Play all audios:
ABSTRACT The excitatory/inhibitory (E/I) imbalance hypothesis posits that imbalance between excitatory (glutamatergic) and inhibitory (GABAergic) mechanisms underlies the behavioral
characteristics of autism. However, how E/I imbalance arises and how it may differ across autism symptomatology and brain regions is not well understood. We used innovative analysis
methods—combining competitive gene-set analysis and gene-expression profiles in relation to cortical thickness (CT) to investigate relationships between genetic variance, brain structure and
autism symptomatology of participants from the AIMS-2-TRIALS LEAP cohort (autism = 359, male/female = 258/101; neurotypical control participants = 279, male/female = 178/101) aged 6–30
years. Using competitive gene-set analyses, we investigated whether aggregated genetic variation in glutamate and GABA gene-sets could be associated with behavioral measures of autism
symptoms and brain structural variation. Further, using the same gene-sets, we corelated expression profiles throughout the cortex with differences in CT between autistic and neurotypical
control participants, as well as in separate sensory subgroups. The glutamate gene-set was associated with all autism symptom severity scores on the Autism Diagnostic Observation Schedule-2
(ADOS-2) and the Autism Diagnostic Interview-Revised (ADI-R) within the autistic group. In adolescents and adults, brain regions with greater gene-expression of glutamate and GABA genes
showed greater differences in CT between autistic and neurotypical control participants although in opposing directions. Additionally, the gene expression profiles were associated with CT
profiles in separate sensory subgroups. Our results suggest complex relationships between E/I related genetics and autism symptom profiles as well as brain structure alterations, where there
may be differential roles for glutamate and GABA. SIMILAR CONTENT BEING VIEWED BY OTHERS QUANTITATIVE TRAIT LOCUS ANALYSIS FOR ENDOPHENOTYPES REVEALS GENETIC SUBSTRATES OF CORE SYMPTOM
DOMAINS AND NEUROCOGNITIVE FUNCTION IN AUTISM SPECTRUM DISORDER Article Open access 24 September 2022 MOLECULAR AND NETWORK-LEVEL MECHANISMS EXPLAINING INDIVIDUAL DIFFERENCES IN AUTISM
SPECTRUM DISORDER Article 09 March 2023 VARIEGATION OF AUTISM RELATED TRAITS ACROSS SEVEN NEUROGENETIC DISORDERS Article Open access 07 April 2022 INTRODUCTION Autism spectrum disorder
(autism) is a neurodevelopmental condition characterized by challenges in social interaction and communication, restricted and repetitive patterns of behavior and/or atypical sensory
processing [1]. One influential hypothesis regarding its underlying mechanisms is the excitatory/inhibitory (E/I) imbalance hypothesis, which suggests that an imbalance between excitatory
(predominantly glutamatergic) and inhibitory (predominantly GABAergic (γ-aminobutyric acid)) mechanisms in the brain underlies symptomatology [2]. Causal links have been suggested, but so
far with suggestions for both overexcitation and overinhibition [2,3,4,5,6]. However, understanding the mechanisms of _how_ E/I imbalance is underlying autism symptomatology is complex. The
heterogeneity and polygenic nature of autism, and previous opposing findings of E/I imbalance, may be evidence of differential involvement across autism characteristics or brain regions.
Mechanisms of E/I imbalance may have genetic underpinnings. Autism is a polygenic condition where several genetic variants together give rise to the expression of the phenotype. Progress in
identifying common genetic variants associated with autism have included genes encoding proteins involved in glutamate and GABA receptors and transporters [7,8,9,10]. De novo mutations are
also known to underlie a significant portion of the prevalence of autism, where additional links between genes involved in excitatory and inhibitory signaling have been found [11]. Several
studies have suggested glutamatergic and GABAergic genetic links to behavioral autism phenotypes [3, 4, 12,13,14]. These phenotypes have been linked to changes in glutamate and GABA
concentrations in the brain as well [4, 15]. Genetic and behavioral changes in autistic individuals can additionally be linked to brain structure, where a role for E/I imbalance seems
plausible. Differences in cortical thickness (CT) have consistently been found in autism, and have been shown to differ throughout development as well [16, 17]. More specifically, both
increased and decreased cortical thickness has been found in autism mainly in fronto-temporal, fronto-parietal, limbic areas and fronto-striatal circuits [16,17,18,19,20,21,22]. However, we
do not yet have a clear understanding of what is causing these differences, although there is strong evidence that genetic factors play a role [21]. Cell-type specific gene-expression has
been shown to be associated with differences in cortical thickness in several neurodevelopmental disorders, among which autism [23, 24]. Some genes within these cell-type specific gene-sets
relate to cellular E/I function, but a similar relation has not yet been investigated focusing specifically on genetic pathways involved in excitatory and inhibitory signaling. Although not
yet investigated directly, it is plausible that alterations in glutamate and GABA functioning relate to morphological differences such as CT. For instance, glutamate and GABA receptors play
a role in dendritic growth, a process with genetic underpinnings found to be altered in autism [25,26,27]. Dendrite growth is also linked to cortical thickness [23, 28]. Altered dendritic
growth, and associated genes, have been linked to autism symptomatology, especially repetitive behaviors [25, 29, 30]. In order to understand mechanistic underpinnings of morphological
differences in autism it is important to get a better understanding of these links between E/I imbalance and how it may relate to structural differences. This has potential to increase
understanding of the links between molecular and genetic mechanisms of autism with macroscopic measures such as cortical thickness, aiding in developing markers for subtyping and developing
targeted treatment options in autism [19]. In the current study we want to integrate parts of the E/I puzzle by taking a multimodal approach focusing on aggregated (common) genetic
variation, different autism phenotypes and their association with brain structure. One relatively understudied part of the autism phenotype comprises sensory symptoms. These are especially
interesting as they have been suggested promising in an attempt to unscramble the autism heterogeneity [31] as well as for their shown link with E/I imbalance using MR spectroscopy [32].
Additionally, previous investigations within our dataset have shown differences in CT between those with severe and low sensory processing deficits in brain regions enriched for genes that
are expressed in excitatory neurons in the developing cortex [19]. Here we used a competitive gene-set approach [33,34,35], investigating the role of aggregated genetic variation in
glutamate and GABA gene-sets in behavioral autism phenotypes and cortical thickness. By considering several (common) genetic variants in the same analysis, the power of the study in
explaining phenotypic variance is increased. In short, this tests whether genes in the gene-set are more strongly correlated with the phenotype of interest than other genes [33]. This method
has shown utility in other neurodevelopmental disorders showing aggregated genetic effects rather than using single candidate-gene associations [29, 36, 37]. Additionally, using the same
gene-sets, we investigated whether their expression profiles across the cortex could be associated with differences in cortical thickness between autistics and neurotypical controls (NTC).
Building on the previous findings focusing on sensory symptoms [18, 31], we further extended these analyses with linking this E/I related gene-expression to cortical thickness profiles in
separate sensory subgroups. By integrating these approaches, we can deepen our understanding of the links between aggregated genetic variation in glutamate and GABA pathway signaling sets
and different behavioral autism phenotypes as well as brain structure. Based on previous findings regarding excitatory or inhibitory alterations in autism, we expect to find differential
involvement of the glutamate and GABA genes across the autism phenotypes, reflected in the competitive gene-set analysis. This may be further confirmed with the exploratory analyses using
gene-expression in association with structural brain differences in both the autistic versus NTC as well as in the sensory symptom subgroups. METHODS AND MATERIALS PARTICIPANTS We included
participants from the Longitudinal European Autism Project (LEAP), part of the AIMS-2-TRIALS clinical research programme (https://www.aims-2-trials.eu/) [38,39,40]. Our sample consisted of
638 participants (_n_ = 359 autistic and _n_ = 279 neurotypical controls) for whom structural MRI data was available that passed quality control [19]. Phenotypic, genetic and brain imaging
data were collected at six study centers across Europe: Institute of Psychiatry, Psychology and Neuroscience, King’s College London (IoPPN/KCL, UK), Autism Research Centre, University of
Cambridge (UCAM, UK), University Medical Centre Utrecht (UMCU, Netherlands), Radboud University Nijmegen Medical Centre (RUNMC, Netherlands), Central Institute of Mental Health (CIMH,
Germany), and the University Campus Bio-Medico (UCBM) in Rome, Italy. Inclusion criteria for the autism group were an existing diagnosis of autism and an age-range between 6 and 30 years.
Symptoms were additionally assessed using the Autism Diagnostic Observation Schedule Second Edition (ADOS-2; [41]) and the Autism Diagnostic Interview-Revised (ADI-R; [42]). For the NTC,
exclusion criterion comprised of parent- or self-report of any psychiatric disorder. Individuals who had a normative T-score of 70 or higher on the Social Responsiveness Scale Second Edition
(SRS-2) were excluded. Some individuals in the autism and NTC groups had intellectual disability (ID) (autism = 53, NTC = 25), defined as an IQ score between 40 and 74. Ethical approval was
obtained through ethics committees at each study site. All participants or legal guardian (where applicable) provided written informed consent. For further details of the recruitment of
participants in this study see [19, 38, 39]. PHENOTYPIC MEASURES The phenotypic measures used were part of a larger test battery (see [39]). Here we included three questionnaires focusing on
the core autism symptoms; the Social Responsiveness Scale-Revised (SRS-2) [43], the Repetitive Behavior Scale-Revised (RBS-R) [44], and the Short Sensory Profile (SSP) [45]. For these
questionnaires, we used self- or parent-report ratings, depending on age and diagnostic group. We additionally made use of sensory symptom subgroups used in this sample previously, created
based on the SSP scores and factor mixture modeling [19, 31]. GENOTYPING Genotyping was performed at the Centre National de Recherche en Génomique Humaine (CNRGH) using the Infinium
OmniExpress-24v1 BeadChip Illumina. Sample quality controls such as sex check (based on the X chromosome homozygosity rate or the median of the Log R ratio of the X and Y chromosomes),
Mendelian errors (transmission errors within full trios) and Identity By State were performed using PLINK 1.90. Imputation of 17 million SNPs was performed using the 700k genotyped SNPs on
the Michigan Imputation Server [46]. The HRC r1.1 2016 reference panel for a European population was used, as the majority of individuals in the LEAP cohort were from European ancestry. Only
autosomes were imputed. Linkage disequilibrium-based SNP pruning was done for SNPs with a MAF > 1% and SNPs with an R2 < 0.1 in windows of 500 kb were selected. This resulted in 546
participants with genotypic data (_n_ = 304 autistic, _n_ = 242 NTC). Selection of the glutamate (_n_ = 72 genes) and GABA (_n_ = 124 genes) gene-sets was based on Ingenuity Pathway Analysis
software (http://www.ingenuity.com), a frequently updated database for genetic pathway analysis. Supplemental tables S1 and S2 show an overview of the included genes. In case of any
significant associations between the aggregated genetic variation in the gene-sets and the phenotypes of interest, we explored smaller gene-sets containing genes encoding glutamate/GABA
receptors and transporters specifically because of their more direct role in neurotransmitter signaling [47]. Those we refer to as glu-RT (_n_ = 32) and GABA-RT (_n_ = 26) and are defined in
the supplemental Tables S1 and S2. NEUROIMAGING DATA Structural brain images were acquired on 3 T MRI scanners at all sites, with T1-weighted MPRAGE sequence (TR = 2300 ms, TE = 2.93 ms, T1
= 900 ms, voxels size = 1.1 × 1.1 × 1.2 mm, flip angle = 9°, matrix size = 256 × 256, FOV = 270 mm, 176 slices). For a summary of scanner details and acquisition parameters at each site,
see Table S3 in the supplemental information. The processing of all neuroimaging data was conducted at one site for all available data. For each image a model of the cortical surface was
computed using FreeSurfer v6.0 (https://surfer.nmr.mgh.harvard.edu/), using a fully automated and validated procedure [48,49,50,51]. Subsequently, each reconstructed surface went through
strict quality assessments, described in detail in [19]. This quality assessment included visual inspection of reconstruction errors by independent raters, manual editing where needed, and
examination of the Euler number of each FreeSurfer surface reconstruction resulting in the conclusion that there were no differences in the complexity of the reconstructed cortical surface
between participant groups. This resulted in parcellated regional CT measures for all the 638 participants included in our study, with 34 regions in each hemisphere using the
Desikan-Killiany atlas [52]. A more detailed description of the processing of the cortical thickness data can be found in a previous publication (see [19]). GENE-EXPRESSION DATA
Gene-expression data were acquired from post-mortem human brains from the Allen Human Brain Atlas (AHBA) [53], using data from six donors (aged 24–57 years, one female) of the left
hemisphere only. These whole-brain gene-expression data are open source and can be downloaded from the Allen Institute for Brain Science; http://www.brain-map.org. For more details on how
these data were obtained, see [53]. Using previously described procedures [23, 24, 54], these gene-expression data were mapped onto the 34 cortical regions defined by FreeSurfer’s
Desikan-Killiany Atlas [52]. These gene-expression profiles were then used in the two-step procedure described by [55] to select the most consistent profiles for inclusion in our analyses.
First, the correlations of gene-expressions to the median expression values across donors were calculated, and the genes showing consistent correlation profiles were selected
(donor-to-median correlation rho >0.446). Secondly, we used data from the BrainSpan Atlas, where gene-expression data in a wide age-range of donors are available (www.brainspan.org).
Donors were selected within the age range of our LEAP dataset (6–30 years), which gave us 9 donors (male/female = 5/4). We calculated correlations to the median expression values in the 11
cortical regions in the AHBA-to-FreeSurfer data that were also included in the BrainSpan Atlas, using methods as described by [24]. We then selected genes that correlated between the
profiles of the two atlases higher than r = 0.52 (one-sided _test p_ < 0.05), which resulted in 2293 genes available in total. The overlap with the gene-sets left 29 genes in our
glutamate pathway gene-set, and 42 genes in our GABA pathway gene-set. The median expression profiles across regions for these genes constitute the interregional gene-expression profiles
used in our analyses. ANALYSES All analyses included the linear effects of age, sex, IQ and site as covariates. All tests were corrected using the false discovery rate (FDR; _q_ < 0.05
was considered significant) unless otherwise described. GENE-SET ANALYSIS To investigate associations between aggregated genetic variation within the glutamate and GABA gene-sets and the
autism phenotypes of interest (SRS-2 total score, RBS-R total score, SSP total score, and ADOS-2 and ADI-R (the last two for the autism group only)) and cortical thickness, we performed
competitive gene-set analysis using MAGMA (Multi-marker Analysis of GenoMic Annotation) software (version 1.10, [33]). The analysis was performed in two steps. First, gene-based _p_-values
were calculated for each gene (excluding genes located on the X-chromosome, see supplemental tables S1 and S2) on our phenotypes of interest, using a multiple linear principal components
regression using F-tests. Second, we tested the association of the set, aggregating the gene-based _p_-values using competitive analysis. This gene-set analysis is done with an
intercept-only linear regression model for the gene-set, which tests whether the aggregated genetic variation of the genes in a gene-set is more strongly associated with the phenotype of
interest than all other genes in the genome [33]. CORTICAL THICKNESS AND CLINICAL PHENOTYPES To test associations between cortical thickness (CT) and our phenotypes of interest (SRS-2 total
score, RBS-R total score, SSP total score, ADOS-2 and ADI-R), we used linear regression models in the R-software package [56]. This was done in the left hemisphere only, due to the
expression profile analysis being performed only in the left hemisphere. In addition to age, sex, IQ and site we added quadratic age effects and _total_ mean cortical thickness as covariates
as well, as described previously in [19]. EXPRESSION PROFILES To investigate associations between expression profiles of the glutamate and GABA gene-sets and brain structure we used
correlation across interregional profiles of CT with interregional profiles of gene-expression [23, 24, 57]. Profiles of CT were created by subtracting the average CT per region in the NTC
group from the average CT in the autism group, as has been done previously [54]. In order to rule out effects being caused by heterogeneity in the sample, the groups were matched for age,
sex and IQ using _matchit_ [58] in R-software [56] performing nearest neighbor matching, resulting in _n_ = 279 participants in each group. As cortical thickness is strongly associated with
age [59], we additionally decided to do these profile correlation analyses for children, adolescents and adults separately. In order to verify any of these associations, analyses were
replicated using structural imaging data of CT from the multi-site open-source ABIDE database [60]. We included participants in the same age range as our own sample (6–30 years), which
resulted in data from 874 participants matched for age, sex and IQ (_n_ = 437 in both groups). Details on these analyses and results can be found in the supplemental information and Fig. S1.
Building upon previous results from our group [19, 31] and to parse some of the autism heterogeneity, we additionally performed these gene-expression analyses with interregional CT profiles
in separate sensory subgroups (low, _n_ = 375; moderate, _n_ = 37; severe, _n_ = 37). These subgroups were defined previously [31]. The interregional expression profiles of the genes in our
glutamate and GABA pathway gene-sets were then correlated with the CT-difference interregional profiles (autism minus NTC across different age-groups and CT-average interregional profiles
in sensory subgroups), which provided a distribution of correlation coefficients per gene-set. The distributions of correlation coefficients between the gene-expression and CT-difference
interregional profiles were then tested for significance using a resampling approach of 10,000 random samples, as described in [23, 24]. In this approach, a random set of genes of the same
size as the set being tested was selected (from the 2293 available) 10,000 times with the average correlation each time being used to create a null distribution. A two-tailed significance
test was used to test the gene-set of interest against the null distribution. RESULTS DEMOGRAPHICS Demographic and clinical characteristics are shown in Table 1. No differences were found
between the autism and NTC groups in age. The autism group had a higher female-to-male ratio compared to NTC and NTC had a higher IQ than the autism group. As expected, the autism group had
significantly higher scores on the SRS-2 and RBS-R and scored lower in the SSP (where lower scores indicate higher sensory sensitivity). Information on medication use can be found in Table
S4 in the supplemental information. GENE-SET ANALYSIS Aggregated genetic variation within the glutamate gene-set (_n_ = 72 genes) was associated with autism symptoms as defined by
significant associations with all the ADI-R and ADOS-2 subscales (all _q_ < 0.05, see Table 2). Repeating these analyses in the smaller glu-RT gene-set did not give the same significant
results. No associations were found for any of the questionnaire scores. Genetic variation within the GABA gene-set (_n_ = 124 genes) was nominally significantly associated with sensory
processing (SSP total scores; _q_ = 0.07) after FDR correction. Repeating these analyses in the smaller, more specific GABA-RT set gave a similar result (_q_ = 0.06), see Table 2. To
investigate these trend associations further, we performed similar post-hoc association analyses with all the SSP subscales. None of these were significantly associated with the genetic
variation within the GABA gene-set (all _q_-values > 0.05). For more details see Table S5 in the supplemental information. Repeating the gene-set analyses with the questionnaires in the
autism group separately did not result in any significant associations. We additionally investigated gene-set association with CT in the FreeSurfer cortical regions in the left hemisphere.
There were some nominally significant (uncorrected _p_-values < 0.05) associations, although none survived FDR-correction. The details of these results can be seen in supplemental Tables
S6 and S7. CORTICAL THICKNESS AND PHENOTYPES Our group previously showed vertex-wise group differences in cortical thickness between autism and NTC in the current sample [19]. Here we did
not repeat these analyses but instead focused on the continuous measures of autism symptoms using the ADI-R and ADOS-2 in the autism group and the SRS-2, RBS-R and SSP questionnaires in the
entire sample. Cortical thickness in the frontal pole was positively associated with restricted and repetitive behaviors as reflected by the RBS-R total score (_b_ = 0.05, _t_ = 3.33, _q_ =
0.03). No other results survived multiple comparisons corrections, although nominally significant negative associations were found between all ADI-R subscales and precuneus CT (communication
_q_ = 0.26, social _q_ = 0.19, restricted and repetitive behaviors _q_ = 0.05) as well as a nominally significant positive relation between the ADOS-2 total score and the social affect
subscale and CT in the insula (_q_ = 0.16, _q_ = 0.13, respectively). GENE-EXPRESSION PROFILES While the interregional profiles of group differences in cortical thickness (autism minus NTC)
were not significantly associated with gene-expression profiles across our glutamate and GABA gene-sets in the full sample (all _q_-values > 0.05), splitting into groups of children,
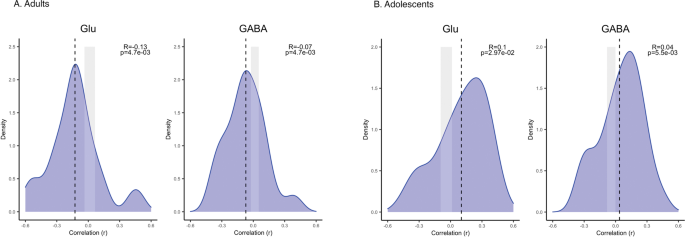
adolescents and adults gave some opposing results. In adolescents (_n_ = 101 autism, _n_ = 100 NTC), the interregional profile of group differences in cortical thickness was positively
associated with interregional variation in expression of both glutamate (t = 2.25, _q_ = 0.030, Cohen’s _d_ = 0.70) and GABA genes (_t_ = 3.28, _q_ = 0.005, Cohen’s _d_ = 1.24). In adults,
on the other hand (_n_ = 124 autism, _n_ = 115 NTC), the group difference profile was negatively associated with expression, again for both gene-sets (glutamate: t = −2.99, _q_ = 0.005, _d_
= −0.93; GABA: _t_ = −3.17, _q_ = 0.005, _d_ = −0.93), reflecting differences in CT between autistic participants and NTC changing with age. In children, no such associations were found. See
Fig. 1 for the distributions of the correlation coefficients and Fig. 2 for CT differences and example genes for each gene set. Interestingly, these results were replicated in the
independent ABIDE sample for adolescents. In adults, however, positive associations between the expression profiles and CT differences were found, as opposed to the negative associations
found in our LEAP sample (see supplemental information and Fig. S1). Investigating the interregional profiles of CT in sensory subgroups separately gave positive associations with the
interregional profiles of gene-expression in all groups (LOW: glutamate: _t_ = 3.02, _q_ = 0.004, Cohen’s _d_ = 0.94; GABA: _t_ = 3.19, _q_ = 0.004, Cohen’s _d_ = 1.21; MODERATE: glutamate:
_t_ = 3.18, _q_ = 0.003, Cohen’s _d_ = 0.99; GABA: _t_ = 3.30, _q_ = 0.003, Cohen’s _d_ = 1.25; SEVERE: glutamate: _t_ = 3.03, _q_ = 0.004, Cohen’s _d_ = 0.95; GABA: _t_ = 3.18, _q_ = 0.004,
Cohen’s _d_ = 1.20), see also Fig. S2. To investigate possible differences _between_ the sensory subgroups we calculated interregional CT-difference profiles between groups as well, however
this gave no significant associations. These results could not be replicated in ABIDE due to unavailability of the SSP questionnaire in that sample. DISCUSSION In the current study, we took
a multimodal approach to investigate the role of E/I imbalance associated gene-sets in relation to behavioral phenotypes and brain structure in autism. The most important takeaway of our
results is that the glutamate and GABA gene-sets were differently associated with autism symptoms, and that the expression profiles of these genes throughout the cortex were associated with
differences in cortical thickness between autistic and NTC participants, depending on age. Aggregated genetic variation in the glutamate gene-set was associated with autism symptom severity
on all core symptom subscales of the ADI-R and ADOS-2 (in autistic participants), while variation in the GABA gene-set showed association with sensory symptoms in the entire group, although
this did not survive strict multiple comparisons correction. In adolescents and adults, but not in children, regions with greater gene-expression of glutamatergic and GABAergic genes showed
greater differences in CT between autism and controls, but in opposite directions. In adolescents, this association was positive, suggesting overall higher cortical thickness in autism than
in NTC, while in adults this was negative, indicating an overall higher CT in controls as opposed to autistic participants. These results provide a better understanding of the mechanistic
underpinnings of the E/I imbalance hypothesis of autism, by supporting the notion that E/I imbalance varies across behavioral autism characteristics and differences in CT between autism and
NTC groups. The findings of associations between genetic variation in the glutamate gene-set and ADI-R and ADOS-2 subscale scores, and the trend associations of these subscale scores with
cortical thickness in the precuneus and insula, areas known to be involved in somatosensory and visuospatial processing, interoception and self-reflection [61, 62], suggest that glutamate
genes may be linked to broader autism characteristics. Additionally, the trend associations of GABA gene-sets on SSP total score and the association with cortical thickness profiles in the
sensory symptom subgroups suggest a particular role for GABAergic genes in sensory processing, which supports previous findings of links between brain GABA concentrations and sensory
deficits [32, 63, 64]. The lack of significant associations between aggregated variation in the glutamate and GABA gene-sets with repetitive behaviors and social responsiveness (RBS-R and
SRS-2) may be considered surprising as previous studies have found links between glutamate and GABA concentrations in several brain regions and/or metabolite altering drugs with these
behaviors [65,66,67,68,69,70]. However, studies investigating in vivo measures of alterations of glutamate and GABA in autism have had inconsistent results that could be due to several
factors; the heterogeneity of autism, differences in study populations and brain regions investigated, or differences in processing pipelines during analysis. Furthermore, here we focused on
behavioral autism characteristics and genetic information, not in vivo brain concentrations of glutamate and GABA. We did however find links between repetitive behaviors (RBS-R) and CT in
the frontal pole, where increased RBS-R scores were associated with increased CT. Measures from the ADI-R and ADOS-2 diagnostic tools (in the autism group only) were differently associated
with CT in the precuneus and insula, although this was only at trend-level. We did not find direct associations of CT with SSP scores, although previous work on this dataset did find
associations of differences in CT between sensory subgroups in right premotor cortex and supplementary motor areas, regions enriched for genes expressed in excitatory neurons in developing
cortex [19]. In support of this, we found that regions with greater expression of genes from both gene-sets also showed greater CT in all sensory subgroups, although there were no
significant differences between sensory subgroups. This show that there are likely associations of glutamatergic and GABAergic gene-expression to alterations in sensory processing but that
differences may be too subtle between groups to show any differences. These associations, combined with the trend significant associations of aggregated genetic variance of the GABA gene-set
are in line with previous work indicating that alterations of GABA are associated with altered sensory processing in autism [32, 71, 72]. It is also possible that we did not see significant
associations with CT-difference scores between these groups due to lower number of participants on the moderate and severe groups (_n_ = 37, _n_ = 18 respectively). Interregional variation
in expression of glutamatergic and GABAergic genes was associated with the group differences in CT in adolescents and adults, but not in children, nor in the overall sample. Regions with
greater expression of both glutamate and GABA genes showed greater differences in CT between autistic and NTC participants. These results taken together suggests possible genetic
underpinnings of excitation/inhibition imbalance affecting autism symptoms. Furthermore, it suggests that there may be important differences in trajectories across development which may be
mediated through altered cortical thickness. This is in line with previous work on this cohort finding differences in CT in regions enriched for genes involved in autism, where degree of
deviance in CT from the NTC range correlated with increased polygenic scores for autism and symptom severity [19]. This needs to be investigated further, and future studies should preferably
include measures of metabolite concentrations to draw further conclusions about these relationships. Our results need to be interpreted with caution, as the presence of glutamate and GABA
protein encoding genes does not directly translate to metabolite concentrations, and genetic alterations might not translate to a common phenotype across individuals [12]. Additionally,
genes differ in coding for loss- or gain-of function, leading to reduced or increased protein function, further complicating any interpretation of _direction_ of glutamate and GABA
involvement in autism symptoms. However, our results strongly indicate critical roles of glutamate and GABA genes in these specific phenotypes and that the link between these measures needs
to be investigated in more detail to increase our understanding of the mechanisms connecting genetics, glutamate and GABA neurotransmitters and autism symptomatology. More direct
investigations of the E/I imbalance hypothesis are needed in order to investigate excitation and inhibition in vivo in relation to brain functioning. Promising new techniques combining
different imaging methods, causal discovery analysis, and pharmacological interventions and longitudinal studies, will allow us to do this in the future through which we hope to further
increase our understanding of how chemical imbalance in the brain is associated with functioning. Ultimately, E/I balance may be manipulated using glutamate- and/or GABA- influencing
pharmacological treatments. One study already showed decreased glutamate and GABA concentrations after bumetanide treatment to be positively associated with autism symptom improvement [71].
Strengths of this study were the combination of genetic, structural and phenotypic data from the same cohort, which gave us the opportunity to for the first time analyze these data together.
Another strength was the relatively large number of participants available giving us more confidence in our results. There were also some limitations. Firstly, there were fewer females than
males included in this study, a common problem in autism research. Furthermore, the gene-expression data were only used in the left hemisphere. However, the gene-expression data used in the
expression profile analyses was robust and only included if the interregional profiles were similar across another dataset (BrainSpan), increasing the confidence in the robustness of these
profiles. Another limitation is that the AHBA donors were all neurotypical controls, and we do not know whether genes are expressed differently in autism. Additionally, there were
differences in ages of participants recruited at different sites, which has been investigated in an initial analysis of the LEAP cohort [38]. We also did not fully replicate our
gene-expression profile results in the independent ABIDE data set (see the supplemental information and Fig. S1). However, the results were largely overlapping showing similar effects,
although in opposite direction in the adult group compared to the adults in our LEAP sample. This shows that heterogeneity of the autism sample and neurotypical controls have a large
influence on the results. In conclusion, we found that glutamate genes are associated with core behavioral autism characteristics and GABA genes may be associated with sensory processing,
and that increased expression of glutamate and GABA genes are associated with larger differences in CT between autistics and NTC in adolescents and adults but not in children. This support
the hypothesis that the influence of E/I imbalance varies across autism phenotypes and brain regions, suggesting that glutamate and GABA genes play different roles underlying different
autism phenotypes and that this may change during development. We also showed the importance of linking structural brain measures, genetic and behavioral phenotype data together to gain a
deeper understanding of possible E/I imbalance mechanisms in autism. REFERENCES * American Psychiatric Association. Diagnostic and statistical manual of mental disorders (5th ed).
Washington, DC:CBS;2013. * Rubenstein JLR, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–67. Article CAS
Google Scholar * Foss-Feig JH, Adkinson BD, Ji JL, Yang G, Srihari VH, McPartland JC, et al. Searching for cross-diagnostic convergence: neural mechanisms governing excitation and
inhibition balance in schizophrenia and autism spectrum disorders. Biol Psychiatry. 2017;81:848–61. Article Google Scholar * Horder J, Petrinovic MM, Mendez MA, Bruns A, Takumi T, Spooren
W, et al. Glutamate and GABA in autism spectrum disorder—a translational magnetic resonance spectroscopy study in man and rodent models. Transl Psychiatry. 2018;8:106. Article Google
Scholar * Nelson SB, Valakh V. Excitatory/Inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 2015;87:684–98. Article CAS Google Scholar * Rosenberg A,
Patterson JS, Angelaki DE. A computational perspective on autism. Proc Natl Acad Sci USA. 2015;112:9158–65. Article CAS Google Scholar * Cross-Disorder Group of the Psychiatric Genomics
Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–9. Article Google Scholar * Grove J, Ripke S,
Als TD, Mattheisen M, Walters RK, Won H, et al. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet. 2019;51:431–44. Article CAS Google Scholar *
Havdahl A, Niarchou M, Starnawska A, Uddin M, van der Merwe C, Warrier V. Genetic contributions to autism spectrum disorder. Psychol Med. 2021;51:2260–73. * Nisar S, Hashem S, Bhat AA, Syed
N, Yadav S, Azeem MW, et al. Association of genes with phenotype in autism spectrum disorder. Aging 2019;11:10742–70. Article CAS Google Scholar * Satterstrom FK, Kosmicki JA, Wang J,
Breen MS, De Rubeis S, An JY, et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 2020;180:568–584.e23. Article
CAS Google Scholar * Heise C, Preuss JM, Schroeder JC, Battaglia CR, Kolibius J, Schmid R, et al. Heterogeneity of cell surface glutamate and GABA receptor expression in shank and CNTN4
autism mouse models. Front Mol Neurosci. 2018;11:212. Article Google Scholar * Lenart J, Augustyniak J, Lazarewicz JW, Zieminska E. Altered expression of glutamatergic and GABAergic genes
in the valproic acid-induced rat model of autism: a screening test. Toxicology 2020;440:152500. Article CAS Google Scholar * Yang S. GABAA receptor subunit gene polymorphisms predict
symptom-based and developmental deficits in Chinese Han children and adolescents with autistic spectrum disorders. Sci Rep. 2017;2017:9. Google Scholar * Ajram LA. Shifting brain inhibitory
balance and connectivity of the prefrontal cortex of adults with autism spectrum disorder. Transl Psychiatry. 2017;7:e1137. Article CAS Google Scholar * Ecker C. The neuroanatomy of
autism spectrum disorder: an overview of structural neuroimaging findings and their translatability to the clinical setting. Autism 2017;21:18–28. Article Google Scholar * Mensen VT,
Wierenga LM, van Dijk S, Rijks Y, Oranje B, Mandl RCW, et al. Development of cortical thickness and surface area in autism spectrum disorder. NeuroImage Clin. 2017;13:215–22. Article Google
Scholar * Ecker C, Ginestet C, Feng Y, Johnston P, Lombardo MV, Lai MC, et al. Brain surface anatomy in adults with autism: The relationship between surface area, cortical thickness, and
autistic symptoms. Arch Gen Psychiatry. 2013;70:59–70. Google Scholar * Ecker C, Pretzsch CM, Jones EJH, Leap Team T, Murphy D. Inter-individual differences in cortical thickness and their
genomic underpinnings in autism spectrum disorder. Am J Psychiatry. 2021;179:242–54. * Ohta H, Nordahl CW, Iosif AM, Lee A, Rogers S, Amaral DG. Increased surface area, but not cortical
thickness, in a subset of young boys wth autism spectrum disorder. Autism Res. 2016;9:232–48. Article Google Scholar * Romero-Garcia R, Warrier V, Bullmore ET, Baron-Cohen S, Bethlehem
RAI. Synaptic and transcriptionally downregulated genes are associated with cortical thickness differences in autism. Mol Psychiatry. 2019;24:1053–64. Article CAS Google Scholar * van
Rooij D, Anagnostou E, Arango C, Auzias G, Behrmann M, Busatto GF, et al. Cortical and subcortical brain morphometry differences between patients with autism spectrum disorder and healthy
individuals across the lifespan: tesults from the ENIGMA ASD working group. Am J Psychiatry. 2018;175:359–69. Article Google Scholar * Patel Y, Shin J, Drakesmith M, Evans J, Pausova Z,
Paus T. Virtual histology of multi-modal magnetic resonance imaging of cerebral cortex in young men. NeuroImage 2020;218:116968. Article CAS Google Scholar * Shin J, French L, Xu T,
Leonard G, Perron M, Pike GB, et al. Cell-specific gene-expression profiles and cortical thickness in the human brain. Cereb Cortex. 2018;28:3267–77. Article Google Scholar * Poelmans G,
Franke B, Pauls DL, Glennon JC, Buitelaar JK. AKAPs integrate genetic findings for autism spectrum disorders. Transl Psychiatry. 2013;3:e270. Article CAS Google Scholar * Previtera ML,
Langhammer CG, Langrana NA, Firestein BL. Regulation of dendrite arborization by substrate stiffness is mediated by glutamate receptors. Ann Biomed Eng. 2010;38:3733–43. * Tyzio R, Represa
A, Jorquera I, Ben-Ari Y, Gozlan H, Aniksztejn L. The establishment of GABAergic and glutamatergic synapses on CA1 pyramidal neurons is sequential and correlates with the development of the
apical dendrite. J Neurosci. 1999;19:10372–82. * Vidal-Pineiro D, Parker N, Shin J, French L, Grydeland H, Jackowski AP, et al. Cellular correlates of cortical thinning throughout the
lifespan [Internet]. Sci Rep. 2020;10:21803. * Bralten J, Van Hulzen KJ, Martens MB, Galesloot TE, Arias Vasquez A, Kiemeney LA, et al. Autism spectrum disorders and autistic traits share
genetics and biology. Mol Psychiatry. 2018;23:1205–12. Article CAS Google Scholar * Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in
neuropsychiatric disorders. Nat Neurosci. 2011;14:285–93. Article CAS Google Scholar * Tillmann J, Uljarevic M, Crawley D, Dumas G, Loth E, Murphy D, et al. Dissecting the phenotypic
heterogeneity in sensory features in autism spectrum disorder: a factor mixture modelling approach. Mol Autism. 2020;11:67. Article CAS Google Scholar * Puts NAJ, Wodka EL, Harris AD,
Crocetti D, Tommerdahl M, Mostofsky SH, et al. Reduced GABA and altered somatosensory function in children with autism spectrum disorder: Abnormal GABA and touch in ASD. Autism Res.
2017;10:608–19. Article Google Scholar * de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11:1–19. Article Google
Scholar * de Leeuw CA, Neale BM, Heskes T, Posthuma D. The statistical properties of gene-set analysis. Nat Rev Genet. 2016;17:353–64. Article Google Scholar * de Leeuw CA, Stringer S,
Dekkers IA, Heskes T, Posthuma D. Conditional and interaction gene-set analysis reveals novel functional pathways for blood pressure. Nat Commun. 2018;9:3768. Article Google Scholar * Mota
NR, Poelmans G, Klein M, Torrico B, Fernàndez-Castillo N, Cormand B, et al. Cross-disorder genetic analyses implicate dopaminergic signaling as a biological link between
attention-deficit/hyperactivity disorder and obesity measures. Neuropsychopharmacology 2020;45:1188–95. Article CAS Google Scholar * Naaijen J, Bralten J, Poelmans G, Glennon JC, Franke
B, Buitelaar JK, et al. Glutamatergic and GABAergic gene sets in attention-deficit/hyperactivity disorder: association to overlapping traits in ADHD and autism. Transl Psychiatry.
2017;7:e999–7. Article CAS Google Scholar * Charman T, Loth E, Tillmann J, Crawley D, Wooldridge C, Goyard D, et al. The EU-AIMS Longitudinal European Autism Project (LEAP): clinical
characterisation. Mol Autism. 2017;8:1–21. Article Google Scholar * Loth E, Charman T, Mason L, Tillmann J, Jones EJH, Wooldridge C, et al. The EU-AIMS Longitudinal European Autism Project
(LEAP): design and methodologies to identify and validate stratification biomarkers for autism spectrum disorders. Mol Autism. 2017;8:24. Article Google Scholar * Murphy D, Spooren W.
EU-AIMS: A boost to autism research. Nat Rev Drug Disco. 2012;11:815–6. Article CAS Google Scholar * Lord C, Risi S, Lambrecht L, Cook EH, Leventhal BL, DiLavore PC, et al. The Autism
diagnostic observation schedule–generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord. 2000;19:205–23. * Rutter M, Le
Couteur A, Lord C. Autism diagnostic interview-revised. West Psychol Serv. 2003. https://doi.org/10.1007/978-1-4419-1698-3_894 * Constantino J, Gruber C. Social responsiveness scale 2nd ed.
Los Angeles: Western Psychilogical Services; 2002. * Bodfish JW, Symons FJ, Parker DE, Lewis MH. Varieties of repetitive behavior in autism: comparisons to mental retardation. J Autism Dev
Disord. 2000. https://doi.org/10.1007/978-1-4419-1698-3_894 * Tomchek SD, Dunn W. Sensory processing in children with and without Autism: a comparative study using the short sensory profile.
Am J Occup Ther. 2007;61:190–200. * Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48:1284–7.
Article CAS Google Scholar * Galvan A, Kuwajima M, Smith Y. Glutamate and GABA receptors and transporters in the basal ganglia: What does their subsynaptic localization reveal about their
function? Neuroscience 2006;143:351–75. Article CAS Google Scholar * Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis. NeuroImage. 1999;9:179–94. * Fischl B. FreeSurfer.
NeuroImage 2012;62:774–81. Article Google Scholar * Fischl B, Sereno MI, Tootell RBH, Dale AM. High-resolution intersubject averaging and a coordinate system for the cortical surface.
1999;13:272–84. * Ségonne F, Dale AM, Busa E, Glessner M, Salat D, Hahn HK, et al. A hybrid approach to the skull stripping problem in MRI. NeuroImage 2004;22:1060–75. Article Google
Scholar * Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based
regions of interest. NeuroImage 2006;31:968–80. Article Google Scholar * Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, et al. An anatomically comprehensive atlas
of the adult human brain transcriptome. Nature 2012;489:391–9. Article CAS Google Scholar * Writing Committee for the Attention-Deficit/Hyperactivity Disorder, Autism Spectrum Disorder,
Bipolar Disorder, Major Depressive Disorder, Obsessive-Compulsive Disorder, and Schizophrenia ENIGMA Working Groups, et al. Virtual histology of cortical thickness and shared neurobiology in
6 psychiatric disorders. JAMA Psychiatry 2021;78:47–63. * French L, Paus T. A FreeSurfer view of the cortical transcriptome generated from the Allen human brain Atlas. Front Neurosci.
2015;9:1–5. Article Google Scholar * Booth DS, Szmidt-Middleton H, King N, Westbrook MJ, Young SL, Kuo A, et al. RStudio: integrated development for R. Nature. 2018;29:3026–38. * Patel Y,
Shin J, Gowland PA, Pausova Z, Paus T. Maturation of the human cerebral cortex during adolescence: myelin or dendritic arbor? Cereb Cortex. 2019;29:3351–62. Article CAS Google Scholar *
Ho DE, Imai K, King G, Stuart EA. MatchIt: Nonparametric preprocessing for parametric causal inference. J Stat Softw. 2011. https://doi.org/10.18637/jss.v042.i08 * Tamnes CK, Østby Y, Fjell
AM, Westlye LT, Due-Tønnessen P, Walhovd KB. Brain maturation in adolescence and young adulthood: regional age-related changes in cortical thickness and white matter volume and
microstructure. Cereb Cortex. 2010;20:534–48. Article Google Scholar * Martino AD, Castellanos FX, Assaf M, Deen B. The autism brain imaging data exchange: towards large-scale evaluation
of the intrinsic brain architecture in autism. Am J Occup Ther. 2014;19:659–67. * Cavanna AE, Trimble MR. The precuneus: a review of its functional anatomy and behavioural correlates. Brain
2006;129:564–83. Article Google Scholar * Uddin LQ, Nomi JS, Hébert-Seropian B, Ghaziri J, Boucher O. Structure and function of the human insula. J Clin Neurophysiol. 2017;34:300–6.
Article Google Scholar * Dumas G, Lefebvre A, Cliquiet F, Amsellem F, Bourgeron T, Delorme R. Clinical & genetic subtypes of sensory processing sensitivities in autism. Montreal:
INSAR; 2019. * Robertson CE, Baron-Cohen S. Sensory perception in autism. Nat Rev Neurosci. 2017;18:671–84. Article CAS Google Scholar * Chadman KK. Fluoxetine but not risperidone
increases sociability in the BTBR mouse model of autism. Pharm Biochem Behav. 2011;97:586–94. Article CAS Google Scholar * Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, et al.
Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 2010;468:263–9. Article CAS Google Scholar * Di J, Li J, O’Hara B, Alberts I, Xiong
L, Li J, et al. The role of GABAergic neural circuits in the pathogenesis of autism spectrum disorder. Int J Dev Neurosci. 2020;80:73–85. Article CAS Google Scholar * Marotta R, Risoleo
MC, Messina G, Parisi L, Carotenuto M, Vetri L, et al. The neurochemistry of autism. Brain Sci. 2020;10:163. Article CAS Google Scholar * Posey DJ, Kem DL, Swiezy NB, Sweeten TL, Wiegand
RE, McDougle CJ. A pilot study of D-cycloserine in subjects with autistic disorder. Am J Psychiatry. 2004;161:2115–7. Article Google Scholar * Urbano M, Okwara L, Manser P, Hartmann K,
Herndon A, Deutsch SI. A Trial of D-cycloserine to treat stereotypies in older adolescents and young adults with autism spectrum disorder. Clin Neuropharmacol. 2014;37:69–72. Article CAS
Google Scholar * Huang Q, Pereira AC, Velthuis H, Wong NML, Ellis CL, Ponteduro FM, et al. GABAB receptor modulation of visual sensory processing in adults with and without autism spectrum
disorder. Sci Transl Med. 2022;14:eabg7859. Article CAS Google Scholar * Mentch J, Spiegel A, Ricciardi C, Robertson CE. GABAergic inhibition gates perceptual awareness during binocular
rivalry. J Neurosci. 2019;39:8398–407. Article CAS Google Scholar Download references FUNDING This work has been supported by the EU-AIMS (European Autism Interventions) and AIMS-2-TRIALS
programmes which receive support from Innovative Medicines Initiative Joint Undertaking Grant No. 115300 and 777394, the resources of which are composed of financial contributions from the
European Union’s FP7 and Horizon2020 Programmes, and from the European Federation of Pharmaceutical Industries and Associations (EFPIA) companies’ in-kind contributions, and AUTISM SPEAKS,
Autistica and SFARI; by the Horizon2020 supported programme CANDY Grant No. 847818 and a Veni grant from the Netherlands organization for scientific research (NWO) under grant number
VI.Veni.194.032 awarded to JN. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the
decision to publish the results. Any views expressed are those of the authors and not necessarily those of the funders. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Cognitive
Neuroscience, Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Center, Nijmegen, the Netherlands Viola Hollestein, Natalie J. Forde, Christian F. Beckmann,
Jan K. Buitelaar & Jilly Naaijen * Department of Human Genetics, Radboud University Medical Center, Nijmegen, The Netherlands Geert Poelmans * Department of Psychology, Institute of
Psychiatry, Psychology & Neuroscience, King’s College London, London, UK Christine Ecker * Department of Child and Adolescent Psychiatry, Psychosomatics and Psychotherapy, University
Hospital Frankfurt am Main, Goethe University, Frankfurt, Germany Christine Ecker, Caroline Mann & Tim Schäfer * Department of Psychiatry and Psychotherapy, Central Institute of Mental
Health, University of Heidelberg, Mannheim, Germany Carolin Moessnang * Child and Adolescent Psychiatry, Central Institute of Mental Health, Medical Faculty Mannheim, University of
Heidelberg, Mannheim, Germany Sarah Baumeister & Tobias Banaschewski * Institut Pasteur, Human Genetics and Cognitive Functions Unit, Paris, France Thomas Bourgeron * Sackler Institute
for Translational Neurodevelopment, Institute of Psychiatry, Psychology & Neuroscience, King’s College London, London, UK Eva Loth, Flavio Dell’Acqua, Declan G. M. Murphy & Nicolaas
A. Puts * Department of Forensic and Neurodevelopmental Sciences, Institute of Psychiatry, Psychology & Neuroscience, King’s College London, London, UK Eva Loth, Flavio Dell’Acqua,
Declan G. M. Murphy & Nicolaas A. Puts * Medical Research Council (MRC) Centre for Neurodevelopmental Disorders, King’s College London, London, UK Declan G. M. Murphy & Nicolaas A.
Puts * Roche Pharma Research and Early Development, Neuroscience and Rare Diseases, Roche Innovation Center Basel, F. Hoffmann–La Roche Ltd., Basel, Switzerland Julian Tillmann * Department
of Psychology, Institute of Psychiatry, Psychology, & Neuroscience, King’s College London, London, UK Tony Charman * Centre for Brain and Cognitive Development, Birkbeck, University of
London, Henry Wellcome Building, London, UK Emily J. H. Jones & Luke Mason * Department of Psychiatry, University Medical Center Utrecht Brain Center, Utrecht University, Utrecht, the
Netherlands Sara Ambrosino * Autism Research Centre, Department of Psychiatry, University of Cambridge, Cambridge, UK Rosemary Holt * Center of Neurodevelopmental Disorders (KIND), Centre
for Psychiatry Research; Department of Women’s and Children’s Health, Karolinska Institutet & Stockholm Health Care Services, Region Stockholm, Stockholm, Sweden Sven Bölte * Child and
Adolescent Psychiatry, Stockholm Health Care Services, Region Stockholm, Stockholm, Sweden Sven Bölte * Curtin Autism Research Group, Curtin School of Allied Health, Curtin University,
Perth, Western Australia Sven Bölte * Karakter Child and Adolescent Psychiatry University Center, Nijmegen, the Netherlands Jan K. Buitelaar Authors * Viola Hollestein View author
publications You can also search for this author inPubMed Google Scholar * Geert Poelmans View author publications You can also search for this author inPubMed Google Scholar * Natalie J.
Forde View author publications You can also search for this author inPubMed Google Scholar * Christian F. Beckmann View author publications You can also search for this author inPubMed
Google Scholar * Christine Ecker View author publications You can also search for this author inPubMed Google Scholar * Caroline Mann View author publications You can also search for this
author inPubMed Google Scholar * Tim Schäfer View author publications You can also search for this author inPubMed Google Scholar * Carolin Moessnang View author publications You can also
search for this author inPubMed Google Scholar * Sarah Baumeister View author publications You can also search for this author inPubMed Google Scholar * Tobias Banaschewski View author
publications You can also search for this author inPubMed Google Scholar * Thomas Bourgeron View author publications You can also search for this author inPubMed Google Scholar * Eva Loth
View author publications You can also search for this author inPubMed Google Scholar * Flavio Dell’Acqua View author publications You can also search for this author inPubMed Google Scholar
* Declan G. M. Murphy View author publications You can also search for this author inPubMed Google Scholar * Nicolaas A. Puts View author publications You can also search for this author
inPubMed Google Scholar * Julian Tillmann View author publications You can also search for this author inPubMed Google Scholar * Tony Charman View author publications You can also search for
this author inPubMed Google Scholar * Emily J. H. Jones View author publications You can also search for this author inPubMed Google Scholar * Luke Mason View author publications You can
also search for this author inPubMed Google Scholar * Sara Ambrosino View author publications You can also search for this author inPubMed Google Scholar * Rosemary Holt View author
publications You can also search for this author inPubMed Google Scholar * Sven Bölte View author publications You can also search for this author inPubMed Google Scholar * Jan K. Buitelaar
View author publications You can also search for this author inPubMed Google Scholar * Jilly Naaijen View author publications You can also search for this author inPubMed Google Scholar
CONTRIBUTIONS VH: formal analysis, software, data curation, writing original draft, writing review and editing, visualization. JKB, CFB, DGMM, EL, CE, TB, TB, TC, EJHJ, SB:
conceptualization, validation, project administration, funding acquisition. GP, NJF, CM, TS, CM, SB, FD, JT, LM, SA, RH: writing review and editing. NAP: Conceptualization, writing review
and editing, project administration. JN: conceptualization, methodology, software, investigation, data curation, writing review and editing, project administration. CORRESPONDING AUTHOR
Correspondence to Viola Hollestein. ETHICS DECLARATIONS COMPETING INTERESTS TB served in an advisory or consultancy role for ADHS digital, Infectopharm, Lundbeck, Medice, Neurim
Pharmaceuticals, Oberberg GmbH, Roche, and Takeda. He received conference support or speaker’s fee by Medice and Takeda. He received royalities from Hogrefe, Kohlhammer, CIP Medien, Oxford
University Press; the present work is unrelated to these relationships. JKB has been in the past 3 years a consultant to/member of advisory board of/and/or speaker for Takeda/Shire, Roche,
Medice, Angelini, Janssen, and Servier. He is not an employee of any of these companies, and not a stock shareholder of any of these companies. He has no other financial or material support,
including expert testimony, patents, royalties. The remaining authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to
jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTAL MATERIAL RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a
Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit
to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are
included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and
your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this
license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Hollestein, V., Poelmans, G., Forde, N.J. _et al._
Excitatory/inhibitory imbalance in autism: the role of glutamate and GABA gene-sets in symptoms and cortical brain structure. _Transl Psychiatry_ 13, 18 (2023).
https://doi.org/10.1038/s41398-023-02317-5 Download citation * Received: 01 June 2022 * Revised: 22 December 2022 * Accepted: 10 January 2023 * Published: 21 January 2023 * DOI:
https://doi.org/10.1038/s41398-023-02317-5 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative