Play all audios:
ABSTRACT Intestinal epithelial cells (IECs) contribute to the regulation of intestinal homeostasis and inflammation through their interactions with the environment and host immune responses.
Yet our understanding of IEC-intrinsic regulatory pathways remains incomplete. Here, we identify the guanine nucleotide exchange factor RABGEF1 as a regulator of intestinal homeostasis and
innate pathways dependent on IECs. Mice with IEC-specific _Rabgef1_ deletion (called _Rabgef1__IEC-KO_ mice) developed a delayed spontaneous colitis associated with the local upregulation of
IEC chemokine expression. In mouse models of colitis based on Interleukin-10 deficiency or dextran sodium sulfate (DSS) exposure, we found that IEC-intrinsic RABGEF1 deficiency exacerbated
development of intestinal pathology and dysregulated IEC innate pathways and chemokine expression. Mechanistically, we showed that RABGEF1 deficiency in mouse IECs in vitro was associated
with an impairment of early endocytic events, an increased activation of the p38 mitogen-activated protein kinase (MAPK)-dependent pathway, and increased chemokine secretion. Moreover, we
provided evidence that the development of spontaneous colitis was dependent on microbiota-derived signals and intrinsic MYD88-dependent pathways in vivo. Our study identifies mouse RABGEF1
as an important regulator of intestinal inflammation, MYD88-dependent IEC-intrinsic signaling, and chemokine production. This suggests that RABGEF1-dependent pathways represent interesting
therapeutic targets for inflammatory conditions in the gut. You have full access to this article via your institution. Download PDF SIMILAR CONTENT BEING VIEWED BY OTHERS NRP1 INSTRUCTS
IL-17-PRODUCING ILC3S TO DRIVE COLITIS PROGRESSION Article 01 January 2025 PRKAR2A DEFICIENCY PROTECTS MICE FROM EXPERIMENTAL COLITIS BY INCREASING IFN-STIMULATED GENE EXPRESSION AND
MODULATING THE INTESTINAL MICROBIOTA Article Open access 04 August 2021 INTERLEUKIN-37 REGULATES INNATE IMMUNE SIGNALING IN HUMAN AND MOUSE COLONIC ORGANOIDS Article Open access 15 April
2021 INTRODUCTION The intestinal mucosa is particularly exposed to microorganisms and foreign antigens. Intestinal homeostasis thus relies on finely tuned interactions between the
environment, intestinal epithelial cells (IECs) and host immune responses, which allow the host to assimilate nutrients and to tolerate commensal microorganisms and dietary antigens, while
mounting appropriate defense responses against pathogens.1,2,3 IECs critically contribute to the regulation of intestinal homeostasis and inflammation.2,3 Indeed, in addition to forming a
physical barrier and eliciting efficient secretory defenses, IECs express a wide range of pattern recognition receptors (PRRs)2,4,5,6 that can sense microbiota-derived signals and integrate
them into appropriate homeostatic or defensive responses.2,3,7 The abundance of symbiotic microorganisms necessitates that IECs express negative regulators of PRR signaling pathways in order
to avoid aberrant immune responses and a break of tolerance, which can be associated with chronic inflammatory conditions such as inflammatory bowel diseases (IBDs). Experimental models of
IBDs have substantially contributed to our understanding of IBD pathogenesis, which is thought to be driven by interactions between microbial, environmental, and host genetic
factors.2,5,8,9,10,11,12 RAB guanine nucleotide exchange factor 1 (RABGEF1) was first identified as a GDP/GTP exchange factor for the small GTPase Rab5, which regulates endocytosis and early
endosome trafficking events.13,14,15,16,17 RABGEF1 contains several functional domains, including a N-terminal A20-like zinc finger domain that can exhibit E3 ubiquitin ligase activity18,19
and a Vps9 domain promoting GEF activity for Rab5.13,14,15 RABGEF1 is critical for health, as globally RABGEF1-deficient mice exhibit perinatal mortality.20 In addition, we showed that
RABGEF1 expression within skin epithelial cells (i.e., keratinocytes) is required to sustain optimal epidermal barrier functions and skin homeostasis in mice.21 Based on evidence suggesting
a homeostatic role of keratinocyte-intrinsic RABGEF1, we have speculated that this protein could also act as a regulator of homeostasis and inflammation at other epithelial surfaces, such as
the intestine. In that site, colonization by microorganisms requires tight regulation of the intestinal epithelium in order to cope with the myriad of microbial stimuli and antigens.
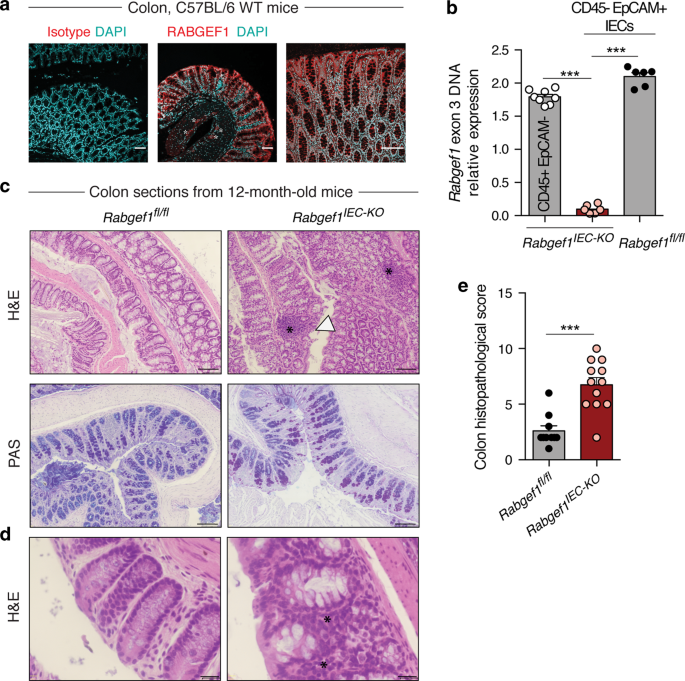
RESULTS IEC-SPECIFIC RABGEF1 DELETION PROMOTES A SPONTANEOUS AND DELAYED COLITIS In the mouse colon, RABGEF1 protein was found to be highly expressed in IECs (Fig. 1a) as well as in discrete
cell clusters located in the submucosa, which may correspond to the myenteric plexus (Fig. 1a, asterisks). To investigate the functions of IEC-intrinsic RABGEF1 in vivo, _Villin-Cre__+__;
Rabgef1__fl/fl_ mice with conditional _Rabgef1_ deletion within IECs were generated (called _Rabgef1__IEC-KO_ mice hereafter) (Fig. 1b). Under conventional conditions of housing,
_Rabgef1__IEC-KO_ mice were born with a Mendelian ratio and did not develop any mortality (Supplementary Fig. 1a), weight loss (Supplementary Fig. 1b) or macroscopic intestinal abnormalities
(Supplementary Fig. 1c) until the age of 12 months. Histopathological analyses of colon sections from 10-week-old mice did not reveal development of intestinal pathology in
_Rabgef1__IEC-KO_ mice (Supplementary Fig. 1d, e). However, similar analyses performed on 12-month-old mice showed signs of moderate inflammation (i.e., mucosal and submucosal inflammatory
infiltrates) and epithelial damage (i.e., hyperplasia and erosion without goblet cell loss) in the colon of _Rabgef1__IEC-KO_ mice as compared to controls (Fig. 1c–e), while no abnormalities
were found in the small intestine (Supplementary Fig. 2a–c). We also analyzed other organs of aged _Rabgef1__IEC-KO_ mice and did not find any obvious histological abnormalities
(Supplementary Fig. 2d–l). EPITHELIAL RABGEF1 DEFICIENCY RESULTS IN IEC-SPECIFIC ALTERATION OF CYTOKINE EXPRESSION WITHOUT SYSTEMIC EFFECTS Next, we characterized IEC-intrinsic immune and
barrier functions and features of systemic immune activation in _Rabgef1__IEC-KO_ and littermate _Rabgef1__fl/fl_ mice. Quantitative reverse transcription polymerase chain reaction (RT-qPCR)
analyses of IECs from 12-month-old mice revealed that RABGEF1-deficient IECs exhibited a higher expression of _Tnfa_, _Ccl2_, _Cxcl1_, and _Cxcl2_ as compared to the littermate
counterparts, while the expression of other cytokines (_Il6_, _Vegfa_, _Ccl11_, and _Ccl5_) and _Muc2_ (coding for Mucin 2) was not affected by RABGEF1 deficiency (Fig. 2a). Such effects
were however not observed in IECs from 10-week-old _Rabgef1__IEC-KO_ mice (Supplementary Fig. 3). To evaluate intestinal permeability to luminal contents, plasma levels of fluorescein
isothiocyanate (FITC) were measured following oral administration of FITC-dextran. Notably, no differences were found between 12-month-old _Rabgef1__IEC-KO_ and littermate controls (Fig.
2b). Third, serum levels of several cytokines were not altered by IEC-restricted RABGEF1 deficiency in such mice (Fig. 2c). These results indicate that epithelial RABGEF1 deficiency was
associated with local changes in IEC innate responses, while no evidence of impaired intestinal permeability or systemic inflammation were observed. EPITHELIAL RABGEF1 LIMITS COLON
INFLAMMATION AND PATHOLOGY IN IL-10-DEFICIENT MICE The genetic model of colitis based on IL-10 deficiency has been extensively used to help dissecting the mechanisms of IBD.22 It is thought
to influence IEC responses23,24,25 and promotes a spontaneous, microbiota-dependent colitis.26,27 To characterize the effects of IL-10 deficiency on IECs in our housing conditions, we
performed mRNA-sequencing (RNA-seq) of FACS-sorted colonic IECs isolated from 10-week-old _Il10__−/−_ and littermate _Il10__+/+_ mice and found that IECs clustered according to their
genotype (i.e., _Il10__−/−_ or _Il10__+/+_ mice) (Supplementary Fig. 4a, b). In addition, we found a significant enrichment, among genes upregulated in _Il10__−/−_ IECs, of transcripts
implicated in oxidative phosphorylation, protein translation and export, and ribosome function (Supplementary Fig. 4c, d), as well as MHC class II-related genes, chemokines, and genes
implicated in the response to lipopolysaccharide (LPS) and the negative regulation of inflammation (Supplementary Fig. 4e). Conversely, genes downregulated in _Il10__−/−_ IECs were shown to
be involved in adherens/apical junctions, digestion or transmembrane transporter activity (Supplementary Fig. 4c, d). These results support the idea that IECs isolated from _Il10__−/−_ mice
exhibit increased metabolic and innate immune activities associated with impaired barrier and digestive functions. In order to address the contribution of IEC-intrinsic RABGEF1 in IL-10
deficiency-induced intestinal inflammation, _Rabgef1__IEC-KO_ mice were generated on the _Il10__−/−_ background, and the development of intestinal pathology was evaluated. At the age of 3–4
months, the weight and the colon lengths were not significantly different between _Il10__−/−__; Rabgef1__IEC-KO_ and control _Il10__−/−__; Rabgef1__fl/fl_ mice (Supplementary Fig. 5).
However, _Il10__−/−__; Rabgef1__IEC-KO_ mice exhibited exacerbated signs of colon inflammation and damage as compared to controls (Fig. 3). Indeed, microscopic analysis of colon sections
revealed that _Il10__−/−__; Rabgef1__IEC-KO_ mice had more severe inflammation (associated with transmural and dense inflammatory infiltrates), as well as more epithelial damage, sometimes
developing into extensive ulcerated areas (Fig. 3a). Immunofluorescent staining of neutrophils with anti-myeloperoxydase (MPO) antibodies showed a higher number of neutrophils infiltrating
the colon of _Il10__−/−__; Rabgef1__IEC-KO_ mice as compared to controls (Fig. 3b, c). In addition, a substantial and consistent loss of mucus-producing goblet cells was observed (Fig. 3d).
As a consequence, a higher histopathological score was observed in the colon of _Il10__−/−__; Rabgef1__IEC-KO_ mice vs. controls (Fig. 3e). Within IECs, RABGEF1 deficiency was associated
with increased expression of genes coding for pro-inflammatory cytokines (_Il6_, _Tnfa_, _Vegfa_, _Ccl11_, _Ccl2_, _Cxcl1_, and _Cxcl2_) and reduced expression of the _Muc2_ gene, consistent
with the loss of goblet cells (Fig. 4a). In addition, like the situation observed in 12-month-old _Rabgef1__IEC-KO_ and littermates, intestinal permeability to FITC-dextran and systemic
levels of cytokines were not affected by RABGEF1 deficiency in this model (Fig. 4b, c). EPITHELIAL RABGEF1 REGULATES SUSCEPTIBILITY TO DSS-INDUCED COLON DAMAGE AND PATHOLOGY Next, we
assessed whether IEC-intrinsic RABGEF1 expression could be involved in the regulation of intestinal inflammation triggered by exogenous stimuli such as DSS. We used a model based on
administration of 3% DSS in drinking water for 7 days, which promoted acute increases in numbers of neutrophils, eosinophils, monocytes/macrophages, and dendritic cells as compared to the
baseline, a profile that was not observed in 10-week-old _Il10__−/−_ mice (Supplementary Fig. 6). Despite these differences between the two models, the transcriptomic profile of DSS-exposed
colonic IECs evaluated by RNA-seq was strikingly similar to the one of IECs isolated from age-matched _Il10__−/−_ animals (Supplementary Fig. 7). Based on such findings, we decided to
evaluate the contribution of epithelial RABGEF1 using this model. Compared to _Rabgef1__fl/fl_ mice, _Rabgef1__IEC-KO_ mice exhibited liquid, sticky and bloody feces (Fig. 5a) and
significantly shorter and highly inflamed colons (Fig. 5b, c). Histopathological examination of colons from DSS-treated _Rabgef1__IEC-KO_ mice revealed increased epithelial hyperplasia and
crypt elongation, the presence of dense granulocyte-rich cell infiltrates in the submucosa, and extensive epithelial ulceration and transmural inflammation when compared to DSS-treated
controls (Fig. 5d, e), resulting in a more severe histopathological score (Fig. 5f). Two days after DSS treatment, when intestinal permeability to FITC-dextran is known to be affected by
DSS,28 permeability to FITC-dextran was, again, not different between _Rabgef1__IEC-KO_ and control mice (Fig. 5g). These results indicate that, similar to its role in the IL-10 deficiency
model, RABGEF1 expression is also critical in limiting DSS-induced inflammation and damage. Altogether, our results so far support the idea that RABGEF1 deficiency promotes development of
intestinal inflammation and locally exacerbates IEC innate responses. RABGEF1 MODULATES INTRINSIC EXPRESSION OF GENES IMPLICATED IN IEC PHYSIOLOGY, ENDOCYTOSIS, AND INNATE IMMUNITY To gain
mechanistic insights into RABGEF1-dependent regulation of IEC responses, we performed RNA-seq analyses of FACS-sorted IECs from 10-week-old _Il10__−/−_; _Rabgef1__IEC-KO_ and littermate
_Il10__−/−_;_Rabgef1__fl/fl_ mice. As shown in Fig. 6a, RABGEF1-deficient IECs were separated from the control counterparts across principal component (PC)1 (explaining 40% of the variance
between samples). Using an adjusted _P_ value (_P_adj) < 10_−_2, a total of 102 genes were found to be differentially expressed (DE) between _Il10__−/−_;_Rabgef1__IEC-KO_ and _Il10__−/−_;
_Rabgef1__fl/fl_ IECs (Fig. 6b and Supplementary Table 1). First, on the basis of MouseNet v2 network29 and a bibliographical search, we found that several genes implicated in transmembrane
transporter activity and metabolic processing of nutrients were downregulated in RABGEF1-deficient IECs (Fig. 6c), supporting the idea that IECs’ physiological functions were impaired when
RABGEF1 was absent. Conversely, genes implicated in the regulation of innate immune pathways, such as the MAPK pathways or the NF-κB pathway, were upregulated in RABGEF1-deficient IECs (Fig.
6c). Second, gene set enrichment analysis (GSEA), used to identify biological signatures in IECs from _Il10__−/−_; _Rabgef1__IEC-KO_ mice as compared to those from _Il10__−/−_;
_Rabgef1__fl/fl_ mice, revealed a significant enrichment, among genes downregulated in RABGEF1-deficient IECs, of genes involved in endocytic events (Fig. 6d). We also assessed the gene
expression profiles of IECs from _Rabgef1__IEC-KO_ and littermate control mice on the wild-type (WT) background by RNA-seq (Supplementary Fig. 8a, b and Supplementary Table 2). Notably, we
found a highly significant similarity in gene expression profiles of IECs from _Rabgef1__IEC-KO_ mice under the IL-10-deficient and those under the WT background (Supplementary Fig. 8c), as
well as a downregulation of endocytosis-related transcripts in IECs from _Rabgef1__IEC-KO_ mice as compared to the control counterparts (Supplementary Fig. 8d). These data support the
conclusion that IECs exhibit alterations of their endolysosomal system when RABGEF1 is absent. RABGEF1-DEFICIENT IECS DISPLAY ENDOSOMAL ABNORMALITIES IN VITRO To support our RNA-seq-based
observations at the protein level, we took advantage of immortalized mouse IECs, which display structural features of primary IECs and can functionally respond to microbial stimulation.30
Lentiviral-based expression of RABGEF1 knockdown was induced in IECs (Supplementary Figs. 9 and 10). Using antibodies directed against the RABGEF1 endocytic effector Rab5, we showed that the
distribution of Rab5 staining at steady-state was markedly different between RABGEF1-sufficient vs. RABGEF1-deficient IECs, with Rab5 being more diffusely distributed and less structurally
organized when RABGEF1 was absent (Fig. 7a). After 60 min of LPS stimulation of RABGEF1-sufficient IECs, Rab5 staining intensity was increased and concentrated in polarized areas of the cell
(Fig. 7a). Conversely, Rab5 staining remained more diffuse and less polarized in LPS-stimulated RABGEF1-deficient IECs (Fig. 7a). In addition, we assessed the levels of early endosome
antigen-1 (EEA-1) protein in RABGEF1-sufficient and RABGEF1-deficient IECs by Western blotting. EEA-1 is an early endosomal marker and effector of Rab5 whose levels have been shown to be
reduced in Rab5-deficient cells17. We found that EEA-1 levels significantly increased following LPS stimulation of RABGEF1-sufficient IECs, supporting the occurrence of early endosomal
events following LPS stimulation (Fig. 7b, c). By contrast, we found that EEA-1 levels did not increase after LPS stimulation in RABGEF1-deficient IECs (Fig. 7b, c). Altogether, these data
indicate that Rab5-dependent endocytic events occur in IECs after LPS stimulation but that RABGEF1 deficiency disrupts such endocytic events. RABGEF1 REGULATES IEC-INTRINSIC P38-DEPENDENT
SIGNALING AND CHEMOKINE PRODUCTION Next, we examined the signaling pathways mediating the interaction of LPS and the Toll like receptor (TLR)4. Upon LPS stimulation, RABGEF1 knockdown had no
effects on the levels of phosphorylation of IκBα, an inhibitor of NF-κB whose phosphorylation is required for NF-κB activation (Fig. 8a, b).31 While increased levels of phosphorylated ERK
were noted in RABGEF1 knockdown IECs, such activation effect was not restored in RABGEF1-rescued IECs (Fig. 8a, b). However, after 5 or 15 min of LPS stimulation, RABGEF1 knockdown IECs
exhibited increased phosphorylation levels of p38 MAPK as compared to those in RABGEF1-sufficient IECs, a signaling activation event that was not observed when RABGEF1 expression was
restored (Fig. 8a, b). Functionally, such activation was associated with an increased ability of RABGEF1 knockdown IECs to produce the chemokines CCL2, CCL11, CXCL1, and CXCL2 as compared to
the control or RABGEF1-rescued IECs (Fig. 8c). Altogether, these data support the idea that RABGEF1 influences the TLR4-mediated p38 MAPK-dependent signaling pathways and their associated
productions of several chemokines in IECs. INTESTINAL INFLAMMATION IN _RABGEF1_ _IEC-KO_ MICE IS MEDIATED BY INTRINSIC MYD88-DEPENDENT SIGNALING AND MICROBE-DERIVED SIGNALS MYD88 mediates
the signaling responses induced by activation of IL-1/IL-18/Toll receptor superfamily members to induce activation of NF-κB and MAPK pathways leading to the release of pro-inflammatory
mediators.32 Since RABGEF1 regulates the p38 MAPK pathway in response to LPS in vitro, which signals through MYD88, we wondered whether MYD88 signaling contributed to the phenotype observed
in aged _Rabgef1__IEC-KO_ mice. In order to test this, we generated and analyzed _Rabgef1/Myd88__IEC-KO_ mice, in which IECs lack both MYD88 and RABGEF1. Notably, deletion of _Myd88_ in
_Rabgef1/Myd88__IEC-KO_ mice largely prevented the colitis observed in _Rabgef1__IEC-KO_ counterparts (Fig. 9a, b), which was associated with a similar IEC cytokine expression profile
between _Rabgef1/Myd88__IEC-KO_ mice and littermate controls (Fig. 9c). These results support the conclusion that MYD88-dependent signaling mediates intestinal inflammation in
_Rabgef1__IEC-KO_ mice. The commensal microorganisms that colonize the colon after birth are critical components of host physiology and immune homeostasis in healthy individuals, but can
also contribute to pathological processes in a dysregulated system1,33,34. IECs express a wide range of TLRs, which sense and transduce microbiota-derived signals via MYD88, except for
TLR34,7,31. Since the delayed colitis of _Rabgef1__IEC-KO_ mice was dependent on MYD88, we sought to determine the contribution of the microbiome in this context. First, we wondered whether
IEC-specific RABGEF1 depletion could induce alterations in the colon microbial composition. Therefore, we performed sequencing of bacterial 16S ribosomal RNA genes (16S rRNA-seq) to compare
the composition of the microbiota found in the colon lumen of _Rabgef1__IEC-KO_ and littermate _Rabgef1__fl/fl_ controls at the age of 10 months. Multidimensional scaling and unsupervised
hierarchical clustering did not reveal any genotype-dependent clustering (Fig. 10a, b), and bacterial diversity was found to be similar in the colon of _Rabgef1__IEC-KO_ as compared to the
control counterparts (Fig. 10c). In addition, colon microbiota composition was not different between _Rabgef1__IEC-KO_ and _Rabgef1__fl/fl_ control mice (Fig. 10d), supporting that
epithelial RABGEF1 deficiency does not induce changes in the gut bacterial composition. Second, we continuously fed 6-month-old _Rabgef1__IEC-KO_ and littermate controls with autoclaved
water with or without antibiotics (AB) for a period of 4 months, and colon histopathological scoring was performed at the end of the treatment. Notably, we found that antibiotic treatment
significantly reduced the features of colitis and the histopathological score in AB-treated _Rabgef1__IEC-KO_ mice as compared to the untreated counterparts (Fig. 10e, f), supporting the
conclusion that microbiota-dependent signals are involved in the delayed colitis that develops in aged _Rabgef1__IEC-KO_ mice. DISCUSSION We have identified herein a novel role for the
guanine exchange factor RABGEF1 in the regulation of IEC functions and intestinal inflammation in vivo. Using mice conditionally deficient for _Rabgef1_ in IECs, we have provided
experimental evidence that epithelial RABGEF1 deficiency promotes development of a microbiota- and MYD88-dependent colitis with age. Such colitis was associated with an upregulation of
IEC-intrinsic transcripts coding for pro-inflammatory cytokines and chemokines. Notably, however, no defects in intestinal barrier permeability, nor features of systemic immune activation,
were observed in 12-month-old _Rabgef1__IEC-KO_ mice. This supports the idea that epithelial RABGEF1 regulates events that are restricted to the gut mucosa, without substantial systemic
effects. Interestingly, it has been reported in some mouse models that intestinal and systemic pathology could be regulated by distinct mechanisms and disconnected from each other.35 To
investigate the role of epithelial RABGEF1 in the context of intestinal inflammation, we used two distinct models of colitis, initiated either by a dysregulated host immune response (i.e.,
IL-10 deficiency) or by an epithelial-eroding agent (i.e., DSS). The fact that IECs from 10-week-old _Il10__−/−_ mice or from DSS-exposed WT mice shared a strikingly similar profile supports
the conclusion that the IEC response to colitogenic triggers, whether they are endogenous (i.e., IL-10 deficiency) or exogenous (i.e., DSS), is conserved. This is particularly interesting,
since the IL-10 deficient and the DSS models are different in several aspects, e.g., chronic vs. acute, and endogenous immune trigger vs. exogenous trigger, respectively. Given the
differences observed in the recruitment of immune cells between the two models, it seems likely that the eroding properties of DSS and the subsequent release of damage-associated signals
from dying epithelial cells initially contribute to the acute immune cell infiltration upon DSS exposure, as opposed to the situation in IL-10-deficient mice. However, the features of
DSS-induced pathology, including the cellular immune profile in the colon, are thought to be influenced by many factors, including the dose and duration of DSS treatment, the gender and
genetic background of the host, and the microbial status or the housing conditions, which makes comparisons between studies difficult.36,37 During either form of colitis, IEC-specific
RABGEF1 deficiency exacerbated several cardinal features of colon pathology and was associated with increased expression of pro-inflammatory cytokines, but did not impact intestinal
permeability and systemic levels of pro-inflammatory cytokines, supporting the existence of RABGEF1-dependent local regulatory mechanisms. Moreover, genes involved in the regulation of the
endolysosomal system were found to be downregulated in IECs from _Rabgef1__IEC-KO_ mice as compared to controls, both under the _Il10__−/−_ and the WT background. This suggests that the GEF
activity of RABGEF1 for the GTPase Rab5, critical for the biogenesis of endosomes17, mediates such effects in IECs in vivo. The transcriptomic analyses of RABGEF1-deficient IECs also
suggested a possible involvement of RABGEF1 in regulating MAPK- or NF-κB-dependent innate signaling. Specifically, we used an in vitro tool to investigate the potential RABGEF1-dependent
regulation of such pathways. Immortalized mouse primary IECs were chosen because they allow analysis of innate immune functions30, but circumvent the restricted availability and homogeneity
of primary cells and organoids, respectively, and also avoid the structural and functional alterations often present in intestinal cell lines. Using this model, we have shown on the one hand
that RABGEF1 knockdown markedly altered Rab5-dependent endocytic events, at steady-state and upon LPS stimulation. On the other hand, we found that, upon LPS stimulation, RABGEF1
specifically prevented activation of p38 MAPK and downregulated the production of several chemokines, including CCL2, CXCL1, and CXCL2, consistent with the transcriptomic profile of
RABGEF1-deficient IECs in vivo. Notably, RABGEF1-sufficient IECs seemed particularly hyporesponsive to LPS stimulation in our experimental setting, which reflects well the
hypo-responsiveness of IECs to LPS and other TLR ligands in vivo.38,39,40 However, when IEC-intrinsic RABGEF1 expression was knocked down, LPS responsiveness was potentiated, leading to
aberrant activation of the p38 MAPK and increased chemokine production. These data are consistent with the hypothesis that RABGEF1 may act as a regulator of IEC innate responsiveness in
vivo, and that such responsiveness is compromised when epithelial RABGEF1 is absent, which would contribute to increased chemokine expression and the exaggerated inflammation observed in
_Rabgef1__IEC-KO_ mice. In this regard, it is interesting to note that the monocyte chemoattractant CCL2 and the neutrophil chemoattractants CXCL1 and CXCL2 are thought to be involved in
human or experimental IBD,41,42,43,44 and the p38 MAPK pathway has been shown to specifically control the expression of chemokines within IECs.45,46 The delayed development of colitis in
_Rabgef1__IEC-KO_ mice suggested that environmental factors may mediate disease susceptibility in those animals, and microbe-derived signals arguably represented appealing candidates.
Indeed, _Rabgef1__IEC-KO_ mice developed intestinal inflammation at a site of heavy microbial exposure (i.e., the colon) but did not have detectable pathology in the small intestine where
the bacterial load is much lower. Moreover, we showed that the spontaneous colitis of _Rabgef1__IEC-KO_ mice was MYD88-dependent, and it is well-known that many microbe-associated patterns
can activate TLRs through the adapter protein MYD8847,48. By analyzing the microbial composition of the colons of _Rabgef1__IEC-KO_ mice and by treating them with AB, we have provided
evidence that the intestinal inflammation that develops in aged _Rabgef1__IEC-KO_ mice depends on microbe-derived commensal signals. In younger mice, it may be that regulatory mechanisms are
more effective in preventing development of colitis. Previous work supports the conclusion that RABGEF1 can regulate many different pathways of innate immunity. In mast cells in vitro,
RABGEF1 has been shown to regulate FcεRI-dependent activation of Ras and ERK20 and SCF-dependent activation of ERK and JNK.49 In the skin, we have demonstrated that keratinocyte RABGEF1 can
maintain skin homeostasis by regulating intrinsic IL1R-MYD88-NF-κB-dependent signaling pathways independently of the skin microbiota.21 The current study indicates that epithelial RABGEF1
can also regulate microbiota- and MYD88-dependent intestinal inflammation and p38 MAPK-dependent signaling and chemokine production in IECs. We cannot exclude the possibility that IL-1β
contributes to MYD88-dependent inflammation in _Rabgef1__IEC-KO_ mice. However, RABGEF1-dependent regulatory pathways appear to exhibit differences in functional and interactive properties
in epidermal keratinocytes, IECs and mast cells, suggesting that there may be tissue- and cell-specific mechanisms mediating different RABGEF1 effects. The precise mechanisms by which the
activities of RABGEF1 regulate the activation of innate immune pathways, and the expression of cytokines, in IECs are still unclear. Nevertheless, our data are in accord with the hypothesis
that the GEF activity for Rab5 may contribute to these effects. Indeed, we have demonstrated that deletion of RABGEF1 can markedly alter Rab5-dependent endocytic events, as well as
TLR4-MYD88-dependent signaling pathways, in IECs. Since activation of TLR4 by LPS is regulated by endosomal-mediated endocytosis, which requires Rab5 activity that is regulated by
RABGEF113,14,15,16,17,48, RABGEF1 deficiency may downregulate LPS-induced receptor-mediated endocytosis. Because RABGEF1 represents an important negative regulator of TLR4 signaling, RABGEF1
deficiency may promote the increased activation of LPS-dependent pathways, resulting in an increased cytokine expression in IECs. Stimulation of the TLR4-MYD88-dependent pathway by LPS also
can activate several transcription factors, including NF-κB, AP-1, and CREB31,48. In our signal transduction analyses, we showed that stimulation of RABGEF1-deficient IECs with LPS did not
increase levels of p-IκBα, suggesting that the NF-κB pathway is not involved in the observed effect elicited by RABGEF1 deficiency. Similarly, our signaling studies also showed that ERK MAPK
was not involved in the LPS-induced effect observed with these cells, suggesting that the AP-1 pathway is likewise not directly involved. However, levels of phosphorylation of p38 MAPK were
significantly enhanced by LPS stimulation in RABGEF1-deficient IECs, suggesting that the CREB transcription factor pathway is involved in mediating the cytokine transcription effects during
the TLR4-dependent activation of IECs31,48. Obviously, future detailed mechanistic studies using epidermal keratinocytes, mast cells and IECs expressing different RABGEF1 domain mutants
will help in investigating how different domains of RABGEF1 are involved in such effects and the precise molecular mechanisms underlying the regulatory functions of RABGEF1. In conclusion,
our study identifies a previously unknown function for RABGEF1 protein in the regulation of MYD88-dependent intestinal inflammation and IEC innate pathways in mice. It is currently unknown
whether dysregulated epithelial RABGEF1 expression also contributes to intestinal pathology in IBD in humans. However, in 2013, Montero-Meléndez and colleagues performed microarray gene
expression profiling of colon biopsies from a small cohort of IBD patients in search of predictive genomic signatures, and identified _RABGEF1_ transcript level as a clinically relevant
predictor for classifying IBD patients, with glucocorticoid-resistant patients exhibiting the lowest RABGEF1 expression.50 Together with our study, these data suggest that RABGEF1-dependent
regulatory mechanisms may represent promising therapeutic targets in the context of IBD. METHODS MICE C57BL/6 _Rabgef1__fl/fl_ mice were previously described.21 Transgenic C57BL/6
_Villin-Cre_ (B6.Cg-Tg(Vil1-cre)997Gum/J),51 _Il10__−/−_ (B6.129P2-_Il10__tm1Cgn_/J) and _Myd88__fl/fl_ (B6.129P2[SJL]-_Myd88__tm1Defr_/J) mice were from The Jackson Laboratory (Bar Harbor,
USA). Mice were housed under specific pathogen free (SPF) conditions and maintained in a 12-h light–dark cycle with food and water ad libitum. Age-matched littermate mice were used for
experiments throughout the study. All animal experiments described in this study were carried out with the approval of the Institutional Animal Care and Use Committee of Liege University.
The “Guide for the Care and Use of Laboratory Animals”, prepared by the Institute of Laboratory Animal Resources, National Research Council, and published by the National Academy Press, as
well as European and local legislations, were followed carefully. REAGENTS AND ANTIBODIES Details about reagents and antibodies can be found in the Supplementary material. IMMUNOSTAINING AND
CONFOCAL MICROSCOPY Paraffin-embedded intestinal sections were deparaffinized, rehydrated, and heat-induced epitope retrieval was performed in sodium citrate buffer. After blocking, samples
were incubated overnight at 4 °C with primary antibodies. Secondary antibody staining was performed at room temperature. For immunostaining of IECs in vitro, IECs were cultured on slides
for 24 h. Slides were methanol-fixed and permeabilized before staining with primary and secondary antibodies. Images were acquired using a Leica SP5 (Wetzlar, Germany) confocal
laser-scanning microscope. Additional Details can be found in the Supplementary material. ASSESSMENT OF INTESTINAL PERMEABILITY To evaluate intestinal permeability, FITC-dextran (FD4, Merck,
Darmstadt, Germany) was administered by gastric gavage to fasting mice (4 h food and water fasting, 0.6 mg/g diluted in phosphate-buffered saline (PBS)). Four hours later, plasma was
collected and plasma levels of FITC were measured by spectrophotometry. QUANTITATIVE RT-PCR ON IECS After IEC FACS sorting, RNA was extracted and reverse-transcribed into cDNA, and PCR
reactions were performed in duplicate. Primer sequences can be found in Supplementary Table 3. Data were normalized to the housekeeping genes _Hprt and Ppib_ in the same sample. Additional
details can be found in the Supplementary material. QUANTIFICATION OF SERUM CYTOKINES Blood was collected from the tail vein and sera were assayed for CXCL1, CXCL2, GM-CSF, IL1-β, IL-6,
VEGF, and TNF-α using Luminex Mouse Magnetic Assay (R&D, Minneapolis, USA), according to the manufacturer’s recommendations. COLON HISTOPATHOLOGICAL SCORING For evaluation of colon
pathology in antibiotic-treated or untreated _Rabgef1__IEC-KO_, _Rabgef1/Myd88__IEC-KO_ and control counterparts at steady-state, on the WT or _Il10__−/−_ background, the scoring system
described in the report of Dieleman et al.52 was adapted as described in the Supplementary material. MOUSE MODEL OF DSS-INDUCED COLITIS Experiments involving DSS exposure involved between 8-
and 12-week-old female mice. Acute colitis was induced by addition of 3% DSS (DSS Colitis grade, MP biomedicals, Santa Ana, USA) to the drinking water for 7 days. Stool consistency was
evaluated at day 7 during 4 h and mice were then sacrificed to perform a macroscopic evaluation of the colons and for histopathological analyses. The severity of DSS-induced colitis was
evaluated, as previously reported53 and as described in the Supplementary material. IN VITRO IEC CULTURE AND STIMULATION Mouse CI-muINTEPI cells (InSCREENeX, Braunschweig, Germany) were
cultured in muINTEPI complete medium (InSCREENeX). Plates were precoated with collagen solution (InSCREENeX) for 2 h and washed with PBS before use. When cultured cells reached 80%
confluence, NT control, RABGEF1-knockdown and RABGEF1-rescued IECs were seeded in 24-well plates (4 × 105 cells). For confocal microscopy analysis, cells were cultured and stimulated as
described above. For Western blot studies, cells were stimulated with LPS (100 ng/ml) for 5, 15, or 60 min; for cytokine measurements, cells were stimulated with LPS for 6 or 24 h and cell
culture supernatants were assayed for CCL2, CCL11, CXCL1, CXCL2, and TNF-α using Luminex Mouse Magnetic Assay (R&D, Minneapolis, USA). WESTERN BLOTTING Mouse IECs were lysed in lysis
Buffer (3.03 g TRIS pH 7.5 + 0.19 g EDTA + 5 mL NP-40 + 6 g NaCl in 500 mL DQ water) supplemented with Complete™ Protease Inhibitor Cocktail (Roche, Basel, Switzerland) and PhosSTOP™
Phosphatase Inhibitor Cocktail (Roche, Basel, Switzerland). Protein concentrations in the resulting lysates were measured using the Pierce™ BCA Protein Assay Kit (ThermoFisher Scientific,
Waltham, USA). Totally, 50 µg proteins were loaded in 4–20% Mini-PROTEAN® TGX™ Gel (Bio-rad, Hercules, USA) and electroblotted onto Invitrolon polyvinylidene fluoride membranes (Novex,
ThermoFisher Scientific, Waltham, USA). Membranes were blocked in 5% nonfat dry milk in Tris-buffered saline-Tween 20 (0.1%) buffer and then probed with primary antibodies in 5% bovine serum
albumin-tris-buffered saline-Tween 20 (0.1%) buffer. Equal loading was confirmed by probing for actin, vinculin or HSP-90α. ANTIBIOTIC TREATMENT Six-month-old _Rabgef1__IEC-KO_ mice and
_Rabgef1__fl/fl_ littermate controls were fed autoclaved water with or without enrofloxacin 1% (Baytril, Bayer) for 4 months. OTHER METHODS See the Methods section in Supplementary material
for cell isolation, staining and flow cytometry, assessment of _Rabgef1_ deletion by quantitative PCR, sample preparation and histology, high-throughput mRNA-sequencing, GSEA,
lentiviral-based RABGEF1 knock down and rescuing of mouse IECs, 16S rRNA gene sequencing and metagenomic analyses. STATISTICAL ANALYSES Respect of the assumptions of normal distribution of
residuals and homoscedasticity were verified. Unless otherwise indicated, data are presented as mean + SEM, as well as individual values, from independent experiments and were pooled for
analysis in each data panel. Statistical analyses were performed using Prism 6 (GraphPad Software, La Jolla, USA), except for RNA-seq data, for which the R package “DESeq2” was used. We
performed the Mantel–Cox test, one-way or two-way ANOVA with Tukey’s post hoc tests for multiple comparisons, and Mann–Whitney or unpaired two-tailed Student’s _t_ tests as mentioned in the
respective figure legends. We considered a _P_ value lower than 0.05 as significant. *_P_ < 0.05; **_P_ < 0.01, ***_P_ < 0.001; ns not significant. DATA AVAILABILITY The RNA-seq
data provided in this manuscript have been deposited in the ArrayExpress database at EMBL-EBI (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-7315) under accession number E-MTAB-7315.
REFERENCES * Kaiko, G. E. & Stappenbeck, T. S. Host-microbe interactions shaping the gastrointestinal environment. _Trends Immunol._ 35, 538–548 (2014). Article CAS PubMed PubMed
Central Google Scholar * Peterson, L. W. & Artis, D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. _Nat. Rev. Immunol._ 14, 141–153 (2014).
Article CAS PubMed Google Scholar * Okumura, R. & Takeda, K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. _Exp. Mol. Med._ 49, e338 (2017). Article
CAS PubMed PubMed Central Google Scholar * McClure, R. & Massari, P. TLR-dependent human mucosal epithelial cell responses to microbial pathogens. _Front. Immunol._ 5, 386 (2014).
Article PubMed PubMed Central CAS Google Scholar * Pastorelli, L., De Salvo, C., Mercado, J. R., Vecchi, M. & Pizarro, T. T. Central role of the gut epithelial barrier in the
pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics. _Front. Immunol._ 4, 280 (2013). Article PubMed PubMed Central CAS Google Scholar
* Gallo, R. L. & Hooper, L. V. Epithelial antimicrobial defence of the skin and intestine. _Nat. Rev. Immunol._ 12, 503–516 (2012). Article CAS PubMed PubMed Central Google Scholar
* Rakoff-Nahoum, S., Paglino, J., Eslami-Varzaneh, F., Edberg, S. & Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis.
_Cell_ 118, 229–241 (2004). Article CAS PubMed Google Scholar * Podolsky, D. K. Inflammatory bowel disease. _N. Engl. J. Med._ 347, 417–429 (2002). Article CAS PubMed Google Scholar
* Strober, W., Fuss, I. & Mannon, P. The fundamental basis of inflammatory bowel disease. _J. Clin. Invest._ 117, 514–521 (2007). Article CAS PubMed PubMed Central Google Scholar *
Jostins, L. et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. _Nature_ 491, 119–124 (2012). Article CAS PubMed PubMed Central Google
Scholar * Hooper, L. V. & Gordon, J. I. Commensal host-bacterial relationships in the gut. _Science_ 292, 1115–1118 (2001). Article CAS PubMed Google Scholar * Ni, J., Wu, G. D.,
Albenberg, L. & Tomov, V. T. Gut microbiota and IBD: causation or correlation? _Nat. Rev. Gastroenterol. Hepatol._ 14, 573 (2017). Article PubMed PubMed Central Google Scholar *
Delprato, A., Merithew, E. & Lambright, D. G. Structure, exchange determinants, and family-wide rab specificity of the tandem helical bundle and Vps9 domains of Rabex-5. _Cell_ 118,
607–617 (2004). Article CAS PubMed Google Scholar * Horiuchi, H. et al. A novel Rab5 GDP/GTP exchange factor complexed to Rabaptin-5 links nucleotide exchange to effector recruitment and
function. _Cell_ 90, 1149–1159 (1997). Article CAS PubMed Google Scholar * Zerial, M. & McBride, H. Rab proteins as membrane organizers. _Nat. Rev. Mol. Cell Biol._ 2, 107–117
(2001). Article CAS PubMed Google Scholar * Mattera, R. & Bonifacino, J. S. Ubiquitin binding and conjugation regulate the recruitment of Rabex-5 to early endosomes. _EMBO J._ 27,
2484–2494 (2008). Article CAS PubMed PubMed Central Google Scholar * Zeigerer, A. et al. Rab5 is necessary for the biogenesis of the endolysosomal system in vivo. _Nature_ 485, 465–470
(2012). Article CAS PubMed Google Scholar * Lee, S. et al. Structural basis for ubiquitin recognition and autoubiquitination by Rabex-5. _Nat. Struct. Mol. Biol._ 13, 264–271 (2006).
Article CAS PubMed PubMed Central Google Scholar * Mattera, R., Tsai, Y. C., Weissman, A. M. & Bonifacino, J. S. The Rab5 guanine nucleotide exchange factor Rabex-5 binds ubiquitin
(Ub) and functions as a Ub ligase through an atypical Ub-interacting motif and a zinc finger domain. _J. Biol. Chem._ 281, 6874–6883 (2006). Article CAS PubMed Google Scholar * Tam, S.
Y. et al. RabGEF1 is a negative regulator of mast cell activation and skin inflammation. _Nat. Immunol._ 5, 844–852 (2004). Article CAS PubMed Google Scholar * Marichal, T. et al.
Guanine nucleotide exchange factor RABGEF1 regulates keratinocyte-intrinsic signaling to maintain skin homeostasis. _J. Clin. Invest._ 126, 4497–4515 (2016). Article PubMed PubMed Central
Google Scholar * Kuhn, R., Lohler, J., Rennick, D., Rajewsky, K. & Muller, W. Interleukin-10-deficient mice develop chronic enterocolitis. _Cell_ 75, 263–274 (1993). Article CAS
PubMed Google Scholar * Barnett, M. P. et al. Changes in colon gene expression associated with increased colon inflammation in interleukin-10 gene-deficient mice inoculated with
Enterococcus species. _BMC Immunol._ 11, 39 (2010). Article PubMed PubMed Central CAS Google Scholar * Hansen, J. J., Holt, L. & Sartor, R. B. Gene expression patterns in
experimental colitis in IL-10 deficient mice. _Inflamm. Bowel Dis._ 15, 890 (2009). Article PubMed Google Scholar * Russ, A. E. et al. Gene expression changes in the colon epithelium are
similar to those of intact colon during late inflammation in interleukin-10 gene deficient mice. _PLoS ONE_ 8, e63251 (2013). Article CAS PubMed PubMed Central Google Scholar * Keubler,
L. M., Buettner, M., Hager, C. & Bleich, A. A Multihit Model: colitis lessons from the interleukin-10-deficient mouse. _Inflamm. Bowel Dis._ 21, 1967–1975 (2015). Article PubMed
Google Scholar * Paul, G., Khare, V. & Gasche, C. Inflamed gut mucosa: downstream of interleukin-10. _Eur. J. Clin. Invest._ 42, 95–109 (2012). Article CAS PubMed Google Scholar *
Yan, Y. et al. Temporal and spatial analysis of clinical and molecular parameters in dextran sodium sulfate induced colitis. _PLoS One_ 4, e6073 (2009). Article PubMed PubMed Central CAS
Google Scholar * Kim, E. et al. MouseNetv2: a database of gene networks for studying the laboratory mouse and eight other model vertebrates. _Nucleic Acids Res._ 44, D848–D854 (2016).
Article CAS PubMed Google Scholar * Schwerk, J. et al. Generation of mouse small intestinal epithelial cell lines that allow the analysis of specific innate immune functions. _PloS ONE_
8, e72700 (2013). Article CAS PubMed PubMed Central Google Scholar * Kondo, T., Kawai, T. & Akira, S. Dissecting negative regulation of Toll-like receptor signaling. _Trends
Immunol._ 33, 449–458 (2012). Article CAS PubMed Google Scholar * O’Neill, L. A. & Bowie, A. G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling.
_Nat. Rev. Immunol._ 7, 353–364 (2007). Article PubMed CAS Google Scholar * Kamada, N., Seo, S. U., Chen, G. Y. & Nunez, G. Role of the gut microbiota in immunity and inflammatory
disease. _Nat. Rev. Immunol._ 13, 321–335 (2013). Article CAS PubMed Google Scholar * Dethlefsen, L., McFall-Ngai, M. & Relman, D. A. An ecological and evolutionary perspective on
human-microbe mutualism and disease. _Nature_ 449, 811–818 (2007). Article CAS PubMed Google Scholar * Uhlig, H. H. et al. Differential activity of IL-12 and IL-23 in mucosal and
systemic innate immune pathology. _Immunity_ 25, 309–318 (2006). Article CAS PubMed Google Scholar * Nunes, N. S. et al. Temporal clinical, proteomic, histological and cellular immune
responses of dextran sulfate sodium-induced acute colitis. _World J. Gastroenterol._ 24, 4341–4355 (2018). Article CAS PubMed PubMed Central Google Scholar * Eichele, D. D. &
Kharbanda, K. K. Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. _World J. Gastroenterol._ 23,
6016–6029 (2017). Article CAS PubMed PubMed Central Google Scholar * Lotz, M. et al. Postnatal acquisition of endotoxin tolerance in intestinal epithelial cells. _J. Exp. Med._ 203,
973–984 (2006). Article CAS PubMed PubMed Central Google Scholar * Otte, J. M., Cario, E. & Podolsky, D. K. Mechanisms of cross hyporesponsiveness to Toll-like receptor bacterial
ligands in intestinal epithelial cells. _Gastroenterol_ 126, 1054–1070 (2004). Article CAS Google Scholar * Rhee, S. H. et al. Pathophysiological role of Toll-like receptor 5 engagement
by bacterial flagellin in colonic inflammation. _Proc. Natl Acad. Sci. USA_ 102, 13610–13615 (2005). Article CAS PubMed PubMed Central Google Scholar * Alkim, C., Alkim, H., Koksal, A.
R., Boga, S. & Sen, I. Angiogenesis in inflammatory bowel disease. _Int J. Inflam._ 2015, 970890 (2015). PubMed PubMed Central Google Scholar * Danese, S. & Gasbarrini, A.
Chemokines in inflammatory bowel disease. _J. Clin. Pathol._ 58, 1025–1027 (2005). Article CAS PubMed PubMed Central Google Scholar * Wang, D., Dubois, R. N. & Richmond, A. The role
of chemokines in intestinal inflammation and cancer. _Curr. Opin. Pharm._ 9, 688–696 (2009). Article CAS Google Scholar * Shea-Donohue, T. et al. Mice deficient in the CXCR2 ligand,
CXCL1 (KC/GRO-alpha), exhibit increased susceptibility to dextran sodium sulfate (DSS)-induced colitis. _Innate Immun._ 14, 117–124 (2008). Article CAS PubMed Google Scholar * Parhar,
K., Ray, A., Steinbrecher, U., Nelson, C. & Salh, B. The p38 mitogen-activated protein kinase regulates interleukin-1beta-induced IL-8 expression via an effect on the IL-8 promoter in
intestinal epithelial cells. _Immunology_ 108, 502–512 (2003). Article CAS PubMed PubMed Central Google Scholar * Waterhouse, C. C., Joseph, R. R., Winsor, G. L., Lacombe, T. A. &
Stadnyk, A. W. Monocyte chemoattractant protein-1 production by intestinal epithelial cells in vitro: a role for p38 in epithelial chemokine expression. _J. Interferon Cytokine Res._ 21,
223–230 (2001). Article CAS PubMed Google Scholar * Kawai, T. & Akira, S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. _Nat. Immunol._
11, 373–384 (2010). Article CAS PubMed Google Scholar * Gay, N. J., Symmons, M. F., Gangloff, M. & Bryant, C. E. Assembly and localization of Toll-like receptor signalling
complexes. _Nat. Rev. Immunol._ 14, 546–558 (2014). Article CAS PubMed Google Scholar * Kalesnikoff, J. et al. RabGEF1 regulates stem cell factor/c-Kit-mediated signaling events and
biological responses in mast cells. _Proc. Natl Acad. Sci. USA_ 103, 2659–2664 (2006). Article CAS PubMed PubMed Central Google Scholar * Montero-Melendez, T., Llor, X.,
Garcia-Planella, E., Perretti, M. & Suarez, A. Identification of novel predictor classifiers for inflammatory bowel disease by gene expression profiling. _PloS ONE_ 8, e76235 (2013).
Article CAS PubMed PubMed Central Google Scholar * Madison, B. B. et al. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal
(duodenum, cecum) axes of the intestine. _J. Biol. Chem._ 277, 33275–33283 (2002). Article CAS PubMed Google Scholar * Dieleman, L. A. et al. Chronic experimental colitis induced by
dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. _Clin. Exp. Immunol._ 114, 385–391 (1998). Article CAS PubMed PubMed Central Google Scholar * Erben, U. et al. A
guide to histomorphological evaluation of intestinal inflammation in mouse models. _Int J. Clin. Exp. Pathol._ 7, 4557–4576 (2014). PubMed PubMed Central Google Scholar Download
references ACKNOWLEDGEMENTS We thank all members of the Laboratories of Cellular and Molecular Immunology and Immunophysiology for discussions; Sandra Ormenese, Raafat Stefan, Alexandre
Hego, and Jean-Jacques Goval from the GIGA Flow Cytometry and Cell Imaging Platform; Pierre Drion, Gaëlle Lambert and all staff members from the GIGA Mouse facility and Transgenics Platform;
Cécile Oury for discussions about colitis induction protocols; Chantal Humblet and staff members from the Immunohistology Platform; Benoît Charloteaux, Manon Deckers, Latifa Karim, and
members from the Genomics Platform; Pauline Maréchal, Cedric François, Raja Fares, and Ilham Sbai for their excellent technical and administrative support. The Cellular and Molecular
Immunology Laboratory is supported by an Excellence Of Science (EOS) program from the F.R.S.-FNRS and FWO. S.J.G., M.T. and S.-Y.T. are supported by National Institutes of Health grant
NIH/NIAMS R01 AR067145. F.B. is supported by the F.R.S.-FNRS for the FRFS-WELBIO under grant CR-2012S-01R. T.M. is a Research Associate of the F.R.S.-FNRS and is supported by an “Incentive
Grant for Scientific Research” of the F.R.S.-FNRS (F.4508.18), by the FRFS-WELBIO under grant CR-2017s-04, by the Acteria Foundation and by an ERC Starting Grant (ERC-StG-2018 IM-ID 801823).
AUTHOR INFORMATION Author notes * These authors contributed equally: Sophie El Abbas, Coraline Radermecker. AUTHORS AND AFFILIATIONS * Laboratory of Immunophysiology, GIGA Institute, Liege
University, Liège, Belgium Sophie El Abbas, Coraline Radermecker, Qiang Bai, Charline Beguin, Joey Schyns, Margot Meunier & Thomas Marichal * Faculty of Veterinary Medicine, Liege
University, Liège, Belgium Sophie El Abbas, Coraline Radermecker, Joey Schyns, Margot Meunier, Dimitri Pirottin, Christophe J. Desmet, Tatiana Art, Fabrice Bureau & Thomas Marichal *
Laboratory of Cellular and Molecular Immunology, GIGA Institute, Liege University, Liège, Belgium Dimitri Pirottin & Fabrice Bureau * Gastroenterology Unit, University Hospital (CHU),
Liege University, Liège, Belgium Marie-Alice Meuwis & Edouard Louis * Laboratory of Translational Gastroenterology, GIGA Institute, Liege University, Liège, Belgium Marie-Alice Meuwis
& Edouard Louis * Department of Pathology, Stanford University School of Medicine, Stanford, CA, USA See-Ying Tam, Mindy Tsai & Stephen J. Galli * Sean N. Parker Center for Allergy
and Asthma Research, Stanford University School of Medicine, Stanford, CA, USA See-Ying Tam, Mindy Tsai & Stephen J. Galli * WELBIO, Walloon Excellence in Life Sciences and
Biotechnology, Wallonia, Belgium Fabrice Bureau & Thomas Marichal * Department of Microbiology and Immunology, Stanford University School of Medicine, Stanford, CA, USA Stephen J. Galli
Authors * Sophie El Abbas View author publications You can also search for this author inPubMed Google Scholar * Coraline Radermecker View author publications You can also search for this
author inPubMed Google Scholar * Qiang Bai View author publications You can also search for this author inPubMed Google Scholar * Charline Beguin View author publications You can also search
for this author inPubMed Google Scholar * Joey Schyns View author publications You can also search for this author inPubMed Google Scholar * Margot Meunier View author publications You can
also search for this author inPubMed Google Scholar * Dimitri Pirottin View author publications You can also search for this author inPubMed Google Scholar * Christophe J. Desmet View author
publications You can also search for this author inPubMed Google Scholar * Marie-Alice Meuwis View author publications You can also search for this author inPubMed Google Scholar * Tatiana
Art View author publications You can also search for this author inPubMed Google Scholar * Edouard Louis View author publications You can also search for this author inPubMed Google Scholar
* See-Ying Tam View author publications You can also search for this author inPubMed Google Scholar * Mindy Tsai View author publications You can also search for this author inPubMed Google
Scholar * Fabrice Bureau View author publications You can also search for this author inPubMed Google Scholar * Stephen J. Galli View author publications You can also search for this author
inPubMed Google Scholar * Thomas Marichal View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS T.M. conceived of the project. S.E.A., C.R.,
D.P., M-A.M., T.A., C.J.D., E.L., S-Y.T., M.T., F.B., S.J.G. and T.M. were involved in experimental design. S.E.A. and C.R. performed most of the experiments and analyzed the data, and
compiled the data with the help of T.M. C.B., J.S., M.M. and M-A.M. helped with experiments. Q.B., D.P. and T.M. performed the bioinformatic analyses with the support of the GIGA Genomics
Platform. S-Y.T., M.T. and S.J.G. provided _Rabgef1__fl/fl_ mice. S.E.A. and T.M. prepared the figures. T.M. secured funding and wrote the original paper. All authors participated in writing
or editing the paper. CORRESPONDING AUTHOR Correspondence to Thomas Marichal. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ETHICS STATEMENT All animal
experiments described in this study were carried out with the approval of the Institutional Animal Care and Use Committee of Liege University (DE1626). The “Guide for the Care and Use of
Laboratory Animals”, prepared by the Institute of Laboratory Animal Resources, National Research Council, and published by the National Academy Press, as well as European and local
legislations, were followed carefully. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional
affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY MATERIAL RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE El Abbas, S., Radermecker, C., Bai, Q. _et
al._ Epithelial RABGEF1 deficiency promotes intestinal inflammation by dysregulating intrinsic MYD88-dependent innate signaling. _Mucosal Immunol_ 13, 96–109 (2020).
https://doi.org/10.1038/s41385-019-0211-z Download citation * Received: 05 December 2018 * Revised: 18 September 2019 * Accepted: 28 September 2019 * Published: 18 October 2019 * Issue Date:
January 2020 * DOI: https://doi.org/10.1038/s41385-019-0211-z SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative