15/Z), F.K. and S.M. were supported by PhD Fellowships from the
National Heart and Lung Institute Foundation (registered charity number 1048073), and C.J. was supported by a grant from the Rosetrees Trust (M370). AUTHOR INFORMATION Author notes *
Spyridon Makris Present address: MRC/UCL Lab for Molecular Cell Biology, London, UK AUTHORS AND AFFILIATIONS * National Heart and Lung Institute, Imperial College London, St Mary’s Hospital,
Norfolk Place, London, W2 1PG, UK Freja C. M. Kirsebom, Fahima Kausar, Rinat Nuriev, Spyridon Makris & Cecilia Johansson Authors * Freja C. M. Kirsebom View author publications You can
also search for this author inPubMed Google Scholar * Fahima Kausar View author publications You can also search for this author inPubMed Google Scholar * Rinat Nuriev View author
publications You can also search for this author inPubMed Google Scholar * Spyridon Makris View author publications You can also search for this author inPubMed Google Scholar * Cecilia
Johansson View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS F.C.M.K. designed, performed, and analyzed the experiments and wrote the paper.
F.K developed techniques, performed experiments, and reviewed the paper. R.N. performed specific experiments and reviewed the paper. S.M. contributed technical expertise and reviewed the
paper. C.J. supervised the project, designed, and analyzed the experiments and wrote the paper. CORRESPONDING AUTHOR Correspondence to Cecilia Johansson. ETHICS DECLARATIONS COMPETING
INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in published maps and
institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0
International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the
source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Kirsebom, F.C.M., Kausar, F., Nuriev, R. _et al._ Neutrophil recruitment and
activation are differentially dependent on MyD88/TRIF and MAVS signaling during RSV infection. _Mucosal Immunol_ 12, 1244–1255 (2019). https://doi.org/10.1038/s41385-019-0190-0 Download
citation * Received: 05 November 2018 * Revised: 14 June 2019 * Accepted: 29 June 2019 * Published: 29 July 2019 * Issue Date: September 2019 * DOI: https://doi.org/10.1038/s41385-019-0190-0
SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy
to clipboard Provided by the Springer Nature SharedIt content-sharing initiative