Play all audios:
ABSTRACT The long non-coding RNA X-inactive specific transcript (lncRNA XIST) and _MUC1_ gene are dysregulated in chronic inflammation and cancer; however, there is no known interaction of
their functions. The present studies demonstrate that MUC1-C regulates XIST lncRNA levels by suppressing the RBM15/B, WTAP and METTL3/14 components of the m6A methylation complex that
associate with XIST A repeats. MUC1-C also suppresses the YTHDF2-CNOT1 deadenylase complex that recognizes m6A sites and contributes to XIST decay with increases in XIST stability and
expression. In support of an auto-regulatory pathway, we show that XIST regulates MUC1-C expression by promoting NF-κB-mediated activation of the _MUC1_ gene. Of significance, MUC1-C and
XIST regulate common genes associated with inflammation and stemness, including (i) miR-21 which is upregulated across pan-cancers, and (ii) TDP-43 which associates with the XIST E repeats.
Our results further demonstrate that the MUC1-C/XIST pathway (i) is regulated by TDP-43, (ii) drives stemness-associated genes, and (iii) is necessary for self-renewal capacity. These
findings indicate that the MUC1-C/XIST auto-regulatory axis is of importance in cancer progression. SIMILAR CONTENT BEING VIEWED BY OTHERS MODULAR SCAFFOLDING BY LNCRNA HOXA10-AS PROMOTES
ORAL CANCER PROGRESSION Article Open access 20 July 2022 LNCRNA TINCR FAVORS TUMORIGENESIS VIA STAT3–TINCR–EGFR-FEEDBACK LOOP BY RECRUITING DNMT1 AND ACTING AS A COMPETING ENDOGENOUS RNA IN
HUMAN BREAST CANCER Article Open access 14 January 2021 THE HOXD9-MEDIATED PAXIP1-AS1 REGULATES GASTRIC CANCER PROGRESSION THROUGH PABPC1/PAK1 MODULATION Article Open access 24 May 2023
INTRODUCTION The lncRNA XIST initiates X chromosome inactivation (XCI) in female placental mammals to balance dosage of X-linked gene expression between the sexes [1]. In executing phases of
XCI, repeat arrays in the XIST RNA interact with diverse effectors that include the m6A methylation complex, epigenetic modifiers, and RNA splicing factors [2]. In addition to initiating
and executing XCI, XIST plays a role in the inflammatory response of post-XCI somatic cells. XIST responds to acute inflammation by regulating NF-κB activity [3, 4]. XIST is also
dysregulated in settings of chronic inflammation, including atherosclerosis [5] and neurodegenerative diseases [6]. Dysregulation of _XIST_ expression has been recognized across numerous
female and male cancers in association with both tumor progression and suppression [7,8,9]. Studies in breast cancer cells have implicated XIST overexpression with loss of the inactive X
chromosome (Xi) or Barr body and increased transcription of the active X chromosome [10,11,12]. Other work has indicated that the XCI transcriptional program can be selectively accessed in
male cancers [13]. Aberrant overexpression of XIST in cancer largely functions in promoting oncogenesis by regulating inflammation and diverse miRNAs that contribute to the CSC state and
drug resistance [9, 14, 15]. In contrast to studies of XCI, less is known about the regulation of XIST, the effectors that bind to the XIST array repeats and downstream target genes in
cancer cells. The potential involvement of XIST in contributing to intrinsic chronic inflammation in cancer progression also remains unclear. The _MUC1_ gene evolved in mammals for the
protection of barrier tissues from loss of homeostasis [16,17,18]. _MUC1_ encodes a transmembrane MUC1-C subunit that is activated by inflammation and contributes to barrier tissue stem cell
functions associated with wound healing [17, 18]. MUC1-C induces lineage plasticity, the epithelial-mesenchymal transition (EMT), epigenetic reprogramming, and chromatin remodeling, which
are of importance for repair and memory responses to inflammation [17,18,19,20]. These responses are in principle reversible with repair; however, persistent MUC1-C activation in settings of
chronic inflammation can promote progression of the cancer stem cell (CSC) state [17, 18]. MUC1-C is activated by the inflammatory IKK→NF-κB pathway in cancer cells [21, 22]. In an
auto-inductive pathway, MUC1-C binds directly to NF-κB and promotes activation of NF-κB target genes that include _MUC1_ itself [21, 22]. In turn, MUC1-C interacts with other transcription
factors (TFs), such as MYC and E2Fs, in activating the (i) Polycomb Repressive Complexes 1/2 (PRC1/2), and (ii) SWI/SNF embryonic stem cell esBAF and poly-bromo PBAF complexes [20,
23,24,25,26]. In this way, MUC1-C drives global changes in chromatin accessibility across the genomes of cancer cells [27]. Moreover, CSCs derived from diverse cancers are dependent on
MUC1-C for chromatin remodeling, self-renewal and treatment resistance [18, 25,26,27,28,29,30,31,32]. Little is known about MUC1-C involvement in the regulation of long non-coding RNAs
(lncRNAs) [33]. MUC1-C and XIST are dysregulated in chronic inflammation and cancer [9, 18]. There is no known association of MUC1-C and XIST in promoting cancer progression. The present
results demonstrate that MUC1-C upregulates XIST expression in male and female cancer cells. Moreover, XIST activates the _MUC1_ gene and thereby increases MUC1-C expression. In support of
this MUC1-C/XIST auto-inductive pathway, we show that MUC1-C and XIST regulate common sets of genes associated with chronic inflammation and cancer progression. RESULTS MUC1-C IS NECESSARY
FOR XIST EXPRESSION IN HUMAN CANCER CELLS XIST is upregulated in male and female cancers by unclear mechanisms [9]. To determine if MUC1-C contributes to XIST dysregulation, we studied
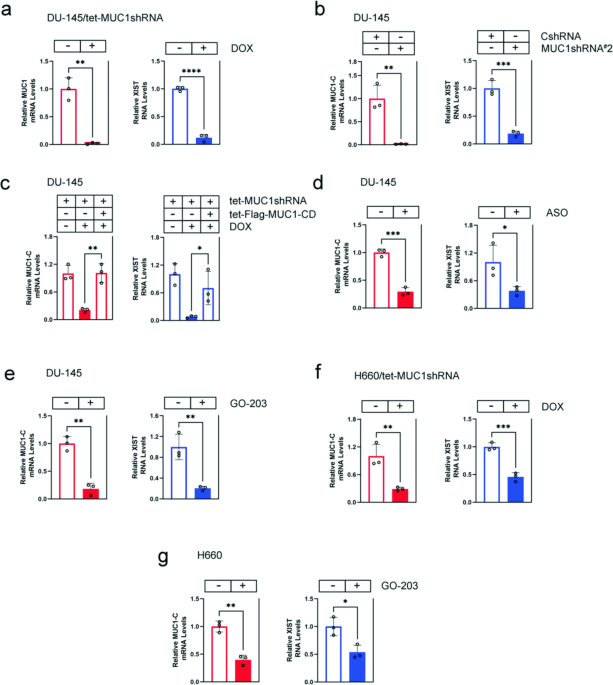
DU-145 castration-resistant prostate cancer (CRPC) cells expressing a tet-CshRNA or tet-MUC1shRNA vector [28]. DOX treatment of DU-145/tet-MUC1shRNA, but not DU-145/tet-CshRNA, cells was
associated with suppression of XIST RNA levels (Fig. 1a; Supplementary Fig. S1a). To exclude potential off-target effects, studies were performed on DU-145 cells expressing a second
MUC1shRNA#2, which similarly demonstrated downregulation of XIST transcripts (Fig. 1b). As a control, rescue of MUC1-C silencing by DOX-inducible expression of a Flag-tagged MUC1-C
cytoplasmic domain (tet-Flag-MUC1-CD) reversed XIST suppression (Fig. 1c). These results were extended by demonstrating that targeting MUC1-C with an anti-sense oligonucleotide (ASO) also
decreases XIST levels (Fig. 1d). The MUC1-C cytoplasmic domain contains a CQC motif that is necessary for MUC1-C homodimerization and function [17]. Treatment of DU-145 cells with the GO-203
inhibitor, which blocks the CQC motif [17], suppressed XIST transcripts, confirming that MUC1-C increases XIST expression (Fig. 1e). These results were not limited to DU-145 cells in that
targeting MUC1-C in male H660 neuroendocrine prostate cancer (NEPC) cells downregulated XIST levels (Fig. 1f, g). In addition, we studied female MDA-MB-436 and MDA-MB-468 triple-negative
breast cancer (TNBC) cells, which express XIST (Supplementary Fig. S1b), and found that silencing MUC1-C decreases XIST expression (Supplementary Fig. S1c, d). These findings indicated that
MUC1-C regulates XIST in cancers derived from both sexes. MUC1-C STABILIZES XIST BY SUPPRESSING XIST M6A METHYLATION In embryonic development, _XIST_ transcription is activated by the lncRNA
JPX, which is encoded in the XIC and evicts the CTCF suppressor of XIST expression [34,35,36]. Here, we sought to understand how MUC1-C increases XIST levels in cancer cells. In contrast to
XIST regulation in embryonic development, silencing MUC1-C in DU-145 and MDA-MB-436 cells had no apparent effect on _XIST_ transcription (Supplementary Fig. S2a, b). Rather, we found that
targeting MUC1-C is associated with decreases in XIST RNA stability in DU-145 (Fig. 2a) and MDA-MB-436 (Fig. 2b) cells, indicating that MUC1-C regulates XIST expression by a
post-transcriptional mechanism. XIST is modified by m6A methylation, which decreases XIST stability [37, 38]. We found that silencing MUC1-C increases XIST m6A methylation (Fig. 2c). RBM15
and its paralogue RBM15B bind to the XIST A-repeat region and recruit the WTAP and METTL3/14 components of the m6A methylation complex [2]. Of interest, silencing MUC1-C increased RBM15 and
RBM15B expression (Fig. 2d). Analogous to these effects, silencing MUC1-C also increased WTAP and METTL3/14 levels (Fig. 2e). In addressing potential off-target effects, we found that
targeting MUC1-C with an ASO similarly increases RBM15/B, WTAP and METTL3/14 levels (Fig. 2f). As confirmation of these results, rescue of MUC1-C silencing with Flag-MUC1-CD abrogated the
increases in RBM15/B, WTAP and METTL3/14 (Fig. 2g). Moreover, silencing MUC1-C in MDA-MB-436 cells increased expression of RBM15, WTAP and METTL3/14 (Supplementary Fig. S2c). These findings
indicated that MUC1-C increases XIST stability by suppressing (i) RBM15/B, WTAP, and METTL3/14 expression, and (ii) XIST m6A methylation in male and female cancer cells. MUC1-C REGULATES THE
YTHDF2-CNOT1 DEADENYLASE COMPLEX The YTHDF2 “reader” recognizes m6A sites and contributes to RNA decay [39]. In this way, m6A-methylated XIST is recognized by YTHDF2 in promoting XIST
degradation [38]. By contrast, the IGF2BP1 “reader” stabilizes m6A-containing RNAs [40, 41]. We found that silencing MUC1-C in DU-145 cells has no significant effect on YTHDF2 and IGF2BP1
transcripts (Fig. 3a). Further analysis demonstrated that silencing MUC1-C regulates YTHDF2 levels and expression of a IGF2BP1 doublet that may be related to post-translational
ubiquitination (Fig. 3b). These results were confirmed in DU-145/MUC1shRNA#2 (Fig. 3c) and in MDA-MB-436/tet-MUC1shRNA cells (Supplementary Fig. S3a, b), indicating that MUC1-C regulates
YTHDF2 expression by a post-transcriptional mechanism. YTHDF2 interacts with the CNOT1 subunit of the CCR4-NOT deadenylase complex [42]. Silencing MUC1-C in DU-145 cells decreased CNOT1
transcripts (Fig. 3d) and increased CNOT1 protein levels (Fig. 3e). These effects were confirmed in MDA-MB-436 cells (Supplementary Fig. S3c, d), indicating that, like YTHDF2, MUC1-C
regulates CNOT1 by a post-transcriptional mechanism. As a control, the effects of MUC1-C silencing on YTHDF2, IGF2BP1 and CNOT1 were reversed by rescue with MUC1-CD (Fig. 3f). These findings
indicated that MUC1-C (i) suppresses the YTHDF2 and CNOT1 proteins that destabilize XIST and (ii) upregulates IGF2BP1, which stabilizes m6A-containing RNAs. MUC1-C REGULATES TDP-43
EXPRESSION The demonstration that MUC1-C regulates the RBM15/B proteins that bind to the XIST A repeats invoked the possibility these findings could extend to other XIST RBPs. In that
regard, MUC1-C regulates components of the PRC1/2 complexes that interact with the XIST B/C-repeat arrays [2, 20, 23, 24]. TDP-43 interacts with the XIST E-repeat, plays a role in XCI [2,
43] and is widely involved in the regulation of mRNA alternative processing, transport and stability [44, 45]. TDP-43 is also overexpressed in pan-cancers by unclear mechanisms [44].
Silencing MUC1-C in DU-145 cells suppressed _TDP-43_ gene transcription and mRNA levels (Fig. 4a). By contrast, MUC1-C had no significant effect on expression of the _CELF1 and MATR3_ genes
that encode other XIST E-repeat binding proteins [2] (Supplementary Fig. S4a, b). We also found that, like _MUC1_, NF-κB is necessary for transcription of the _TDP-43_ gene and TDP-43 mRNA
levels (Fig. 4b). The _TDP-43_ gene has a putative NF-κB binding motif in the promoter region, which we found is occupied by MUC1-C and NF-κB (Fig. 4c). Silencing MUC1-C decreased NF-κB
occupancy of that region (Fig. 4c). Moreover, silencing MUC1-C was associated with decreases in (i) H3K27ac and H3K4me3 levels (Fig. 4c) and chromatin accessibility of the _TDP-43_ promoter
region (Fig. 4d). Consistent with these results, silencing MUC1-C and NF-κB decreased TDP-43 protein levels (Fig. 4e), which were rescued with MUC1-CD (Fig. 4f). In addition, we found that,
as shown for MUC1-C and NF-κB, silencing XIST decreases TDP-43 expression (Supplementary Fig. S4c). In support of an auto-regulatory loop, TDP-43 was also necessary for expression of MUC1-C
and p-NF-κB (Fig. 4g). MUC1-C AND XIST FUNCTION IN AN AUTO-INDUCTIVE PATHWAY THAT REGULATES COMMON GENE SIGNATURES MUC1-C has been linked to certain effectors through the formation of
auto-inductive pathways [31]. To determine if XIST regulates MUC1-C, we silenced XIST with a tet-XISTshRNA (Supplementary Fig. S5a) and found downregulation of _MUC1_ gene transcription and
mRNA levels (Fig. 5a). Targeting XIST with a second XISTsiRNA similarly downregulated MUC1-C expression (Supplementary Fig. S5b, c). XIST regulates NF-κB activity in the inflammatory
response [3, 4]. MUC1-C binds directly to NF-κB p65 and promotes activation of NF-κB target genes [21, 22]. Here, silencing XIST decreased MUC1-C and NF-κB levels (Fig. 5b), indicating that
XIST may regulate MUC1-C expression by promoting NF-κB-driven activation of the _MUC1_ gene. Analysis of the _MUC1_ enhancer region, which includes an NF-κB binding motif [22], demonstrated
that silencing XIST decreases MUC1-C and NF-κB occupancy (Fig. 5c). These results in support of a MUC1-C/XIST auto-inductive pathway suggested that MUC1-C and XIST may regulate common sets
of genes. To address this notion, we performed RNA-seq on XIST-silenced DU-145 cells and identified 478 downregulated and 289 upregulated genes (Supplementary Fig. S5d). Comparison of this
RNA-seq dataset with one derived from MUC1-C-silenced DU-145 cells uncovered 56 and 19 common downregulated and upregulated genes, respectively (Fig. 5d; Supplementary Table S3). MUC1-C and
XIST have been linked to the regulation of diverse miRNAs associated with cancer progression [9, 33]. Among common miRNAs identified in in the RNA-seq datasets, we confirmed that MUC1-C and
XIST are necessary for expression of miR-21 (Fig. 5e), which was of interest in that miR-21 is overexpressed across pan-cancers and is associated with the regulation of NF-κB and
inflammation [46, 47]. By extension, GSEA demonstrated that MUC1-C and XIST significantly regulate the GABRIELLY MIR21 TARGETS gene signature (Fig. 5f; Supplementary Fig. S5d). GSEA also
uncovered that MUC1-C and XIST regulate the MIR146A TARGETS gene signature, which like miR-21, miR-146a associates with inflammation and cancer [46, 47]. Moreover, we found that MUC1-C and
XIST commonly regulate the TP53 (Supplementary Fig. S5f) and mRNA destabilization (Supplementary Fig. S5g) gene signatures. Focusing here on other selected genes associated with inflammation
and stemness, we confirmed that MUC1-C and XIST regulate expression of ATF2 [48], SOX2 [49], BMI1 [20] and WNT5A [50] (Fig. 5g) genes. We also found that the MUC1-C/XIST pathway regulates
expression of the NEAT1 and MALAT1 lncRNAs (Fig. 5h), which have been linked to inflammation and cancer progression [51, 52]. MUC1-C/XIST PATHWAY REGULATES CSC SELF-RENEWAL DU-145 CSCs are
dependent on MUC1-C for self-renewal as evidenced by the capacity for tumorsphere formation [25, 26, 28]. Analysis of DU-145 cells grown as 2D monolayers vs 3D tumorspheres demonstrated
marked increases in MUC1-C and XIST, as well as TDP-43 expression (Fig. 6a). Silencing MUC1-C in DU-145 3D cells was associated with downregulation of XIST RNA levels (Fig. 6b; Supplementary
Fig. S6a). Silencing XIST in DU-145 3D cells also suppressed MUC1-C and TDP-43 expression (Fig. 6c), consistent with activation of the MUC1-C/XIST auto-inductive pathway in CSCs. Moreover,
we found that MUC1-C and XIST are necessary for expression of the NOTCH1, CD44, and BMI1 CSC markers (Fig. 6d, e), indicating functional involvement in driving the CSC state. Along these
lines and as reported for MUC1-C [25, 26, 28], DU-145 3D cells were also dependent on XIST for tumorsphere formation (Fig. 6f). Rescue of MUC1-C silencing with Flag-MUC1-CD was sufficient to
reestablish self-renewal capacity (Fig. 6g). Importantly, rescue of DU-145/XISTshRNA cells with Flag-MUC1-CD also recovered the capacity for self-renewal and markedly increased size of the
tumorspheres (Fig. 6h). Moreover, rescue of XIST expression in MUC1-C-silenced DU-145 cells reversed in part loss of tumorsphere formation (Supplementary Fig. S6b, c). These results indicate
that MUC1-C-dependent self-renewal is in part conferred by XIST and that other pathways play a role. DISCUSSION Transcription of the _XIST_ gene is activated by the JPX lncRNA in embryonic
development [34,35,36]; whereas little is known about XIST regulation in somatic cells. XIST is aberrantly upregulated in diverse human cancers from both sexes by unclear mechanisms [9].
Studies in female and male cancer cells have indicated that XIST overexpression is associated with transcription of the active X chromosome [10,11,12,13]. The present studies demonstrate
that MUC1-C increases XIST expression in male and female cancer cells. Notably, however, MUC1-C had no apparent effect on transcription of the _XIST_ gene, in support of distinct regulation
from that associated with induction of the active X chromosome. Rather, our results demonstrate that MUC1-C increases XIST stability and thereby XIST lncRNA levels (Fig. 7). The _MUC1_ gene
is activated in barrier tissues in response to loss of homeostasis [18]. Persistent _MUC1_ activation and dysregulation of MUC1-C expression in settings of chronic inflammation contribute to
cancer progression [18]. Of interest in this regard, dysregulation of XIST in somatic cells has been linked to the inflammatory response [3,4,5,6, 48]. These findings supported potential
involvement of MUC1-C in increasing XIST expression and regulating inflammation of somatic cells. This MUC1-C/XIST pathway should, in principle, be reversible with resolution of
inflammation; however, if irreversibly established in cancer progression, represents a mechanistic explanation for the upregulation of XIST in pan-cancers. The XIST transcript is highly
modified by m6A methylation [37], which plays a role in regulating lncRNA stability [53]. Consistent with increasing XIST levels, we found that MUC1-C suppresses XIST m6A methylation.
RBM15/B recruit the WTAP and METTL3/14 components of the m6A methylation complex to the XIST A repeat region [37]. Interactions of the XIST A repeat with RBM15B and METTL3 are required for
XIST m6A methylation and XIST-mediated XCI [37]. In cancer cells, we found that MUC1-C decreases expression of (i) RBM15/B, (ii) WTAP, and (iii) METTL3/14 (Fig. 7). These results supported a
model in which MUC1-C suppresses function of the m6A methylation complex in association with downregulation of XIST m6A methylation (Fig. 7). YTHDF2 recognizes m6A sites and contributes to
RNA instability by interacting with the CNOT1 subunit of the CCR4-NOT deadenylase complex [39, 42]. We found that MUC1-C suppresses YTHDF2 and CNOT1 expression (Fig. 7). By contrast, MUC1-C
increased the expression of IGF2BP1 (Fig. 7), which stabilizes m6A-containing RNAs [40, 41], indicating that MUC1-C coordinates a program of suppressing XIST m6A methylation, recognition and
degradation (Fig. 7). Importantly, the effects of targeting MUC1-C on the (i) m6A methylation complex components, (ii) m6A “readers” YTHDF2 and IGF2BP1, and (iii) CNOT1 subunit of the
deadenylase complex were rescued by the MUC1-CD cytoplasmic domain, confirming dependence on MUC1-C for their expression (Fig. 7). Of note, IGF2BP1 plays a role in activating NF-κB p65 in
chronic inflammation [54,55,56] and therefore could also contribute to regulation of the MUC1-C/NF-κB auto-inductive pathway and XIST expression. Our results further demonstrate that MUC1-C
and NF-κB are necessary for expression of TDP43, which binds to the XIST E-repeat region where it forms condensates essential for the XIST-independent phase of XCI [43]. TDP-43 is
overexpressed in cancer and promotes the dysregulation of mRNA, miRNA, and ncRNA metabolism [44, 57, 58]. We found that MUC1-C and NF-κB occupy the _TDP-43_ PLS and that MUC1-C is necessary
for (i) NF-κB occupancy and chromatin accessibility of that region and (ii) driving _TDP-43_ transcription (Fig. 7). In addition, we identified MUC1-C-dependent increases in H3K27ac and
H3K4me3 of the _TDP-43_ PLS, consistent with functions of MUC1-C in regulating p300/CBP and the SET1A/WDR5 COMPASS Complex [59]. These findings demonstrate that MUC1-C/NF-κB signaling
integrates the regulation of XIST and TDP-43 in cancer cells. Unexpectedly, we found that XIST is necessary for _MUC1_ gene activation and MUC1-C expression, in support of an auto-regulatory
MUC1-C/XIST pathway. Mechanistically, silencing XIST decreased expression of NF-κB p65, which activates the _MUC1_ gene in an auto-inductive MUC1-C/NF-κB circuit [18, 21, 22]. In further
support of the MUC1-C/XIST pathway, we found that MUC1-C and XIST regulate common genesets associated with activation of (i) miRNAs, such as miR-21, (ii) lncRNAs NEAT1 and MALAT1, and
protein encoding genes that are linked to inflammation and dysregulated in cancer. These findings collectively supported involvement of MUC1-C in driving a previously unrecognized an
auto-inductive MUC1-C/XIST pathway in cancer cells (Fig. 7). Our results do not exclude the possibility that the interaction between MUC1-C and XIST is also mediated by the PRC1 and PRC2
repressive complexes, which are activated by MUC1-C and are of importance in XIST-directed deposition of H2AK119ub and H3K27me3 in gene silencing [2, 18]. Our studies performed on male CRPC
and female TNBC cancer cells demonstrate that effects of MUC1-C on XIST are consistent across sexes. These results also suggest that MUC1-C co-opts involvement of XIST in post-XCI somatic
cells to promote cancer progression (Fig. 7). In support of this notion, we found that the MUC1-C/XIST pathway is necessary for CSC self-renewal. Silencing of MUC1-C and XIST was rescued by
MUC1-CD to reestablish self-renewal capacity, in support of the importance of MUC1-CD in driving the CSC state. MUC1-C/CD is a 72 aa disordered protein that is devoid of a kinase function
[18]. MUC1-CD is modified by RTKs at the cell membrane and interacts with effectors of the IKK→NF-κB pathway in integrating chronic inflammation with cancer progression. In this way, MUC1-C
drives a program of inflammatory memory that is necessary for CSC self-renewal capacity [18, 32]. The present results indicate that the MUC1-C/XIST pathway in somatic cells may also
contribute to the establishment of inflammatory memory, which is essential for protection of barrier tissues and has been co-opted by cancer cells in driving the CSC state, DNA damage
resistance and immune evasion. Additional evidence supporting involvement of the MUC1-C/XIST pathway in promoting chronic inflammation and inflammatory memory will require further
investigation. MATERIALS AND METHODS CELL CULTURE Human DU-145 CRPC cells (ATCC) were cultured in RPMI1640 medium (Corning Life Sciences, Corning, NY, USA) containing 10% heat-inactivated
FBS. Human NCI-H660 NEPC cells (ATCC) were cultured in RPMI1640 medium containing 5% heat-inactivated FBS, 10 nM β-estradiol (Millipore Sigma), 10 nM hydrocortisone, 1%
insulin-transferrin-selenium (ThermoFisher Scientific, Waltham, MA, USA) and 2 mM L-glutamine (ThermoFisher Scientific). Human MDA-MB-436 TNBC cells (ATCC) were cultured in Leibovitz’s L-15
medium (ThermoFisher Scientific) containing 10% heat-inactivated FBS. Human MDA-MB-468 TNBC cells (ATCC) were cultured in Leibovitz’s L-15 medium (ThermoFisher Scientific) containing 10%
FBS. Cells were treated with the MUC1-C inhibitor GO-203 [17]. Cells were maintained in culture for 3–4 months. Authentication of the cells was performed by short tandem repeat (STR)
analysis. Cells were monitored for mycoplasma contamination using the MycoAlert Mycoplasma Detection Kit (Lonza, Rockland, ME, USA). GENE SILENCING AND RESCUE MUC1shRNA (MISSION shRNA
TRCN0000122938), and a control scrambled shRNA (CshRNA)(Millipore Sigma) were inserted into pLKO-tet-puro (Plasmid #21915; Addgene, Cambridge, MA, USA). XISTshRNA was purchased from
GenePharma (Shanghai, China). Control, XIST, and TDP-43 targeted siRNAs (ThermoFisher Scientific) were transfected into cells using Lipofectamine 3000 (ThermoFisher Scientific).
DOX-inducible lentiviral shRNA targeting XIST (GE Dharmacon, V3SH11258_245457769) was obtained from Horizon Discovery (Cambridge, UK). DOX-inducible DYRK1A/pTRE3G-FL-hXIST vector was
obtained from Addgene (Plasmid #149608; RRID:Addgene_149608). The CshRNA, MUC1shRNA, MUC1shRNA#2 (MISSION shRNA TRCN0000430218), and NF-κBshRNA (MISSION shRNA TRCN0000014687) were produced
in HEK293T cells as described [28]. Flag-tagged MUC1-CD [60] was inserted into pInducer20 (Plasmid #44012, Addgene) [61]. Transduced cells were selected for growth in 1–2 μg/ml puromycin or
100 μg/ml geneticin. For inducible gene silencing, cells were treated with 0.1% DMSO as the vehicle control or 500 ng/ml doxycycline (DOX; Millipore Sigma). Cells were transfected with a
MUC1/ASO (LG00788741; Qiagen, Hilden, Germany) or a control C/ASO (LG00000001; Qiagen) in the presence of Lipofectamine 3000 Reagent. IMMUNOBLOT ANALYSIS Whole-cell lysates were prepared in
RIPA buffer containing protease inhibitor cocktail (ThermoFisher Scientific). Immunoblotting was performed with anti-MUC1-C (#16564S, 1:1000 dilution; Cell Signaling Technology (CST),
Danvers, MA, USA), anti-NF-κB p65 (#8242S, 1:1000 dilution; CST), anti-phospho-NF-κB p65 (#3037s, 1:1000 dilution; CST), anti-GAPDH (5174, 1:1000 dilution; CST), anti-RBM15B (22249-1-AP,
1:1000 dilution; Proteintech, Rosemont, IL, USA), anti-RBM15 (10587-1-AP, 1:5000 dilution; Proteintech), anti-YTHDF2 (24744-1-AP, 1:5000 dilution; Proteintech), anti-CNOT1 (14276-1-AP,
1:1000 dilution; Proteintech), anti-WTAP (#56501, 1:1000 dilution; CST), anti-METTL3 (#96391, 1:1000 dilution; CST), anti-METTL14 (#51104, 1:1000 dilution; CST), anti-IGF2BP1 (22803-1-AP,
1:5000 dilution; Proteintech), anti-TDP-43 (#32654, 1:1000 dilution; CST), anti-NOTCH1 (#3608S, 1:1000 dilution; CST), anti-BMI1 (#6964P, 1:1000 dilution; CST), anti-CD44 (#5640S, 1:1000
dilution; CST), anti-β-actin (A5441; 1:50000 dilution; Sigma, St. Louis, MO, USA) and anti-tubulin (#2144S, 1:1000 dilution; CST). Signals shown in immunoblots and in a separate biologic
replicate were each scanned in triplicate. The results (mean ± SD of six determinations) are expressed as relative signal intensity compared to that obtained for GAPDH. QUANTITATIVE
REVERSE-TRANSCRIPTION PCR (QRT-PCR) Total cellular RNA was isolated using Trizol reagent (ThermoFisher Scientific). cDNAs were synthesized using the High-Capacity cDNA Reverse Transcription
Kit (Applied Biosystems, Grand Island, NY, USA). The cDNA samples were amplified as described [62]. Primers used for qRT-PCR are listed in Supplementary Table 1. The primers for MIR21
(#000397) and relative control U6 (#001093) were purchased from ThermoFisher Scientific. The cDNA samples were amplified using TaqMan MicroRNA Reverse Transcription Kit (ThermoFisher
Scientific) according to the manufacturer’s protocol. CLICK-IT NASCENT RNA ASSAY Nascent RNA labeling with EU was performed using the Click-iT Nascent RNA Capture kit (Invitrogen, Carlsbad,
CA, USA) according to the manufacturer’s protocol. Briefly, cells were pulsed with 0.5 mM EU for 24 h. Total RNA was isolated and the nascent transcripts were captured on streptavidin
magnetic beads. cDNA synthesis was performed directly on the beads using the High-Capacity cDNA Reverse Transcription Kit followed by analysis with qRT-PCR. RNA STABILITY ASSAY Cells were
incubated with 5 μg/ml actinomycin D (11421, Cayman, Ann Arbor, MI, USA) for 0, 3, and 6 h. Total RNA was analyzed by qRT-PCR. N6-METHYLADENOSINE-RNA IMMUNOPRECIPITATION–QUANTITATIVE
POLYMERASE CHAIN REACTION M6A RNA ENRICHMENT (MERIP) MeRIP experiments were performed using the EpiQuik CUT&RUN m6A RNA Enrichment Kit (#P-9018-24, Epigentek, New York, USA) according to
the manufacturer’s protocol. Briefly, RNA containing target m6A-modified regions were cleaved into fragments pulled down with beads bound to an m6A capture antibody. The enriched RNA was
transcribed to cDNA using the Superscript VILO cDNA synthesis kit (Invitrogen) followed by analysis with qRT-PCR. CHROMATIN IMMUNOPRECIPITATION (CHIP) ChIP was performed on cells crosslinked
with 1% formaldehyde for 5 min at 37 °C, quenched with 2 M glycine, washed with PBS, and then sonicated in a Covaris E220 sonicator to generate 300–600 bp DNA fragments. Immunoprecipitation
was performed using a control IgG (3900S, CST) and antibodies against MUC1-C (#16564S, CST), NF-κB p65 (#ab16502, Abcam), H3K27ac (#ab4729, RRID:AB_2118291, Abcam), and H3K4me3 (#ab8580;
RRID:AB_306649, Abcam). Precipitated DNAs were detected by PCR using primers listed in Supplementary Table S2. Quantitation was performed on immunoprecipitated DNA using SYBR-green and the
CFX384 real-time PCR machine (Bio-Rad, Berkeley, CA, USA). Data are reported as a percentage of input DNA for each sample. RNA-SEQ ANALYSIS Total RNA from cells cultured in triplicates was
isolated using Trizol reagent (Invitrogen) as described [28, 30, 63]. TruSeq Stranded mRNA (Illumina, San Diego, CA, USA) was used for library preparation. Raw sequencing reads were aligned
to the human genome (GRCh38.74) using STAR. Raw feature counts were normalized and differential expression analysis using DESeq2 as described [28, 30, 63]. Differential expression rank order
for subsequent Gene Set Enrichment Analysis (GSEA) was performed using the fgsea (v1.8.0) package in R. ATAC-SEQ ATAC-seq libraries were generated from three biologically independent
replicates per condition. Library preparation and quality control were performed as described [27, 64]. The raw ATAC-seq data was processed using the pipeline:
(https://github.com/macs3-project/genomics-analysis-pipelines). MACS2 was used to generate signal tracks for Integrative Genome Browser (IGV) snapshots as described [27]. CHROMATIN
ACCESSIBILITY ASSAY DNAse1 chromatin accessibility assays were performed on chromatin isolated as described [27]. Aliquots of chromatin were left untreated or digested with 3 U/100 μl DNase
I (Promega, Madison, WI, USA) for 5 min at room temperature as described [27]. DNA was purified and amplified by qPCR using primers listed in Supplementary Table S2. qPCR results were
analyzed according to the formula 100/2Ct (DNase I) −Ct (no DNase I). The data were normalized to input DNA without DNase I treatment as described [27]. TUMORSPHERE FORMATION ASSAYS
Single-cell suspensions were cultured in MammoCult Human MediumKit (Stemcell Technologies, Cambridge, MA, USA) at a density of 5,000 cells per well of a 6-well ultralow attachment culture
plate (Corning) for 10 days as described [27]. Tumorspheres with a diameter >50 microns were counted under an inverted microscope in triplicate wells. STATISTICAL ANALYSIS Each experiment
was performed with at least three independent biologic replicates. Data are expressed as the mean ± SD. The unpaired Student’s _t_-test was used to examine differences between two groups. A
_p_ value of <0.05 was considered a statistically significant difference. Graphpad Prism 8 was used for all statistical analyses. Asterisks represent *_P_ ≤ 0.05, **_P_ ≤ 0.01, ***_P_ ≤
0.001, ****_P_ ≤ 0.0001 with CI = 95%. DATA AVAILABILITY The RNA-seq data have been deposited in the GEO database under accession codes GSE164141 and GSE203055. Data in this study is
available upon reasonable request from the corresponding author at [email protected]. REFERENCES * Dossin F, Heard E. The molecular and nuclear dynamics of X-chromosome
inactivation. Cold Spring Harb Perspect Biol. 2022;14:a040196. * Jacobson EC, Pandya-Jones A, Plath K. A lifelong duty: how Xist maintains the inactive X chromosome. Curr Opin Genet Dev.
2022;75:101927. Article CAS PubMed PubMed Central Google Scholar * Shenoda BB, Ramanathan S, Gupta R, Tian Y, Jean-Toussaint R, Alexander GM, et al. Xist attenuates acute inflammatory
response by female cells. Cell Mol Life Sci. 2021;78:299–316. Article CAS PubMed Google Scholar * Zhou Z, Ni H, Li Y, Jiang B. LncRNA XIST promotes inflammation by downregulating GRalpha
expression in the adenoids of children with OSAHS. Exp Ther Med. 2021;21:500. Article CAS PubMed PubMed Central Google Scholar * Yang K, Xue Y, Gao X. LncRNA XIST promotes
atherosclerosis by regulating miR-599/TLR4 axis. Inflammation. 2021;44:965–73. Article CAS PubMed Google Scholar * Wang H, Wang L, Luo Y, Gui Y, Wu W, Zhao J, et al. LncRNA XIST: a
breakthrough in inflammation-related diseases. Curr Med Chem. 2024; Online ahead of print. * Yang Z, Jiang X, Jiang X, Zhao H. X-inactive-specific transcript: a long noncoding RNA with
complex roles in human cancers. Gene. 2018;679:28–35. Article CAS PubMed Google Scholar * Chen YK, Yen Y. The ambivalent role of lncRNA Xist in carcinogenesis. Stem Cell Rev Rep.
2019;15:314–23. Article CAS PubMed Google Scholar * Yang J, Qi M, Fei X, Wang X, Wang K. Long non-coding RNA XIST: a novel oncogene in multiple cancers. Mol Med. 2021;27:159. Article
PubMed PubMed Central Google Scholar * Sirchia SM, Ramoscelli L, Grati FR, Barbera F, Coradini D, Rossella F, et al. Loss of the inactive X chromosome and replication of the active X in
BRCA1-defective and wild-type breast cancer cells. Cancer Res. 2005;65:2139–46. Article CAS PubMed Google Scholar * Sirchia SM, Tabano S, Monti L, Recalcati MP, Gariboldi M, Grati FR, et
al. Misbehaviour of XIST RNA in breast cancer cells. PLoS ONE. 2009;4:e5559. Article PubMed PubMed Central Google Scholar * Chaligne R, Popova T, Mendoza-Parra MA, Saleem MA, Gentien D,
Ban K, et al. The inactive X chromosome is epigenetically unstable and transcriptionally labile in breast cancer. Genome Res. 2015;25:488–503. Article CAS PubMed PubMed Central Google
Scholar * Sadagopan A, Nasim IT, Li J, Achom M, Zhang CZ, Viswanathan SR. Somatic XIST activation and features of X chromosome inactivation in male human cancers. Cell Syst.
2022;13:932–44.e5. Article CAS PubMed Google Scholar * Liu JH, Li C, Cao L, Zhang CH, Zhang ZH. Cucurbitacin B regulates lung cancer cell proliferation and apoptosis via inhibiting the
IL-6/STAT3 pathway through the lncRNA XIST/miR-let-7c axis. Pharm Biol. 2022;60:154–62. Article CAS PubMed Google Scholar * Ma Y, Zhu Y, Shang L, Qiu Y, Shen N, Wang J, et al. LncRNA
XIST regulates breast cancer stem cells by activating proinflammatory IL-6/STAT3 signaling. Oncogene. 2023;42:1419–37. Article CAS PubMed PubMed Central Google Scholar * Kufe D. Mucins
in cancer: function, prognosis and therapy. Nat Rev Cancer. 2009;9:874–85. Article CAS PubMed PubMed Central Google Scholar * Kufe D. MUC1-C in chronic inflammation and carcinogenesis;
emergence as a target for cancer treatment. Carcinogenesis. 2020;41:1173–83. Article CAS PubMed PubMed Central Google Scholar * Kufe D. Emergence of MUC1 in mammals for adaptation of
barrier epithelia. Cancers. 2022;14:4805. Article CAS PubMed PubMed Central Google Scholar * Rajabi H, Kufe D. MUC1-C oncoprotein integrates a program of EMT, epigenetic reprogramming
and immune evasion in human carcinomas. BBA. Rev Cancer. 2017;1868:117–22. CAS Google Scholar * Rajabi H, Hiraki M, Kufe D. MUC1-C activates polycomb repressive complexes and downregulates
tumor suppressor genes in human cancer cells. Oncogene. 2018;37:2079–88. Article CAS PubMed PubMed Central Google Scholar * Ahmad R, Raina D, Trivedi V, Ren J, Rajabi H, Kharbanda S,
et al. MUC1 oncoprotein activates the IκB kinase β complex and constitutive NF-κB signaling. Nat Cell Biol. 2007;9:1419–27. Article CAS PubMed PubMed Central Google Scholar * Ahmad R,
Raina D, Joshi MD, Kawano T, Kharbanda S, Kufe D. MUC1-C oncoprotein functions as a direct activator of the NF-kappaB p65 transcription factor. Cancer Res. 2009;69:7013–21. Article CAS
PubMed PubMed Central Google Scholar * Hiraki M, Maeda T, Bouillez A, Alam M, Tagde A, Hinohara K, et al. MUC1-C activates BMI1 in human cancer cells. Oncogene. 2017;36:2791–801. Article
CAS PubMed Google Scholar * Rajabi H, Hiraki M, Tagde A, Alam M, Bouillez A, Christensen CL, et al. MUC1-C activates EZH2 expression and function in human cancer cells. Sci Rep.
2017;7:7481. Article PubMed PubMed Central Google Scholar * Hagiwara M, Yasumizu Y, Yamashita N, Rajabi H, Fushimi A, Long MD, et al. MUC1-C activates the BAF (mSWI/SNF) complex in
prostate cancer stem cells. Cancer Res. 2021;81:1111–22. Article CAS PubMed Google Scholar * Hagiwara M, Fushimi A, Yamashita N, Battacharya A, Rajabi H, Long M, et al. MUC1-C activates
the PBAF chromatin remodeling complex in integrating redox balance with progression of human prostate cancer stem cells. Oncogene. 2021;40:4930–40. Article CAS PubMed PubMed Central
Google Scholar * Bhattacharya A, Fushimi A, Yamashita N, Hagiwara M, Morimoto Y, Rajabi H, et al. MUC1-C dictates JUN and BAF-mediated chromatin remodeling at enhancer signatures in cancer
stem cells. Mol Cancer Res. 2022;20:556–67. Article CAS PubMed PubMed Central Google Scholar * Yasumizu Y, Rajabi H, Jin C, Hata T, Pitroda S, Long MD, et al. MUC1-C regulates lineage
plasticity driving progression to neuroendocrine prostate cancer. Nat Commun. 2020;11:338. Article CAS PubMed PubMed Central Google Scholar * Hagiwara M, Fushimi A, Bhattacharya A,
Yamashita N, Morimoto Y, Oya M, et al. MUC1-C integrates type II interferon and chromatin remodeling pathways in immunosuppression of prostate cancer. OncoImmunol. 2022;11:e2029298. Article
Google Scholar * Yamashita N, Long M, Fushimi A, Yamamoto M, Hata T, Hagiwara M, et al. MUC1-C integrates activation of the IFN-gamma pathway with suppression of the tumor immune
microenvironment in triple-negative breast cancer. J Immunother Cancer. 2021;9:e002115. Article PubMed PubMed Central Google Scholar * Yamashita N, Morimoto Y, Fushimi A, Ahmad R,
Bhattacharya A, Daimon T, et al. MUC1-C dictates PBRM1-mediated chronic induction of interferon signaling, DNA damage resistance and immunosuppression in triple-negative breast cancer. Mol
Cancer Res. 2023;21:274–89. Article CAS PubMed Google Scholar * Kufe D. Dependence on MUC1-C in progression of neuroendocrine prostate cancer. Int J Mol Sci. 2023;24:3719. Article CAS
PubMed PubMed Central Google Scholar * Li W, Han Y, Sun C, Li X, Zheng J, Che J, et al. Novel insights into the roles and therapeutic implications of MUC1 oncoprotein via regulating
proteins and non-coding RNAs in cancer. Theranostics. 2022;12:999–1011. Article CAS PubMed PubMed Central Google Scholar * Tian D, Sun S, Lee JT. The long noncoding RNA, Jpx, is a
molecular switch for X chromosome inactivation. Cell. 2010;143:390–403. Article CAS PubMed PubMed Central Google Scholar * Sun S, Del Rosario BC, Szanto A, Ogawa Y, Jeon Y, Lee JT. Jpx
RNA activates Xist by evicting CTCF. Cell. 2013;153:1537–51. Article CAS PubMed PubMed Central Google Scholar * Li J, Ming Z, Yang L, Wang T, Liu G, Ma Q. Long noncoding RNA XIST:
mechanisms for X chromosome inactivation, roles in sex-biased diseases, and therapeutic opportunities. Genes Dis. 2022;9:1478–92. Article CAS PubMed PubMed Central Google Scholar *
Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537:369–73. Article CAS PubMed
PubMed Central Google Scholar * Yang X, Zhang S, He C, Xue P, Zhang L, He Z, et al. METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long
non-coding RNA XIST. Mol Cancer. 2020;19:46. Article CAS PubMed PubMed Central Google Scholar * Wang J-Y, Lu A-Q. The biological function of m6A reader YTHDF2 and its role in human
disease. Cancer Cell Int. 2021;21:109. Article PubMed PubMed Central Google Scholar * Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP
proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285–95. Article CAS PubMed PubMed Central Google Scholar * Zhu P, He F, Hou Y, Tu G, Li Q, Jin T, et al. A
novel hypoxic long noncoding RNA KB-1980E6.3 maintains breast cancer stem cell stemness via interacting with IGF2BP1 to facilitate c-Myc mRNA stability. Oncogene. 2021;40:1609–27. Article
CAS PubMed PubMed Central Google Scholar * Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT
deadenylase complex. Nat Commun. 2016;7:12626. Article CAS PubMed PubMed Central Google Scholar * Pandya-Jones A, Markaki Y, Serizay J, Chitiashvili T, Mancia Leon WR, Damianov A, et
al. A protein assembly mediates Xist localization and gene silencing. Nature. 2020;587:145–51. Article CAS PubMed PubMed Central Google Scholar * Ma X, Ying Y, Xie H, Liu X, Wang X, Li
J. The regulatory role of RNA metabolism regulator TDP-43 in human cancer. Front Oncol. 2021;11:755096. Article CAS PubMed PubMed Central Google Scholar * Linares AJ, Lin CH, Damianov
A, Adams KL, Novitch BG, Black DL. The splicing regulator PTBP1 controls the activity of the transcription factor Pbx1 during neuronal differentiation. Elife. 2015;4:e09268. Article PubMed
PubMed Central Google Scholar * Olivieri F, Prattichizzo F, Giuliani A, Matacchione G, Rippo MR, Sabbatinelli J, et al. miR-21 and miR-146a: the microRNAs of inflammaging and age-related
diseases. Ageing Res Rev. 2021;70:101374. Article CAS PubMed Google Scholar * Rhim J, Baek W, Seo Y, Kim JH. From molecular mechanisms to therapeutics: understanding microRNA-21 in
cancer. Cells. 2022;11:2791. * Chen P, Jiang P, Chen J, Yang Y, Guo X. XIST promotes apoptosis and the inflammatory response in CSE-stimulated cells via the miR-200c-3p/EGR3 axis. BMC Pulm
Med. 2021;21:215. Article PubMed PubMed Central Google Scholar * Ding LN, Yu YY, Ma CJ, Lei CJ, Zhang HB. SOX2-associated signaling pathways regulate biological phenotypes of cancers.
Biomed Pharmacother. 2023;160:114336. Article CAS PubMed Google Scholar * Asem MS, Buechler S, Wates RB, Miller DL, Stack MS Wnt5a signaling in cancer. Cancers (Basel). 2016;8:79. *
Smith NE, Spencer-Merris P, Fox AH, Petersen J, Michael MZ. The long and the short of it: NEAT1 and cancer cell metabolism. Cancers. 2022;14. * Zhang Y, Yang M, Yang S, Hong F. Role of
noncoding RNAs and untranslated regions in cancer: a review. Medicine. 2022;101:e30045. * He Y, Du X, Chen M, Han L, Sun J. Novel insight into the functions of N(6)‑methyladenosine modified
lncRNAs in cancers (Review). Int J Oncol. 2022;61:152. * Xie Z, Zhang H, Wang J, Li Z, Qiu C, Sun K. LIN28B-AS1-IGF2BP1 association is required for LPS-induced NFkappaB activation and
pro-inflammatory responses in human macrophages and monocytes. Biochem Biophys Res Commun. 2019;519:525–32. Article CAS PubMed Google Scholar * Xie J, Li Q, Zhu XH, Gao Y, Zhao WH.
IGF2BP1 promotes LPS-induced NFkappaB activation and pro-inflammatory cytokines production in human macrophages and monocytes. Biochem Biophys Res Commun. 2019;513:820–6. * Feng Y, Dong H,
Zheng L. Ligustrazine inhibits inflammatory response of human endometrial stromal cells through the STAT3/IGF2BP1/RELA axis. Pharm Biol. 2023;61:666–73. Article CAS PubMed PubMed Central
Google Scholar * Ke H, Zhao L, Zhang H, Feng X, Xu H, Hao J, et al. Loss of TDP43 inhibits progression of triple-negative breast cancer in coordination with SRSF3. PNAS USA.
2018;115:E3426–E35. Article CAS PubMed PubMed Central Google Scholar * Guo L, Ke H, Zhang H, Zou L, Yang Q, Lu X, et al. TDP43 promotes stemness of breast cancer stem cells through CD44
variant splicing isoforms. Cell Death Dis. 2022;13:428. * Bhattacharya A, Fushimi A, Yamashita N, Morimoto Y, Ishikawa S, Daimon T, et al. MUC1-C activates the SET1A/WDR5 compass complex
and H3K4 trimethylation in human cancer cells. Commun Biol. 2023;6:1030. Article CAS PubMed PubMed Central Google Scholar * Huang L, Liao X, Beckett M, Li Y, Khanna KK, Wang Z, et al.
MUC1-C oncoprotein interacts directly with ATM and promotes the DNA damage response to ionizing radiation. Genes Cancer. 2010;1:239–50. * Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang
JE, et al. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc Natl Acad Sci USA. 2011;108:3665–70. * Rajabi H, Hata T, Li W, Long M, Hu Q, Liu S, et
al. MUC1-C represses the RASSF1A tumor suppressor in human carcinoma cells. Oncogene. 2019;38:7266–77. * Hata T, Rajabi H, Takahashi H, Yasumizu Y, Li W, Jin C, et al. MUC1-C activates the
NuRD complex to drive dedifferentiation of triple-negative breast cancer cells. Cancer Res. 2019;79:5711–22. Article CAS PubMed PubMed Central Google Scholar * Buenrostro JD, Wu B,
Chang HY, Greenleaf WJ. ATAC-seq: a method for assaying chromatin accessibility genome-wide.Curr Protoc Mol Biol. 2015;109:1–9. Download references FUNDING Funding Research reported in this
publication was supported by the National Cancer Institute of the National Institutes of Health under award numbers CA97098, CA262991, and CA233084. AUTHOR INFORMATION Author notes * These
authors contributed equally: Keyi Wang, Atrayee Bhattacharya. AUTHORS AND AFFILIATIONS * Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA, USA Keyi Wang, Atrayee
Bhattacharya, Naoki Haratake, Tatsuaki Daimon, Ayako Nakashoji, Hiroki Ozawa & Donald Kufe * Department of Urology, Shanghai Tenth People’s Hospital, Tongji University, Shanghai, China
Bo Peng & Wei Li Authors * Keyi Wang View author publications You can also search for this author inPubMed Google Scholar * Atrayee Bhattacharya View author publications You can also
search for this author inPubMed Google Scholar * Naoki Haratake View author publications You can also search for this author inPubMed Google Scholar * Tatsuaki Daimon View author
publications You can also search for this author inPubMed Google Scholar * Ayako Nakashoji View author publications You can also search for this author inPubMed Google Scholar * Hiroki Ozawa
View author publications You can also search for this author inPubMed Google Scholar * Bo Peng View author publications You can also search for this author inPubMed Google Scholar * Wei Li
View author publications You can also search for this author inPubMed Google Scholar * Donald Kufe View author publications You can also search for this author inPubMed Google Scholar
CONTRIBUTIONS Conceptualization, KW, AB, DK; Methodology, KW, AB; Investigation, KW, AB, NH, TD, AN, HO, BP; Bioinformatics analysis, KW, AB; Writing-original draft, DK; Writing-review and
editing, KW, AB, DK; Funding acquisition, WL, DK. CORRESPONDING AUTHORS Correspondence to Wei Li or Donald Kufe. ETHICS DECLARATIONS COMPETING INTERESTS DK has equity interests in Genus
Oncology and is a paid consultant to CanBas. The other authors declared no potential conflicts of interest. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with
regard to jurisdictional claims in published maps and institutional affiliations. Edited by Roberto Mantovani SUPPLEMENTARY INFORMATION SUPPLEMENTAL MATERIAL ORIGINAL DATA FILE RIGHTS AND
PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any
medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The
images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not
included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly
from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Wang, K.,
Bhattacharya, A., Haratake, N. _et al._ XIST and MUC1-C form an auto-regulatory pathway in driving cancer progression. _Cell Death Dis_ 15, 330 (2024).
https://doi.org/10.1038/s41419-024-06684-9 Download citation * Received: 10 October 2023 * Revised: 10 April 2024 * Accepted: 15 April 2024 * Published: 13 May 2024 * DOI:
https://doi.org/10.1038/s41419-024-06684-9 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative