Play all audios:
ABSTRACT Recently, inhibitor of apoptosis proteins (IAPs) and some IAP antagonists were found to regulate autophagy, but the underlying mechanisms remain unclear. WX20120108 is an analogue
of GDC-0152 (a known IAP antagonist) and displays more potent anti-tumor and autophagy-regulating activity in tumor cells, we investigated the regulatory mechanisms underlying
WX20120108-induced autophagy. Using molecular docking and fluorescence polarization anisotropy (FPA) competitive assay, we first demonstrated that WX20120108, acting as an IAP antagonist,
bound to the XIAP-BIR3, XIAP BIR2-BIR3, cIAP1 BIR3, and cIAP2 BIR3 domains with high affinities. In six cancer cell lines, WX20120108 inhibited the cell proliferation with potencies two to
ten-fold higher than that of GDC-0152. In HeLa and MDA-MB-231 cells, WX20120108 induced caspase-dependent apoptosis and activated TNFα-dependent extrinsic apoptosis. On the other hand,
WX20120108 induced autophagy in HeLa and MDA-MB-231 cells in dose- and time-dependent manners. We revealed that WX20120108 selectively activated Foxo3, evidenced by Foxo3 nuclear
translocation in both gene modified cell line and HeLa cells, as well as the upregulated expression of Foxo3-targeted genes (_Bnip3_, _Pik3c3_, _Atg5_, and _Atg4b_), which played a key role
in autophagy initiation. WX20120108-induced autophagy was significantly suppressed when _Foxo3_ gene was silenced. WX20120108 dose-dependently increased the generation of reactive oxygen
species (ROS) in HeLa cells, and WX20120108-induced Foxo3 activation was completely blocked in the presence of catalase, a known ROS scavenger. However, WX20120108-induced ROS generation was
not affected by _cIAP1/2_ or _XIAP_ gene silencing. In conclusion, WX20120108-induced autophagy relies on activating ROS-Foxo3 pathway, which is independent of IAPs. This finding provides a
new insight into the mechanism of IAP antagonist-mediated regulation of autophagy. You have full access to this article via your institution. Download PDF SIMILAR CONTENT BEING VIEWED BY
OTHERS CUR5G, A NOVEL AUTOPHAGY INHIBITOR, EXHIBITS POTENT SYNERGISTIC ANTICANCER EFFECTS WITH CISPLATIN AGAINST NON-SMALL-CELL LUNG CANCER Article Open access 31 October 2022 PHY34 INHIBITS
AUTOPHAGY THROUGH V-ATPASE V0A2 SUBUNIT INHIBITION AND CAS/CSE1L NUCLEAR CARGO TRAFFICKING IN HIGH GRADE SEROUS OVARIAN CANCER Article Open access 10 January 2022 ELAIOPHYLIN TRIGGERS
PARAPTOSIS AND PREFERENTIALLY KILLS OVARIAN CANCER DRUG-RESISTANT CELLS BY INDUCING MAPK HYPERACTIVATION Article Open access 12 September 2022 INTRODUCTION Induction of tumor cell apoptosis
is considered to be an important therapeutic strategy for cancer [1]. Overexpression of X-linked inhibitor of apoptosis protein (XIAP) and cellular IAP1/2 (cIAP1/2), the three major members
of the IAP family, has been reported in a variety of cancer tissues and relevant cancer cells [2, 3]. They inhibit apoptosis by directly or indirectly hindering the activation of caspase-3/7
(executioner caspases for apoptosis) through intrinsic and extrinsic apoptotic pathways, respectively [4,5,6,7]. Therefore, anticancer therapies that target IAPs have attracted extensive
attention [8], and several small-molecule IAP antagonists mimicking the endogenous antagonist Smac (second mitochondria-derived activator of caspases) protein have been designed and
synthesized. Among these molecules, GDC-0152, SM-406, LCL161, TL32711, and others have reached Phase I or II clinical trials [9,10,11]. The anticancer mechanism of the known IAP antagonists
has been studied systematically since the discovery of the first antagonist. All IAP antagonists can selectively bind to the BIR domains of XIAP, cIAP1, cIAP2, or ML-IAP (melanoma inhibitor
of apoptosis protein), but with different affinities. They abrogate XIAP-mediated and cIAP1/2-dependent caspase inhibition and thus increase caspase-3, caspase-7, and caspase-9 activity to
induce apoptosis [12]. Meanwhile, IAP antagonists induce cIAP1/2 degradation, augment TNFα-induced cell death [13], and activate the NF-κB pathway to stimulate the adaptive antitumor immune
response in cancers [14]. Recently, several IAP antagonists, including embelin, GDC-0152, and APG-1387, have been reported to exhibit autophagy-regulating activity
[15,16,17,18,19,20,21,22,23,24,25]. These IAP antagonists showed autophagy-inducing activity in different cell lines; however, the mechanisms underlying this activity appear to be different
and the information available regarding them is less. Meanwhile, XIAP and cIAPs have also been found to be involved in the regulation of autophagy [26, 27]. However, the underlying
regulatory mechanisms are diverse, including IAP-dependent and IAP-independent mechanisms [15]. Normally, autophagy is a cell survival pathway that plays essential roles in maintaining
cellular homeostasis via degradation and recycling of damaged organelles and toxic macromolecules. However, excessive autophagy is implicated in autophagic cell death [28, 29]. Links between
autophagy and apoptotic pathways have already been validated [30]. Autophagy has been speculated to have an antiapoptotic effect; however, under some circumstances, it also demonstrates an
apoptosis-promoting effect [31]. Therefore, clarifying the mechanism of autophagy regulation by an IAP antagonist could not only facilitate further disclosure of the mutual relationship
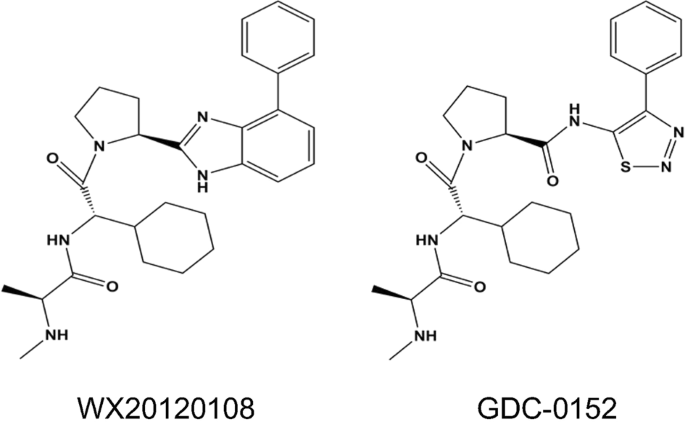
between apoptosis and autophagy but also contribute to the development of novel anticancer drugs. WX20120108 is a novel IAP-targeted compound designed and synthesized at our institute. As an
analogue of GDC-0152 (a known IAP antagonist, Fig. 1), WX20120108 shows more potent antitumor activity and autophagy-regulating activity in tumor cells than its parent molecule in addition
to exhibiting the profile of an IAP antagonist. Here, WX20120108-mediated autophagy induction and the underlying regulatory mechanism were studied, and the ROS-FOXO pathway was confirmed as
a key mediator involved in the WX20120108-induced tumor cell autophagy. MATERIALS AND METHODS COMPOUNDS AND REAGENTS Compound WX20120108 was designed and synthesized in our institute. The
structure (Fig. 1) was validated using nuclear magnetic resonance/mass spectrometry (1H-NMR/MS), and the purity (>98%) was determined via high-performance liquid chromatography. GDC-0152
(Cat. no. S7010, Fig. 1) and wortmannin (S2758) were purchased from Selleck (TX, USA). Z-VAD-FMK (ab120382) was purchased from Abcam (MA, USA). Chloroquine (CQ, P36235) was purchased from
Thermo Scientific (MA, USA). Catalase was purchased from Beyotime (Shanghai, China). Hoechst 33342 (H21492), rabbit antibodies against human LC3B (L10382), donkey anti-rabbit Alexa Fluor 488
(A21206) and donkey anti-mouse Alexa Fluor 546 (A10036) secondary antibodies were purchased from Life Technologies (CA, USA). Rabbit antibodies against human Foxo3 (2497), caspase-3 (9662),
and cleaved caspase-3 (9664) were purchased from CST (MA, USA). Mouse antibodies against human p62 (ab109012) were purchased from Abcam. Rabbit antibodies against human XIAP (sc-11426),
cIAP1 (sc-7943), and cIAP2 (sc-7944) were purchased from Santa Cruz (Texas, USA). The Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit (V13241) and CM-H2DCFDA (C6827) were purchased from
Thermo Scientific. The cathepsin B (CTSB) (K140-100) and cathepsin D (CTSD) (K143-100) activity assay kits were purchased from BioVision (CA, USA). Mouse antibodies against human GAPDH,
horseradish peroxidase-labeled goat anti-rabbit (ZB-2301) and goat anti-mouse (ZB-2305) secondary antibodies were purchased from ZSGB-Bio (Beijing, China). CELL LINES AND CULTURE CONDITIONS
HepG2, HeLa, Skov3, MDA-MB-231, and HLF cells were purchased from the Basic Medical Center of the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences (Beijing, China).
HeLa-bcl2 and Skov3/T cells were established and cultured in our laboratory. HeLa, HeLa-bcl2, MDA-MB-231, and HLF cells were cultured in RPMI-1640 medium (GIBCO, USA) at 37 °C in a
humidified 5% CO2 incubator. HepG2, Skov3, and Skov3/T cells were maintained in high-glucose Dulbecco’s modified Eagle’s medium (DMEM, GIBCO, USA). All media were supplemented with 10% fetal
bovine serum (FBS, GIBCO, USA), 100 U/mL penicillin–streptomycin–ampicillin, and for Skov3/T cells, 10 nmol/L Taxol (Merck, Darmstadt, Germany) was added. Fluorescently labeled reporter
cell lines were purchased from Thermo Scientific, except for the Akt1-EGFP_CHO cell line, which was purchased from GE Healthcare (Fairfield, CT, USA). All cell lines were cultured following
the manufacturer’s recommendation. To be specific, Foxo3-EGFP_U2OS cell lines were cultured in DMEM supplied with 10% FBS and 1% G418. DOCKING METHODOLOGY Molecular docking was performed
using the program Discovery Studio from BIOVIA Inc. (San Diego, CA, USA). Discovery Studio (http://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/) is an exhaustive
flexible docking algorithm with a scoring function that incorporates both empirical and knowledge-based features and was used for active site detection and docking. The pictures were
attained via PyMOL (V 2.0) from DeLano Scientific LLC (San Carlos, CA, USA). FPA COMPETITION ASSAY The binding affinities of WX20120108 for XIAP BIR3, XIAP BIR2-BIR3, and cIAP1/2 BIR3 were
determined using a fluorescence polarization anisotropy (FPA) competition assay developed by the Wang laboratory [32]. Primarily, 5 μL of various concentrations of WX20120108 was added to
each well of black 96-well round-bottom plates (Microfluor 2, Thermo Scientific) and then incubated with 120 μL assay buffer (100 mmol/L potassium phosphate, pH 7.5; 100 μg/mL bovine
γ-globulin; 0.02% sodium azide, Invitrogen) containing target protein and SM-F2 (a fluorescently tagged compound that specifically binds to the BIR3 domain) at room temperature for 2–3 h
with gentle shaking. The polarization values in millipolarization units were measured at an excitation wavelength of 485 nm and an emission wavelength of 530 nm using an Infinite M-1000
plate reader (Tecan U.S., Research Triangle Park, NC). For each experiment, the concentrations of target protein and SM-F2 in different target protein competitive binding tests were
determined according to a previous study [33], and each 96-well plate had a negative control and a positive control group. The negative control group contains the complex of protein and
compound SM-F2, which has an inhibitory rate of 0%. The positive control group contains free protein only, and the inhibitory rate equals 100%. The _K_i value for each compound was
calculated based upon the IC50 value, the _K_d value of the protein–probe complex, and the concentrations of the protein and probe in the competition assay, using a previously reported
algorithm and the associated computer program [34]. The equation is _K_i = [_I_]50/([_L_]50/_K_d + [_P_]0/_K_d + 1), in which [_I_]50 denotes the concentration of the free inhibitor at 50%
inhibition, [_L_]50 is the concentration of SM-F2 at 50% inhibition, [_P_]0 is the concentration of the free protein at 0% inhibition and _K_d is the dissociation constant of the
protein–SM-F2 complex. The IC50 value, the inhibitor concentration at which 50% of the bound tracer was displaced, was determined from the plot using nonlinear least-squares analysis. Curve
fitting was performed using Prism software (GraphPad Software, Inc., San Diego, CA, USA) [34]. ANTIPROLIFERATIVE ACTIVITY ASSAY A sulforhodamine B (SRB) assay was used to assess the
antiproliferative activity of the tested compounds [35]. Briefly, tumor cells (2.5 × 103–5.0 × 103 cells/well depending on cell type) were seeded into 96-well plates and cultured overnight
before compound exposure. After 72 h of compound treatment, the cells were fixed with trichloroacetic acid (TCA, 10%) at 4 °C for 1 h, washed with double-distilled water five times and dried
at room temperature. Then, the cells were dyed with SRB (0.4% in 1% acetic acid) at room temperature for 30 min, washed with 1% acetic acid five times, and dried at room temperature
successively. Finally, 10 mmol/L Tris base solution was added, and the absorbance at 510 nm was measured in the microplate reader (EL800, Bio-TEK, USA). The data were processed as described
[36]. The results were expressed as the means ± SD of at least three independent experiments. The antiproliferative activity was expressed as the inhibitory rate. The inhibitory rate (%) =
(_A_control,72 h − _A_compound,72 h)/(_A_control,72 h − _A_control,0 h) × 100, where _A_ is the absorbance at 510 nm. FCM ANALYSIS To detect cellular apoptosis induced by the tested
compounds, we used an Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit. In brief, HeLa and MDA-MB-231 cells seeded in six-well plates at a density of 8 × 104 cells/mL were cultured
overnight and then treated with test compounds for 24 h before harvesting via centrifugation. The cells were stained with Annexin V and propidium iodide (PI) according to the manufacturer’s
instructions and measured using a FACSCalibur Cytometer (BD Biosciences, CA, USA). Cells were classified as “survival (PI−/Annexin V−)”, “early apoptosis (PI−/Annexin V+)”, “late apoptosis
(PI+/Annexin V+)”, or “necrosis (PI+/Annexin V−)” according to the extent of staining by Annexin V or/and PI. The values of apoptotic cells (%) were the total number of early apoptotic cells
and late apoptotic cells. CELLULAR IMMUNOFLUORESCENCE Immunofluorescence staining for LC3B and Foxo3 was conducted as described below. Briefly, cells were plated in Corning 3603 plates
(blackwall, clear bottom 96-well plates; Cat. no. 3603, Corning, NY, USA), treated with different compounds, fixed with 4% formaldehyde, and washed twice with 1× phosphate-buffered saline.
After permeabilizing the cell membranes using 0.1% Triton X-100 and blocking with 5% bovine serum albumin, target proteins were visualized using primary antibodies and fluorescently labeled
secondary antibodies. Lysosomes were stained with 0.05 μmol/L LysoTracker Red before cells were fixed. Cell nuclei were labeled with 1 μmol/L Hoechst 33342 and subjected to image acquisition
on a high-content analysis (HCA) platform or other detection systems, and the cell count was measured to reflect cell viability. HCS FOR SIGNALING PATHWAYS OR TARGET PROTEINS Twelve
genetically modified reporter cell lines were used in cell-based signaling pathways or target protein screening. For each cell line, we followed the screening procedures recommended by the
manufacturer, and the key information is briefly summarized in Fig. 6a. The concentrations of WX20120108 used for screening were 1, 3, 10, 30, and 100 μmol/L. Detailed information regarding
the Foxo3 assays is provided below. Foxo3-EGFP_U2OS cells (U2OS cells stably expressing the Foxo3-EGFP fusion protein) were seeded in Corning 3603 plates at a density of 6 × 103 cells/well
for 24 h to allow adhesion. Then, different concentrations of WX20120108 were added, and the plates were incubated for 1 h. Finally, cells were fixed with 4% formaldehyde, and the nuclei
were dyed with 1 μmol/L Hoechst 33342 for 30 min at 37 °C. The cells were then subjected to HCA acquisition and analysis using an IN Cell Analyzer 2000 platform. The activity of WX20120108
in Foxo3 pathway assays was expressed as the activation rate relative to that of the positive compound (300 nmol/L wortmannin) and negative control (0.2% DMSO). CATHEPSIN ACTIVITY ASSAY The
catalytic activity of cathepsins was determined by CTSB and CTSD activity fluorometric assay kits. Briefly, 2 × 106 cells were harvested by centrifugation and lysed in 200 μL of chilled cell
lysis buffer for 20 min. Then, 50 μL of supernatant cell lysate was collected via centrifugation at 4 °C for 5 min at top speed and transferred into 96-well plates, mixed with reaction
buffer and substrate, and incubated at 37 °C for 2 h. The samples were read in a fluorometer with 400 nm excitation and 505 nm emission filters. The activity was normalized to the samples’
protein concentration. INTRACELLULAR ROS MEASUREMENT Intracellular ROS levels were measured via HCA after staining with CM-H2DCFDA as described previously [37]. Briefly, 8 × 103 cells were
plated in black 96-well culture plates, allowed to attach overnight, and exposed to DMSO (control) or the indicated concentrations of WX20120108 for 6 h. The cells were stained with 1 μmol/L
CM-H2DCFDA and 1 μmol/L Hoechst 33342 for 45 min at 37 °C, and the fluorescence was measured using an IN Cell Analyzer 2000. In some experiments, the cells were pretreated for 6 h with
siRNA against IAPs prior to WX20120108 exposure and analysis of ROS generation. All settings were identical between the different experimental groups. HIGH-CONTENT IMAGE ACQUISITION AND
ANALYSIS The images were acquired with an IN Cell Analyzer 2000 (GE HealthCare, USA) under a 20× objective lens for the assays [38]. For all measurements, filters were set according to the
wavelength of the fluorescent dyes, and nine fields from each well and no fewer than 200 cells were obtained. The acquired images were analyzed using an IN Cell Analyzer Workstation 3.5 and
the Multi Target Analysis Module (GE Healthcare). The final measurements were averaged over the population of cells detected in the fields of view. COLOCALIZATION ANALYSIS HeLa cells were
grown on coverslips overnight and incubated with tested compounds for a certain time. Then, lysosomes and LC3B were labeled as described above. All samples were examined under an UltraVIEW
VoX confocal microscope (PerkinElmer, MA, USA) equipped with a 63× oil immersion objective. The confocal images were acquired using Improvision Volocity software. The Pearson correlation
coefficient (PCC) was calculated to quantify colocalization. This calculation was performed by Volocity Demo 2.0 software between the stacks of images from two channels. WESTERN BLOTTING
ANALYSIS Cells were lysed after the indicated treatments. Approximately, 40 μg of total protein for each sample was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis on
10% gels and then transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% fat-free milk and incubated with the appropriate primary antibodies at 4 °C
overnight, followed by incubation with horseradish peroxidase-conjugated secondary antibodies at room temperature for 1 h. The immunostaining signal was visualized using ECL Plus reagent
(Applygen, Beijing, China), imaged, and analyzed with an Alpha Imager 5500 (Alpha Innotech, San Leandro, CA, USA). TRANSMISSION ELECTRON MICROSCOPY (TEM) HeLa cells were seeded in 6-well
plates and treated with WX20120108 for 16 h. Cells were harvested, collected and prefixed in 2.5% glutaraldehyde overnight at 4 °C. The samples were then postfixed, dehydrated, embedded,
sectioned, and stained as previously described [39]. Finally, electron micrographs were captured using a transmission electron microscope. QUANTITATIVE POLYMERASE CHAIN REACTION HeLa cells
were exposed to the WX20120108 (30 μmol/L) for the indicated times. Total RNA was extracted using a BioTeke Total RNA Extraction Kit (Cat. no. RP1201, BioTeke, Beijing, China). RNA was then
reverse transcribed into single-stranded cDNA using TransScript SuperMix (Cat. no. AT311-03, TransGen, Beijing, China). The resulting cDNA was used as a template for quantitative polymerase
chain reaction (qPCR). The primers used in this study are listed in Table 1. qPCR was conducted using the ABI Prism 7900 sequence detection system (Applied Biosystems, Foster City, CA, USA),
as previously described [40]. The relative change in mRNA levels was determined using the 2−ΔΔCT method, and β-actin was used as the internal standard. SMALL INTERFERING RNA (SIRNA) AND
TRANSIENT TRANSFECTION The scrambled RNAi oligonucleotides and siRNAs targeting _Foxo3_, _XIAP_, _cIAP1_, and _cIAP2_ (GenePharma, Shanghai, China) were transfected into HeLa cells using
Lipofectamine RNAiMAX (Cat. no. 13778-150, Invitrogen) according to the manufacturer’s protocol. The siRNA sequences were as follows: _Foxo3_, 5′-CAAGCACAGAGUUGGAUGATT-3′; _XIAP_,
5′-GGUCAGUACAAAGUUGAAATT-3′; _cIAP1_, 5′-CCCUACAGUUAUGCAAUGATT-3′; _cIAP2_, 5′-CGUGGCUCUUAUUCAAACUTT-3′. After the designated treatments, cell lysates were prepared for Western blotting.
DATA PROCESSING AND STATISTICAL ANALYSIS All data from the in vitro experiments were expressed as the means ± SD of three independent experiments. IC50 values were calculated with Origin 6.1
software. Statistical analyses were performed using one-way analysis of variance (ANOVA) with GraphPad Prism 5 software (*_P_ < 0.05, **_P_ < 0.01, ***_P_ < 0.001). RESULTS
WX20120108 BINDS TO THE BIR3 DOMAINS OF CIAP1, CIAP2, AND XIAP WITH HIGH AFFINITY Based on the key structural features of the XIAP-BIR3 domain interacting with Smac-Ala-Val-Pro-Ile (AVPI,
the binding peptide of endogenous IAP antagonist Smac), WX20120108 was designed. Docking results of this compound with XIAP-BIR3, as shown in Fig. 2a, b, indicated that WX20120108 inserts
well in the pockets of XIAP-BIR3, and several hydrogen bonds with W323, E314, T308, and Q319 residues in the backbone of BIR3 were formed, which were previously also observed in the binding
mode of Smac-AVPI [41]. An FPA competitive binding experiment was used to further analyze the binding ability of WX20120108 with XIAP BIR3, XIAP BIR2-BIR3, or cIAP1/2 BIR3 domains.
Saturation binding experiments were performed to determine the binding affinity of the fluorescent probes to the IAP constructs of interest, as reported [33]. As shown in Table 2, WX20120108
displayed high affinities with XIAP BIR3, XIAP BIR2-BIR3, cIAP1 BIR3, and cIAP2 BIR3 domains. Meanwhile, the IC50 values of WX20120108 on the competitive inhibitor SM-F2 for the BIR3
domains of cIAP1 and cIAP2 were comparable and were slightly higher than that for XIAP-BIR3. These data showed that WX20120108 binds to cIAP1, cIAP2, and XIAP with high affinity. WX20120108
INHIBITS THE PROLIFERATION OF CANCER CELLS The anticancer activity of WX20120108 was first screened in HepG2, Skov3, Skov3/T, MDA-MB-231, HeLa, and HeLa-bcl2 cancer cell lines by detecting
the antiproliferative activity of WX20120108 using an SRB assay. Compared to GDC-0152, the IAP antagonist that has been entered into phase I clinical trial, WX20120108 exhibited stronger
inhibitory activity in all tested cancer cell lines (Table 3); the potency of WX20120108 was two to tenfold higher than that of GDC-0152. In contrast to the differential antiproliferative
effect of GDC-0152 on these cancer cells, WX20120108 showed nearly comparable potency except on the HeLa-bcl2 cells. Moreover, both GDC-0152 and WX20120108 exhibited reduced cytotoxicity in
human lung fibroblast (HLF) cells; the IC50 values were higher than those in the tested cancer cells. WX20120108 EXHIBITS THE ANTICANCER PROPERTIES TYPICAL OF IAP ANTAGONISTS Overexpression
of IAPs has been correlated with certain types of advanced progressive cancer, poor prognosis, or low response to treatment [42, 43], and IAP antagonists are currently under clinical
considerations for these solid cancers (e.g., cervical and breast cancers) [11]. Herein, HeLa and MDA-MB-231 cells were chosen as the target cells for validating the IAP antagonist-like
characteristics of WX20120108. First, flow cytometry (FCM) following Annexin V-FITC/PI dual labeling showed that WX20120108 increased the proportion of apoptotic and dead cells in these two
cell lines in a dose-dependent manner (Fig. 3a), and this effect was found to be almost completely counteracted by cotreatment with the pan-caspase inhibitor Z-VAD-FMK (Fig. 3b). Second,
Western blotting assays indicated that WX20120108 reduced the levels of IAPs, especially cIAP1, in a dose-dependent manner (Fig. 3c). p-NF-κB and p-IκB-α levels were increased after ~0.5–6 h
of WX20120108 treatment (Fig. 3d), and cleaved caspase-3 levels increased in a dose-dependent manner after 24 h of WX20120108 treatment (Fig. 3e). Finally, WX20120108-mediated promotion of
the TNFα-induced cell death pathway was validated using a cell survival experiment with HCA. WX20120108 significantly reduced the number of viable cells when cotreated with TNFα (Fig. 3f),
which is consistent with previous reports that IAP antagonists synergistically promote TNFα-induced cell death [13, 44]. In addition, in accordance with the above results, Z-VAD-FMK
supplementation also rescued the TNFα-induced reduction in cell viability (Fig. 3f). The above results indicated that WX20120108 promotes caspase-dependent apoptosis mainly through the cIAP
and TNFα pathways and exhibits the same cancer cell inhibitory profile as typical IAP antagonists. WX20120108 REGULATES CANCER CELL AUTOPHAGY IN A DOSE- AND TIME-DEPENDENT MANNER The impact
of WX20120108 on autophagy was further studied in HeLa and MDA-MB-231 cells using fluorescence image-based HCA, Western blot, and transmission electron microscopy (TEM). Similar to the
autophagy inhibitor CQ, WX20120108 apparently increased the green fluorescence intensity and puncta number of antibody-labeled LC3B (as shown in Fig. 4a), which is visualized either as the
cytoplasmic localization or as punctate structures that primarily represent autophagosomes [45, 46]. Quantitative analysis indicated that WX20120108 can increase the content of LC3B puncta
in a dose- and time-dependent manner in both HeLa and MDA-MB-231 cells (Fig. 4a). However, the number of cells decreased as the concentrations of WX20120108 increased, particularly from 3 to
30 μmol/L. There was a drastic increase in LC3B puncta and a dramatic decrease in cell viability after 20 h incubation, which suggested that a dynamic “switch” from autophagy to cell death
occurred with the increase in WX20120108 concentration and incubation time. Corroborating the HCA results, Western blotting confirmed that WX20120108 treatment led to significantly increased
formation of LC3B-II in a dose- and time-dependent manner; meanwhile, p62 was also increased in both cells (Fig. 4b). It is noteworthy that the mRNA levels of _p_62 also increased with
prolonged WX20120108 incubation time (Fig. 4c). More importantly, autophagosomes were observed in WX20120108-treated HeLa cells (Fig. 4d). These results clearly demonstrated that WX20120108
regulated autophagy in HeLa and MDA-MB-231 cells. WX20120108 INDUCES AUTOPHAGIC FLUX The impact of WX20120108 on autophagic flux was assayed to further elucidate the mechanism by which
WX20120108 modulates autophagy, as both the induction and the suppression of autophagy can lead to an increase in the abundance of LC3B and autophagosomes [47]. In the presence of CQ, the
formation of LC3B-II was significantly enhanced in HeLa and MDA-MB-231 cells after WX20120108 treatment for 6 and 16 h, respectively (Fig. 5a). The colocalization of autophagosomes and
lysosomes revealed that WX20120108-treated cells exhibited a significant degree of fusion of autophagosomes and lysosomes. In contrast, there was almost no fusion of the fluorescent signals
for autophagosomes and lysosomes in CQ-treated cells, which is consistent with the autophagic flux blocker characteristics of CQ. RGB profile analysis by ImageJ (V 1.8.0) and PCC values
further confirmed the confocal microscopy results (Fig. 5b). The PCC value of CQ-treated cells (0.28 ± 0.01) was significantly lower than that of the control group (0.32 ± 0.02) (_P_ <
0.05), whereas it was much higher in the WX20120108 treatment group (0.59 ± 0.05) than that of the control group (_P_ < 0.001), indicating that colocalization was abolished by CQ
treatment, while WX20120108 treatment induced colocalization. Since the p62 level increased in the above experiment, the activities of the major lysosomal proteases CTSB and CTSD were
further assayed using the fluorometric assay to investigate whether WX20120108 regulates autophagy at this stage. Both CTSB and CTSD activity levels were increased in a time-dependent manner
upon WX20120108 treatment in HeLa and MDA-MB-231 cells (Fig. 5c). Taken together, these results suggest that WX20120108 induces autophagic flux. WX20120108 SELECTIVELY ACTIVATES FOXOS,
ESPECIALLY FOXO3 Given that WX20120108 induced autophagy, the main signaling pathways or target proteins related to autophagy were screened on twelve EGFP-labeled reporter cell lines
representing different signaling pathways or targets in an unbiased screening assay using HCA. The Z′ factors for these assays were greater than 0.5, confirming that these cellular models
were eligible for high-content screening (HCS) and that the screening system was reliable [48]. Apart from the weak disturbance in phosphatidylinositol 3-kinase (PI3K)-Foxo1, PI3K-Foxo3,
PI3K-Foxo4, and PI3K-FYVE pathway assays, 30 and 100 μmol/L GDC-0152 showed almost no effect in the other eight cell lines (Fig. 6a). Similarly, treatment with 3 and 30 μmol/L WX20120108 did
not affect the majority of signaling pathways or target proteins, except for PI3K-Foxo3, PI3K-Foxo1, PI3K-Foxo4, and PI3K-FYVE. Significant dose-dependent effects were observed in the
PI3K-Foxo3 pathway after WX20120108 treatment (Fig. 6a, b); the EC50 value was 28.87 ± 4.87 μmol/L. Since PI3K-PDK, PI3K-Rac1, and PI3K-Ras were not affected by WX20120108, the FOXO
activation, particularly Foxo3 nuclear translocation, was speculated to be the selective action of WX20120108. To further investigate whether this effect exists in tumor cells, the same
assay was performed in HeLa cells using immunofluorescence staining with anti-Foxo3 antibody to label Foxo3 (Fig. 6c). As expected, WX20120108 promoted the nuclear translocation of Foxo3 in
HeLa cells and dose-dependently induced Foxo3 activation without significantly affecting cell viability at concentrations less than 10 μmol/L, which suggested that WX20120108-induced Foxo3
activation is an early and selective event. WX20120108-INDUCED AUTOPHAGY DEPENDS ON FOXO3 ACTIVATION It has already been demonstrated that FOXO activation is involved in autophagy induction
via the upregulation of the expression of downstream target genes, such as _Bnip3_, _Pik3c3_, _Atg4b_, and _Atg5_, which are relevant to the formation of the autophagy initiation complex
[49, 50]. To confirm the role of Foxo3 in WX20120108-mediated autophagy, _Foxo3_ expression was knocked down in HeLa cells using the RNAi technique. As expected, LC3B puncta and LC3B-II
protein levels were reduced remarkably (Fig. 7a, b), indicating that _Foxo3_ knockdown markedly reduced WX20120108-induced autophagy. Moreover, WX20120108 time-dependently increased the
expression of genes involved in the induction of the autophagic pathway (_Bnip3_ and _Pik3c3_) and the formation of autophagosomes (_Atg4b_ and _Atg5_), which occurred as early as 2 h after
WX20120108 treatment (Fig. 7c). Taken together, these data suggest that Foxo3 is an essential regulator of cellular autophagy induced by WX20120108. WX20120108-MEDIATED FOXO3 ACTIVATION
DEPENDS ON ROS PRODUCTION While searching for the reason behind the induction of Foxo3 by WX20120108, we first examined the impact of IAPs. Knockdown of _XIAP_, _cIAP1_, and _cIAP2_ did not
affect WX20120108-induced Foxo3 nuclear translocation (Fig. 8a, b), indicating that the target proteins of WX20120108 were not involved in Foxo3 activation. Then, the upstream regulator of
Foxo3 activation was explored. We assessed intracellular ROS generation after WX20120108 treatment via HCA using CM-H2DCFDA, an indicator for ROS, which is cleaved by nonspecific cellular
esterases and is oxidized in the presence of peroxides to yield fluorescent 2′,7′-dichlorofluorescein (DCF) [51]. The results showed that WX20120108 significantly induced ROS generation in
HeLa cells in a dose-dependent manner (Fig. 8c). Moreover, the known ROS scavenger catalase almost completely blocked WX20120108 induced-Foxo3 nuclear translocation (Fig. 8d, e) and
suppressed the LC3B-II formation induced by WX20120108 (Fig. 8f). In addition, knockdown of _XIAP_ or _cIAP1_ and _cIAP2_ did not affect ROS release induced by 30 μmol/L WX20120108 (Fig.
8g). These results indicated that WX20120108-induced Foxo3 activation depends on ROS but not XIAP or cIAP1 and cIAP2. DISCUSSION Several studies have demonstrated that small-molecule IAP
antagonists induce tumor cell apoptosis by binding to IAPs, particularly XIAP, cIAP1, or cIAP2 to activate caspases [2, 14]. Notwithstanding the recently reported relationship between IAP
antagonists and autophagy, the understanding of the underlying mechanism is still shallow [17]. This study focused on a novel IAP antagonist, WX20120108, and its potent autophagy regulatory
activity. The autophagy induction mechanism was systematically unraveled based on the validation that WX20120108 exhibits the properties of an IAP antagonist and shows more potent antitumor
activity than other such compounds. WX20120108 is a new compound targeting the XIAP–BIR3 structure. Virtual molecular docking showed that WX20120108 strongly interacted with XIAP–BIR3 via
several hydrogen bonds at W323, E314, T308, and Q319 residues in the backbone of the BIR3 domain, which shares the same binding sites of Smac-AVPI, the binding peptide of an endogenous IAP
antagonist, Smac [9, 52]. Furthermore, a molecular affinity assay revealed that WX20120108 exhibited high affinity with XIAP, cIAP1, and cIAP2, and the IC50 values on SM-F2 for the BIR3
domains of XIAP, cIAP1, and cIAP2 reached nanomolar levels. This is consistent with many known IAP antagonists, such as GDC-0152 and SM-406 [11, 32]. Correspondingly, WX20120108 induced
rapid degradation of cIAP-1, cleavage of pro-caspase-3 and caspase-dependent tumor cell apoptosis in a dose-dependent manner and activated the NF-κB pathway and potentially promoted
caspase-dependent and TNFα-induced cell death synergistically as revealed by cellular functional assays. These phenomena are unique effects mediated by IAP antagonists through interaction
with the target pathway [14, 44, 53]. More interestingly, WX20120108 exhibited much lower IC50 values than the known IAP inhibitor GDC-0152 in six different tumor cell lines, including HeLa,
HeLa-bcl2, Skov3, Skov3/T, MDA-MB-231, and HepG2, and showed less cytotoxicity to normal cells than to cancer cells, just as GDC-0152 does. This evidence clearly indicated that WX2012018 is
a novel IAP antagonist with high in vitro anticancer activity. Autophagic cell death, or type II programmed cell death (PCD), and apoptosis, or type I PCD, can act as partners to induce
cell death in a coordinated or cooperative fashion, although they have different processes. Several signaling pathways that are induced by common cellular stressors regulate both autophagy
and apoptosis, such as Bcl-2, p53, NF-κB, and Beclin1, among others [30, 54]. The potent activity of the novel IAP antagonist WX20120108 on LC3B accumulation in HepG2 cells attracted our
attention. In screening experiments, treatment with 30 μmol/L WX2012018 produced even more LC3B puncta than treatment with 100 μmol/L CQ, a well-known autophagy inhibitor, which prevents
lysosomal endosomal acidification and blocks fusion of autophagosomes with lysosomes and therefore leads to accumulation of LC3B puncta [55]. This effect has been confirmed in multiple tumor
cell types (Supplementary Fig. 1). To systematically investigate the autophagy-regulating characteristics of WX20120108, HeLa, and MDA-MB-231 cells, the main targeting cancers of IAP
antagonists, were chosen for this study. Imaging-based HCA, Western blot assay and transmission electron microscopy observation revealed that WX20120108 dose- and time-dependently increased
LC3B puncta or LC3B-II formation and formed well-defined autophagosomes in cancer cells, and CQ coincubation significantly enhanced LC3B-II levels after both short-term and long-term
WX20120108 exposure. Moreover, colocalization of autophagosomes and lysosomes and lysosomal protease analysis demonstrated that, unlike CQ, WX20120108 promotes the fusion of autophagosomes
and lysosomes and significantly increases the activities of CTSB and CTSD, which are the most abundant lysosomal proteases participating directly in the proteolytic degradation of cellular
macromolecules in the execution of autophagy [56]. Therefore, apart from being an apoptosis inducer, WX20120108 is also an autophagy inducer, promoting autophagic flux. This was in agreement
with the results of previous studies on IAP antagonists, embelin, GDC-0152, and APG-1387, which exhibited autophagy-inducing activity [15, 16, 25]. Since WX20120108 has been demonstrated to
be an autophagy inducer, we first investigated the underlying mechanism with respect to the mammalian target of rapamycin upstream regulatory cascades, including PI3K and its associated
proteins, such as protein kinase B (PKB)/Akt [57,58,59,60], AMPK, and Ras/PKA [58], which are widely accepted as core pathways and targets during autophagy induction. The screening results
of the 12 EGFP-tagged reporter cell lines representing PI3K-Akt, PI3K-Btk, JAK-STAT1, p38 MAPK, PI3K-Foxo1, PI3K-Foxo3, PI3K-Foxo4, PI3K-FYVE, PI3K-PDK, PI3K-Rac1, PI3K-Ras and TULP3
signaling pathways or targets are interesting. WX20120108 dose-dependently activated PI3K-Foxo3 and activated PI3K-Foxo1, PI3K-Foxo4 and PI3K-FYVE only at high concentration (30 μmol/L) but
did not affect any other PI3K-related proteins or pathways. Thus, FOXOs activation seems to be a selective action of WX20120108. Foxo3 is a member of the FOXO subfamily of transcription
factors [61]. When Foxo3 is activated, it is translocated into the nucleus, where it upregulates the expression of _Bnip3_, _Pik3c3_, _BECN1_, _Ulk2_, _Atg4B_, and _Atg5_, genes that are
involved in autophagic pathway induction and autophagosome formation [50, 62]. Foxo3 activation-regulated autophagy has previously been described in various cell types [63]. Here,
WX20120108-induced Foxo3 activation and its relationship with autophagy were further validated and explored in HeLa cells. Consistent with the results of the EGFP-reporter cell lines,
WX20120108 dose-dependently activated Foxo3 in HeLa cells and upregulated the expression of Foxo3-target genes after treatment for 2 h. Silencing of _Foxo3_ significantly suppressed LC3B-II
formation produced by WX20120108. Therefore, WX20120108-induced Foxo3 activation is an important mediator of sustained autophagy induced by WX20120108. As a transcription factor, FOXOs can
be regulated by some regulatory factors, including Akt [64], p38 pathway [65], as well as oxidative stress [49], especially ROS. ROS could modulate the activities of FOXOs at multiple
levels, including posttranslational modifications of FOXOs (such as phosphorylation and acetylation), interaction with coregulators, alterations in the subcellular localization of FOXOs, and
protein synthesis and stability [49, 66]. Considering that WX20120108 did not affect the Akt and p38 pathways, we investigated cellular ROS levels. It is interesting that WX20120108
dose-dependently promoted ROS release after 6 h of incubation, and catalase (a known ROS scavenger) suppressed Foxo3 activation and LC3B-II formation induced by WX20120108. These results
indicated that ROS-mediated Foxo3 activation is involved in the WX20120108-induced autophagy. Although some studies have explored the relationship between IAPs (including XIAP and cIAP1/2)
or their antagonists and autophagy, the exact role of XIAP or cIAP1/2 in autophagy is still controversial. Huang et al. [26] reported that XIAP inhibited autophagy via the XIAP-Mdm2-p53
pathway, whereas Lin et al. [27] found that XIAP and cIAP1/2 induced autophagy via NF-κB activation. Whether IAP antagonist-induced autophagy depends on XIAP and cIAP1/2 has not yet been
reported. In this study, knockdown of _XIAP_ or _cIAP1/2_ affected neither the release of ROS nor Foxo3 activation induced by WX20120108. Transfection with siRNA against _cIAP1/2_ and not
against _XIAP_ partially impaired WX20120108-induced autophagy (Supplementary Fig. 2), which is similar to GDC-0152 (unpublished). This indicated that apart from the cIAP1/2-dependent
pathway, the XIAP and cIAP1/2-independent ROS-Foxo3 pathway plays an important role in WX20120108-induced autophagy. In addition, we have observed in the unbiased screening of
autophagy-related signaling pathways that GDC-0152 also exhibited similar selectivity on PI3K-Foxo1, Foxo3, Foxo4, and FYVE; however, the effect is very weak, which might be the reason that
WX20120108 exhibits stronger autophagy activity. p62 has been proposed to regulate the packaging and delivery of polyubiquitinated, misfolded, and aggregated proteins and dysfunctional
organelles for their degradation via autophagy in mammalian cells [67]. Therefore, p62 level is used as an indicator for identifying autophagy inducers and blockers. Autophagy inducers
decrease the p62 level, while autophagy blockers or inhibitors increase the p62 level [46]. In this study, the p62 level increased after WX20120108 treatment in a time- and dose-dependent
manner in cancer cells, which seemed to be in contrast to the general opinion. Recently, however, a p62-independent autophagic pathway has been proposed [57], and p62-upregulation has been
reported in some studies while autophagic flux increased [68,69,70]. In addition, p62 is a multifunctional factor involved in several cellular pathways. The p62 level is regulated not only
by autophagic degradation but also by autophagy-independent transcriptional regulation [71,72,73,74,75]. For example, the transcription of p62 is modulated by oxidative stress (Nrf2), the
Ras/MAPK pathway, and the JNK/c-Jun pathway [76]. Based on these findings, we further assessed _p62_ mRNA levels after WX20120108 treatment. The results indicated that WX20120108 treatment
increased _p62_ mRNA levels in both HeLa and MDA-MB-231 cells in a time-dependent manner, which might explain why the p62 level increased instead of decreasing after WX20120108 treatment.
This finding is consistent with the WX20120108-mediated regulation of oxidative stress. Promotion of apoptosis has been speculated to be the major way by which IAP antagonists cause cancer
cell death [14]. In this study, WX20120108 exhibited the profiles of IAP antagonists by inducing caspase-dependent apoptosis, which accounts for approximately 80%–90% of total cell death in
both HeLa and MDA-MB-231 cells after treatment with 30–100 μmol/L WX20120108. On the other hand, the parallel dynamic observations on cellular autophagy and cell survival showed that as the
concentration of WX20120108 increased or the incubation time prolonged, the cellular LC3B level increased, and there was an evident “switch” from autophagy to cell death, and this autophagy
is not affected by Z-VAD-FMK, a pan-caspase inhibitor. These results indicated that WX20120108-induced cell death embraced apoptosis and autophagy, and autophagy took place earlier than
apoptosis. A similar phenomenon was reported by You-Jin Lee et al. They found that the XIAP inhibitor embelin induces autophagic and apoptotic cell death in human oral squamous cell
carcinoma cells [15]. However, the mechanism by which IAP antagonist or WX20120108 switches autophagy to apoptosis remains to be studied. In summary, based on the characterizations of the
novel IAP antagonist WX2012018, this study revealed, for the first time, that the IAP-independent ROS-Foxo3 pathway is involved in the tumor cell autophagy induced by WX20120108. This
finding provides important insight into the molecular regulatory mechanism of autophagy induced by an IAP antagonist and provides more information for the development of a novel IAP
antagonist. REFERENCES * Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. Article CAS PubMed Google Scholar * Hunter AM, LaCasse EC, Korneluk RG. The inhibitors of
apoptosis (IAPs) as cancer targets. Apoptosis. 2007;12:1543–68. Article CAS PubMed Google Scholar * Tamm I, Kornblau SM, Segall H, Krajewski S, Welsh K, Kitada S, et al. Expression and
prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin Cancer Res. 2000;6:1796–803. CAS PubMed Google Scholar * Sun C, Cai M, Gunasekera AH, Meadows RP,
Wang H, Chen J, et al. NMR structure and mutagenesis of the inhibitor-of-apoptosis protein XIAP. Nature. 1999;401:818–22. Article CAS PubMed Google Scholar * Chai J, Shiozaki E,
Srinivasula SM, Wu Q, Datta P, Alnemri ES, et al. Structural basis of caspase-7 inhibition by XIAP. Cell. 2001;104:769–80. Article CAS PubMed Google Scholar * Riedl SJ, Renatus M,
Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, et al. Structural basis for the inhibition of caspase-3 by XIAP. Cell. 2001;104:791–800. Article CAS PubMed Google Scholar * Wang CY, Mayo MW,
Korneluk RG, Goeddel DV, Baldwin AS Jr. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–3. Article CAS
PubMed Google Scholar * de Almagro MC, Vucic D. The inhibitor of apoptosis (IAP) proteins are critical regulators of signaling pathways and targets for anti-cancer therapy. Exp Oncol.
2012;34:200–11. PubMed Google Scholar * Fulda S. Smac mimetics as IAP antagonists. Semin Cell Dev Biol. 2015;39:132–8. Article CAS PubMed Google Scholar * Brunckhorst MK, Lerner D,
Wang S, Yu Q. AT-406, an orally active antagonist of multiple inhibitor of apoptosis proteins, inhibits progression of human ovarian cancer. Cancer Biol Ther. 2012;13:804–11. Article CAS
PubMed PubMed Central Google Scholar * Flygare JA, Beresini M, Budha N, Chan H, Chan IT, Cheeti S, et al. Discovery of a potent small-molecule antagonist of inhibitor of apoptosis (IAP)
proteins and clinical candidate for the treatment of cancer (GDC-0152). J Med Chem. 2012;55:4101–13. Article CAS PubMed PubMed Central Google Scholar * Srinivasula SM, Hegde R, Saleh A,
Datta P, Shiozaki E, Chai J, et al. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature. 2001;410:112–6. Article CAS PubMed
Google Scholar * Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007;131:682–93. Article CAS
PubMed Google Scholar * Gyrd-Hansen M, Meier P. IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer. 2010;10:561–74. Article CAS PubMed
Google Scholar * Lee YJ, Park BS, Park HR, Yu SB, Kang HM, Kim IR. XIAP inhibitor embelin induces autophagic and apoptotic cell death in human oral squamous cell carcinoma cells. Environ
Toxicol. 2017;32:2371–8. Article CAS PubMed PubMed Central Google Scholar * Li BX, Wang HB, Qiu MZ, Luo QY, Yi HJ, Yan XL, et al. Novel smac mimetic APG-1387 elicits ovarian cancer cell
killing through TNF-alpha, Ripoptosome and autophagy mediated cell death pathway. J Exp Clin Cancer Res. 2018;37:53. Article PubMed PubMed Central CAS Google Scholar * Wang YF, Zhang
W, He KF, Liu B, Zhang L, Zhang WF, et al. Induction of autophagy-dependent cell death by the survivin suppressant YM155 in salivary adenoid cystic carcinoma. Apoptosis. 2014;19:748–58.
Article CAS PubMed PubMed Central Google Scholar * Wang Q, Chen Z, Diao X, Huang S. Induction of autophagy-dependent apoptosis by the survivin suppressant YM155 in prostate cancer
cells. Cancer Lett. 2011;302:29–36. Article CAS PubMed Google Scholar * Ding YH, Fan XD, Wu JJ, Deng ZK, Wei B, Li YF. Effect of YM155 on apoptosis and autophagy of K562 Cells. Zhongguo
Shi Yan Xue Ye Xue Za Zhi. 2015;23:375–80. CAS PubMed Google Scholar * Zhang L, Zhang W, Wang YF, Liu B, Zhang WF, Zhao YF, et al. Dual induction of apoptotic and autophagic cell death by
targeting survivin in head neck squamous cell carcinoma. Cell Death Dis. 2015;6:e1771. Article CAS PubMed PubMed Central Google Scholar * Cheng SM, Chang YC, Liu CY, Lee JY, Chan HH,
Kuo CW, et al. YM155 down-regulates survivin and XIAP, modulates autophagy and induces autophagy-dependent DNA damage in breast cancer cells. Br J Pharmacol. 2015;172:214–34. Article CAS
Google Scholar * Vequaud E, Seveno C, Loussouarn D, Engelhart L, Campone M, Juin P, et al. YM155 potently triggers cell death in breast cancer cells through an autophagy-NF-κB network.
Oncotarget. 2015;6:13476–86. Article PubMed PubMed Central Google Scholar * Pennati M, Sbarra S, De Cesare M, Lopergolo A, Locatelli SL, Campi E, et al. YM155 sensitizes triple-negative
breast cancer to membrane-bound TRAIL through p38 MAPK- and CHOP-mediated DR5 upregulation. Int J Cancer. 2015;136:299–309. Article CAS PubMed Google Scholar * Jane EP, Premkumar DR,
Sutera PA, Cavaleri JM, Pollack IF. Survivin inhibitor YM155 induces mitochondrial dysfunction, autophagy, DNA damage and apoptosis in Bcl-xL silenced glioma cell lines. Mol Carcinog.
2017;56:1251–65. Article CAS PubMed Google Scholar * Peiqi L, Rong H, Hongming D, Zhuogang L, Wei J, Miao M. GDC-0152-induced autophagy promotes apoptosis in HL-60 cells. Mol Cell
Biochem. 2018;445:135–43. Article PubMed CAS Google Scholar * Huang X, Wu Z, Mei Y, Wu M. XIAP inhibits autophagy via XIAP-Mdm2-p53 signalling. EMBO J. 2013;32:2204–16. Article CAS
PubMed PubMed Central Google Scholar * Lin F, Ghislat G, Luo S, Renna M, Siddiqi F, Rubinsztein DC. XIAP and cIAP1 amplifications induce Beclin 1-dependent autophagy through NFkappaB
activation. Hum Mol Genet. 2015;24:2899–913. Article CAS PubMed PubMed Central Google Scholar * Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological
functions of autophagy. Dev Cell. 2004;6:463–77. Article CAS PubMed Google Scholar * Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene.
2004;23:2891–906. Article CAS PubMed Google Scholar * Gump JM, Thorburn A. Autophagy and apoptosis- what’s the connection? Trends Cell Biol. 2011;21:387–92. Article CAS PubMed PubMed
Central Google Scholar * Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16:966–75.
Article CAS PubMed Google Scholar * Cai Q, Sun H, Peng Y, Lu J, Nikolovska-Coleska Z, McEachern D, et al. A potent and orally active antagonist (SM-406/AT-406) of multiple inhibitor of
apoptosis proteins (IAPs) in clinical development for cancer treatment. J Med Chem. 2011;54:2714–26. Article CAS PubMed PubMed Central Google Scholar * Wang S. Design of small-molecule
Smac mimetics as IAP antagonists. Curr Top Microbiol Immunol. 2011;348:89–113. CAS PubMed Google Scholar * Nikolovska-Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, et al. Development
and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal Biochem. 2004;332:261–73. Article CAS PubMed Google Scholar * Skehan P, Storeng R,
Scudiero D, Monks A, McMahon J, Vistica D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82:1107–12. Article CAS PubMed Google Scholar
* Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–6. Article CAS PubMed Google Scholar * Xiao D, Vogel V, Singh SV.
Benzyl isothiocyanate-induced apoptosis in human breast cancer cells is initiated by reactive oxygen species and regulated by Bax and Bak. Mol Cancer Ther. 2006;5:2931–45. Article CAS
PubMed Google Scholar * Li F, Wei A, Bu L, Long L, Chen W, Wang C, et al. Procaspase-3-activating compound 1 stabilizes hypoxia-inducible factor 1alpha and induces DNA damage by
sequestering ferrous iron. Cell Death Dis. 2018;9:1025. Article PubMed PubMed Central CAS Google Scholar * Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced
by histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2004;101:18030–5. Article CAS PubMed PubMed Central Google Scholar * Chen W, Fan S, Xie X, Xue N, Jin X, Wang L. Novel PPAR
pan agonist, ZBH ameliorates hyperlipidemia and insulin resistance in high fat diet induced hyperlipidemic hamster. PLoS One. 2014;9:e96056. Article PubMed PubMed Central CAS Google
Scholar * Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, et al. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408:1008–12. Article CAS PubMed Google Scholar * Zhang Y,
Zhu J, Tang Y, Li F, Zhou H, Peng B, et al. X-linked inhibitor of apoptosis positive nuclear labeling: a new independent prognostic biomarker of breast invasive ductal carcinoma. Diagn
Pathol. 2011;6:49. Article PubMed PubMed Central Google Scholar * Liu SS, Tsang BK, Cheung AN, Xue WC, Cheng DK, Ng TY, et al. Anti-apoptotic proteins, apoptotic and proliferative
parameters and their prognostic significance in cervical carcinoma. Eur J Cancer. 2001;37:1104–10. Article CAS PubMed Google Scholar * Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X,
Harran PG. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science. 2004;305:1471–4. Article CAS PubMed Google Scholar * Klionsky DJ. Guidelines for the
use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;25:151–75. * Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A,
et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. Article PubMed PubMed Central Google Scholar * Mizushima N,
Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–26. Article CAS PubMed PubMed Central Google Scholar * McGovern SL, Caselli E, Grigorieff N, Shoichet
BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–22. Article CAS PubMed Google Scholar * Webb AE, Brunet A.
FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci. 2014;39:159–69. Article CAS PubMed PubMed Central Google Scholar * Zhao J, Brault JJ, Schild
A, Cao P, Sandri M, Schiaffino S, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab.
2007;6:472–83. Article CAS PubMed Google Scholar * Ameziane El Hassani R, Dupuy C. Detection of intracellular reactive oxygen species (CM-H2DCFDA). Bio-Protocol. 2013;3:e313. Article
Google Scholar * Liu Z, Sun C, Olejniczak ET, Meadows RP, Betz SF, Oost T, et al. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature. 2000;408:1004–8. Article CAS
PubMed Google Scholar * Finlay D, Vamos M, Gonzalez-Lopez M, Ardecky RJ, Ganji SR, Yuan H, et al. Small-molecule IAP antagonists sensitize cancer cells to TRAIL-induced apoptosis: roles
of XIAP and cIAPs. Mol Cancer Ther. 2014;13:5–15. Article CAS PubMed Google Scholar * Thorburn A. Apoptosis and autophagy: regulatory connections between two supposedly different
processes. Apoptosis. 2008;13:1–9. Article CAS PubMed PubMed Central Google Scholar * Steinman RM, Mellman IS, Muller WA, Cohn ZA. Endocytosis and the recycling of plasma membrane. J
Cell Biol. 1983;96:1–27. Article CAS PubMed Google Scholar * Turk B, Stoka V, Rozman-Pungercar J, Cirman T, Droga-Mazovec G, Oresic K, et al. Apoptotic pathways: involvement of lysosomal
proteases. Biol Chem. 2002;383:1035–44. CAS PubMed Google Scholar * Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Molecular definitions of autophagy
and related processes. EMBO J. 2017;36:1811–36. Article CAS PubMed PubMed Central Google Scholar * Noda NN, Inagaki F. Mechanisms of autophagy. Annu Rev Biophys. 2015;44:101–22.
Article CAS PubMed Google Scholar * He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. Article CAS PubMed PubMed Central
Google Scholar * Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–31. Article CAS PubMed Google Scholar *
Sengupta A, Molkentin JD, Yutzey KE. FoxO transcription factors promote autophagy in cardiomyocytes. J Biol Chem. 2009;284:28319–31. Article CAS PubMed PubMed Central Google Scholar *
Yang J, Carra S, Zhu WG, Kampinga HH. The regulation of the autophagic network and its implications for human disease. Int J Biol Sci. 2013;9:1121–33. Article PubMed PubMed Central CAS
Google Scholar * Lv P, Huang J, Yang J, Deng Y, Xu J, Zhang X, et al. Autophagy in muscle of glucose-infusion hyperglycemia rats and streptozotocin-induced hyperglycemia rats via selective
activation of m-TOR or FoxO3. PLoS One. 2014;9:e87254. Article PubMed PubMed Central CAS Google Scholar * Hay N. Interplay between FOXO, TOR, and Akt. Biochim Biophys Acta.
2011;1813:1965–70. Article CAS PubMed PubMed Central Google Scholar * Ho KK, McGuire VA, Koo CY, Muir KW, de Olano N, Maifoshie E, et al. Phosphorylation of FOXO3a on Ser-7 by p38
promotes its nuclear localization in response to doxorubicin. J Biol Chem. 2012;287:1545–55. Article CAS PubMed Google Scholar * Klotz LO, Sanchez-Ramos C, Prieto-Arroyo I, Urbanek P,
Steinbrenner H, Monsalve M. Redox regulation of FoxO transcription factors. Redox Biol. 2015;6:51–72. Article CAS PubMed PubMed Central Google Scholar * Moscat J, Diaz-Meco MT. p62 at
the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001–4. Article CAS PubMed PubMed Central Google Scholar * Toepfer N, Childress C, Parikh A, Rukstalis D, Yang W.
Atorvastatin induces autophagy in prostate cancer PC3 cells through activation of LC3 transcription. Cancer Biol Ther. 2011;12:691–9. Article CAS PubMed Google Scholar * Colosetti P,
Puissant A, Robert G, Luciano F, Jacquel A, Gounon P, et al. Autophagy is an important event for megakaryocytic differentiation of the chronic myelogenous leukemia K562 cell line. Autophagy.
2009;5:1092–8. Article CAS PubMed Google Scholar * Zheng Q, Su H, Ranek MJ, Wang X. Autophagy and p62 in cardiac proteinopathy. Circ Res. 2011;109:296–308. Article CAS PubMed PubMed
Central Google Scholar * Fujita K, Maeda D, Xiao Q, Srinivasula SM. Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and
autophagic degradation. Proc Natl Acad Sci USA. 2011;108:1427–32. Article CAS PubMed PubMed Central Google Scholar * Nakaso K, Yoshimoto Y, Nakano T, Takeshima T, Fukuhara Y, Yasui K,
et al. Transcriptional activation of p62/A170/ZIP during the formation of the aggregates: possible mechanisms and the role in Lewy body formation in Parkinson’s disease. Brain Res.
2004;1012:42–51. Article CAS PubMed Google Scholar * Trocoli A, Bensadoun P, Richard E, Labrunie G, Merhi F, Schlafli AM, et al. p62/SQSTM1 upregulation constitutes a survival mechanism
that occurs during granulocytic differentiation of acute myeloid leukemia cells. Cell Death Differ. 2014;21:1852–61. Article CAS PubMed PubMed Central Google Scholar * B’Chir W, Maurin
AC, Carraro V, Averous J, Jousse C, Muranishi Y, et al. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41:7683–99. Article
PubMed PubMed Central CAS Google Scholar * Cnop M, Abdulkarim B, Bottu G, Cunha DA, Igoillo-Esteve M, Masini M, et al. RNA sequencing identifies dysregulation of the human pancreatic
islet transcriptome by the saturated fatty acid palmitate. Diabetes. 2014;63:1978–93. Article CAS PubMed Google Scholar * Puissant A, Fenouille N, Auberger P. When autophagy meets cancer
through p62/SQSTM1. Am J Cancer Res. 2012;2:397–413. CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We would like to thank Professor Shaomeng Wang at the
University of Michigan in the U.S. for his assistance in the FPA competition assay. This work was supported by the National Natural Science Foundation of China (Grant nos. 81803567 and
81773790) and the National Science and Technology Major Project of the Ministry of Science and Technology of China (Grant nos. 2012ZX09301-001 and 2012ZX09301-003) and partially supported by
a project of the State Key Laboratory of Functions and Applications of Medicinal Plants, Guizhou Medical University (FAMP201708K). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Beijing
Institute of Pharmacology and Toxicology, State Key Laboratory of Toxicology and Medical Countermeasures, Beijing, 100850, China Rui Ding, Xin Wang, Wei Chen, Zhi Li, Ai-li Wei, Qing-bin
Wang, Ai-hua Nie & Li-li Wang * State Key Laboratory of Functions and Applications of Medicinal Plants, Guizhou Medical University, Guiyang, 550014, China Rui Ding & Li-li Wang
Authors * Rui Ding View author publications You can also search for this author inPubMed Google Scholar * Xin Wang View author publications You can also search for this author inPubMed
Google Scholar * Wei Chen View author publications You can also search for this author inPubMed Google Scholar * Zhi Li View author publications You can also search for this author inPubMed
Google Scholar * Ai-li Wei View author publications You can also search for this author inPubMed Google Scholar * Qing-bin Wang View author publications You can also search for this author
inPubMed Google Scholar * Ai-hua Nie View author publications You can also search for this author inPubMed Google Scholar * Li-li Wang View author publications You can also search for this
author inPubMed Google Scholar CONTRIBUTIONS RD, AHN and LLW designed the research; RD, XW, WC, ZL, ALW and QBW performed the research; RD and LLW wrote the paper; XW and AHN provided the
testing compound; LLW, WC, ZL and AHN reviewed and modified the paper. CORRESPONDING AUTHOR Correspondence to Li-li Wang. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no
competing interests. SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURE 1 SUPPLEMENTARY FIGURE 2 RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ding, R.,
Wang, X., Chen, W. _et al._ WX20120108, a novel IAP antagonist, induces tumor cell autophagy via activating ROS-FOXO pathway. _Acta Pharmacol Sin_ 40, 1466–1479 (2019).
https://doi.org/10.1038/s41401-019-0253-5 Download citation * Received: 24 December 2018 * Accepted: 19 May 2019 * Published: 17 July 2019 * Issue Date: November 2019 * DOI:
https://doi.org/10.1038/s41401-019-0253-5 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * IAP antagonists * GDC-0152 * WX20120108 * apoptosis *
autophagy * carcinoma cells * Foxo3 * ROS * catalase