Play all audios:
ABSTRACT Gallbladder cancer (GBC) is the most common malignant tumor of the biliary tract and is often prone to early distant metastasis. However, the mechanisms underlying GBC’s invasive
metastasis remain unclear. This study identified that F-box only protein 33 (FBXO33) expression is significantly elevated in GBC and is negatively associated with patient prognosis. In vivo
and in vitro experiments demonstrated that knockdown of FBXO33 inhibits epithelial-mesenchymal transition (EMT) progression in GBC, while overexpression of FBXO33 promotes EMT progression.
Mechanistically, FBXO33 regulates EMT progression by modulating the polyubiquitination of p53 at K291 and K292. Moreover, the upregulation of FBXO33 in GBC is driven by transcriptional
regulation mediated by Yin Yang-1 (YY1). The lactylation modification of YY1 at K183 was found to be essential for the transcriptional activation of FBXO33. These findings underscore the
role of the lactylation-driven FBXO33-p53 axis in promoting the invasive metastasis of GBC. SIMILAR CONTENT BEING VIEWED BY OTHERS THE E3 UBIQUITIN LIGASE, FBXW5, PROMOTES THE MIGRATION AND
INVASION OF GASTRIC CANCER THROUGH THE DYSREGULATION OF THE HIPPO PATHWAY Article Open access 24 February 2022 FBXO28 SUPPRESSES LIVER CANCER INVASION AND METASTASIS BY PROMOTING
PKA-DEPENDENT SNAI2 DEGRADATION Article Open access 18 August 2023 DEUBIQUITINASE USP39 AND E3 LIGASE TRIM26 BALANCE THE LEVEL OF ZEB1 UBIQUITINATION AND THEREBY DETERMINE THE PROGRESSION OF
HEPATOCELLULAR CARCINOMA Article Open access 01 March 2021 INTRODUCTION GBC is the most common malignant tumor of the biliary tract, characterized by a high mortality rate and poor
prognosis [1, 2]. Despite extensive research, no significant improvement in GBC prognosis has been achieved [3]. Early metastasis is a hallmark of GBC, often making surgical treatment
unfeasible at diagnosis [4]. Thus, identifying effective early biomarkers and therapeutic targets for GBC is essential. Studies suggest that EMT plays a critical role in initiating distant
metastasis in malignant tumors [5]; however, the molecular mechanisms driving EMT in GBC remain unclear. This highlights an urgent need to identify novel EMT-related biomarkers to improve
patient outcomes. Protein ubiquitination involves the covalent attachment of a 76-amino-acid ubiquitin protein to target proteins, mediated by ubiquitin-activating enzymes (E1),
ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3) [6, 7]. This process regulates the biological functions of target proteins and is integral to key signaling pathways,
influencing metastasis and treatment responses in malignant tumors [8, 9]. The F-box family of E3 ubiquitin ligases has been shown to play a pivotal role in EMT progression [10]. For
example, FBXO11 targets Snail for ubiquitination and degradation, thereby inhibiting EMT and metastasis in breast cancer models [11], and also regulates EMT via ubiquitination of SAMD1 [12].
Conversely, FBXO32 facilitates EMT by ubiquitinating and degrading substrates such as CTBP1 and PTEN [13, 14]. Recent studies have identified that FBXO33, a member of the F-box family, acts
as a tumor suppressor by promoting ubiquitination and degradation of MYC, thereby inhibiting metastasis in non-small cell lung cancer [15]. However, the role of FBXO33 in other tumor types,
including GBC, remains unclear. This study demonstrates that FBXO33, a novel E3 ubiquitin ligase, is significantly upregulated in GBC. The effects of FBXO33 on EMT in GBC and its underlying
mechanisms were evaluated. The findings indicate that FBXO33 functions as an oncogene in GBC. Overexpression of FBXO33 promotes EMT progression, while its downregulation inhibits EMT
progression. Mechanistically, ubiquitination at K291 and K292 of p53 is critical for FBXO33-mediated regulation of EMT in GBC. Furthermore, transcriptional activation of FBXO33 in GBC
depends on regulation by the transcription factor YY1. This activation is further reliant on the lactylation modification at K183 of the YY1 protein. MATERIALS AND METHODS PATIENT DATA
COLLECTION Clinical samples were collected in strict accordance with the approved protocol by the Ethics Committee of Fujian Medical University’s Medical College, China. Eighty-two
paraffin-embedded tumor specimens diagnosed as GBC by the Clinical Pathology Department of Fujian Medical University Union Hospital from January 2013 to January 2024, along with paired
benign tissues, were collected. Informed consent was obtained from all patients. None of the patients received any radiotherapy or chemotherapy prior to tumor resection. CELL LINES AND CELL
CULTURE Human GBC cell lines GBC-SD and SGC-996 were purchased from the Shanghai Institute of Life Sciences, China. The 293T cell line was provided by the Department of Oncology, Tongji
University School of Medicine, China. All cell lines underwent mycoplasma detection and STR cell identification to ensure their authenticity and purity. All cells were maintained in
high-glucose Dulbecco’s Modified Eagle Medium (DMEM, Hyclone, USA) supplemented with 10% fetal bovine serum (FBS, Hyclone, USA) and cultured at 37 °C in a humidified atmosphere containing 5%
CO2. All cells were tested negative for mycoplasma contamination. NEXT-GENERATION SEQUENCING Three random 0.8 mm tumor tissue cores were selected to collect tumor DNA. For samples with low
tumor volume, 5-10 tissue slides were used to microdissect the selected tumor regions for DNA enrichment. Although DNA was isolated from the same tumor block, the p53 IHC slides were not
used to guide the microdissection areas. The samples were sequenced using the AmpliSeq Cancer Hotspot Panel version 5. All p53 mutation calls were cross-referenced against publicly available
databases (ClinVar, available at https://www.ncbi.nlm.nih.gov/clinvar/ and COSMIC, available at https://cancer.sanger.ac.uk/cosmic) [16, 17]. The criteria for mutation reporting were a
minimum read depth of 500, an allele ratio ≥5%, a base quality score ≥30, a probability score for single nucleotide changes ≥0.90, and a quality score for insertions/deletions ≥1000.
REAL-TIME QUANTITATIVE POLYMERASE CHAIN REACTION (RT-QPCR) Total RNA was extracted from GBC tissues or cells using TRIzol (15596-026, Invitrogen, USA). mRNA was reverse transcribed using the
All-In-One 5×RT MasterMix (Abm, Canada). Subsequently, PCR was performed using Fast Start Universal SYBR Green Master Mix (Roche, Basel, Switzerland) and fluorescence quantification was
conducted according to the operation manual of the ABI 7500 real-time system. GAPDH was used as an internal control for normalization. The relative expression levels of mRNA were evaluated
using the 2 − ΔΔCt method. Detailed information on primer sequences is provided in Table 1. WESTERN BLOT ANALYSIS (WB) AND ANTIBODIES Total protein was extracted from GBC tissues or cells
using pre-cooled RIPA (Radioimmunoprecipitation assay) buffer (Beyotime, Shanghai, China). Subsequently, the protein lysates were quantified using the BCA (Bicinchoninic acid) protein assay
kit (Thermo Fisher Scientific). After separating the proteins by SDS-PAGE electrophoresis, they were transferred onto PVDF membranes. The membranes were then blocked with 5% non-fat milk for
2 h at room temperature and incubated with primary antibodies overnight at 4 °C. After incubation with secondary antibodies at room temperature for 1 h, the antibody-antigen complexes were
detected using the ECL assay kit (Advansta, USA). The following primary antibodies were purchased: rabbit anti-YY1 (ab227269), rabbit anti-YY1 (ab109237), rabbit anti-p53 (ab32049), mouse
anti-p53 (ab308609), rabbit anti-Flag (ab205606), mouse anti-Flag (ab125243), rabbit anti-His (ab213204), mouse anti-His (ab18184), rabbit anti-HA (ab9110), rabbit anti p65(ab32536), rabbit
anti Myc-tag(ab9106), mouse anti Myc-tag(ab32), rabbit anti-Ubiquitin (ab134953) from Abcam; lactylation pan-antibody (PTM-1401RM) from PTM BIO (Hangzhou, China); rabbit anti-FBXO33
(PA5-61275) from Thermo Fisher Scientific; rabbit anti-E-Cadherin (#3195), rabbit anti-Vimentin (#5741), rabbit secondary antibody (#7074), and mouse secondary antibody (#7076) from Cell
Signaling Technology. Rabbit anti-GAPDH (#5174) was used as an internal control for normalization. CHEMICAL INHIBITORS The following exogenous drugs used in this study were purchased from
MCE (MedChemExpress, USA): MG132 (HY-13259), Cycloheximide (CHX, HY-12320), and Lactate (HY-B2227). IMMUNOHISTOCHEMICAL ANALYSIS (IHC) Paraffin-embedded sections were deparaffinized in
xylene, dehydrated in graded ethanol, and subjected to antigen retrieval. The sections were washed with TBS-T and quenched in a 3% hydrogen peroxide TBS solution for 10 minutes to block
endogenous peroxidase/biotin activity. Non-specific binding was blocked with a serum-free blocking reagent, followed by overnight incubation with primary antibodies. Primary antibodies
specific for FBXO33 (1:500, Thermo Fisher Scientific), p53 (1:400, Abcam), E-Cadherin (1:200, #3195), and Vimentin (1:300, #5741) were incubated overnight at 4 °C. The sections were then
incubated with secondary antibodies for 20 min at room temperature in a humid chamber. Visualization was achieved using DAB (3,3’-Diaminobenzidine) solution, and the sections were
counterstained with hematoxylin. Digital pathology slide scanner (Olympus VS200, Japan) was used to scan the images, and Image-Pro Plus 6.0 (Media Cybernetics, USA) was employed for
analysis. The quantification of immunohistochemical evaluation was based on the product of staining intensity and the percentage of positively stained cells. The percentage of positively
stained cells was scored on a scale from 0 to 4 (0 = 0–10%; 1 = 11–25%; 2 = 26–50%; 3 = 51–75%; 4 = 76–100%), while staining intensity was rated on a scale from 0 to 3 (0 = no staining; 1 =
weak; 2 = moderate; 3 = strong). The overall protein expression in each sample was expressed as histoscore, which was multiplication product of the staining intensity score(0–3) and
percentage of positively stained cells score(0–4) and is between 0 and 12. Low expression is scored 0–4. High expression is scored 6–12. The staining score was evaluated by two independent
pathologists. LENTIVIRUS CONSTRUCTION AND TRANSFECTION Lentiviral particles carrying Flag-tagged FBXO33-GFP (Flag-FBXO33) and its control vector lentivirus (Vector 1), as well as lentiviral
particles containing the YY1 gene coding region for overexpression (OE-YY1) and its control vector lentivirus (Vector 2), were obtained from GENE (Shanghai, China). GBC cells were infected
with lentivirus for 24 hours. Stable clones were selected with puromycin (5 μg/ml; Sigma). Lentivector-mediated short-hairpin FBXO33 (sh-FBXO33) and non-targeting plasmids (sh-Scr 1) and
lentivector-mediated short-hairpin YY1 (sh-YY1) and non-targeting plasmids (sh-Scr 2)were designed and synthesized by GENE (Shanghai, China). Stable transfer GBC cells with FBXO33 and YY1
knockdown were generated in the same way. PLASMID TRANSFECTION All plasmids used for cell transfection were obtained from GENE (Shanghai, China). Cells were seeded in 6-well plates or 10 cm2
culture dishes. When cells reached 70–80% confluence, transfection was performed using Lipofectamine 3000 reagent (Invitrogen Thermo Fisher Scientific, USA) according to the manufacturer’s
instructions. Cells were collected 48 h post-transfection for subsequent experiments. GENERATION OF P53-KNOCKOUT (P53-KO) GBC CELL LINES p53-knockout GBC cell lines were constructed using
the CRISPR–Cas9 gene-editing system. Lentiviruses containing Cas9-guide RNA targeting sequences were designed and synthesized by GENE (Shanghai, China). Lentivirus infection was performed on
GBC cells at 80% confluency. The cells were selected after culture for one week in a medium containing 5 μg/ml puromycin. Monocolonies were picked, and the knockout efficiency was
determined by western blotting. The sequence of single guide RNAs (sgRNAs) targeting human p53: 5ʹ-CCATTGTTCAATATCGTCCG-3ʹ and 5ʹ-CGGACGATATTGAACAATGG-3ʹ. The non-specific control sgRNA in
the same vector were defined as Vector-KO cells. DUAL-LUCIFERASE REPORTER ASSAY Bioinformatics analysis based on the JASPAR database indicated binding sites for YY1 in the FBXO33 promoter
region. Plasmids containing the wild-type (WT) and mutant (MUT) FBXO33 promoter regions were obtained from GENE (Shanghai, China) based on the sequences of these binding sites. The WT and
MUT FBXO33 promoter region plasmids were separately transfected into different experimental and corresponding control group cells. After 48 h, firefly luciferase (F-luc) activity was
measured using the Dual-Luciferase Reporter Assay System (Promega E2920, USA) according to the manufacturer’s instructions, with Renilla luciferase (R-luc) used as an internal control for
normalization. XENOGRAFT TUMOR MODEL Female athymic BALB/c nude mice (4–6 weeks old) were purchased from SIPEIFU (Beijing, China). All mice were housed under specific pathogen-free
conditions, strictly in accordance with the approved protocol of the Ethical Committee of Fujian Medical University’s Medical College (Approval No: IACUC FJMU 2023-0197). Mice were randomly
divided into experimental groups. GBC SGC-996 cells (sh-FBXO33 group and corresponding sh-Scr group (4–6 weeks old, _n_ = 6) or Flag-FBXO33 group and corresponding Vector group (4–6 weeks
old, _n_ = 5), 1 × 107 cells per group) were suspended in 100 μl of sterile PBS. The cells were subcutaneously implanted into the right hind limb of the mice using a syringe to establish the
xenograft tumor model. Tumor growth was monitored weekly, and tumor volume was calculated using the formula: (length × width2)/2. After 4 weeks of tumor formation, the mice were euthanized,
and xenograft specimens were collected by surgical excision and weighed. PROTEIN STABILITY CHX (final concentration 50 μg/ml) was added to the cells. After specified incubation times,
protein levels of p53 were assessed using western blotting. CHROMATIN IMMUNOPRECIPITATION (CHIP-QPCR) ANALYSIS The ChIP assay was conducted using a SimpleChIP Plus Enzymatic Chromatin IP Kit
(CST, 9004S). Briefly, GBC cells (1 × 107 cells per group) were prepared according to the kit instructions and resuspended in ChIP buffer. Chromatin DNA fragments were enzymatically
sheared, and antibody-magnetic bead complexes were used to pull down DNA fragments at 4 °C. Target DNA fragments were then detected using RT-qPCR. CO-IMMUNOPRECIPITATION (CO-IP) When cell
confluency reached 80–90% in 10 cm2 dishes, Co-IP was performed according to the instructions of the protein immunoprecipitation kit (Geneseed, Guangzhou, China). In brief, cell lysates were
prepared and protein concentrations were quantified using BCA protein assay. Subsequently, 1 mg of total protein was incubated overnight at 4 °C with antibody-magnetic bead complexes. The
following day, antibody-antigen complexes were eluted, and subsequent protein blotting was performed for relevant detection. Anti-rabbit IgG for IP (RA1009-01) was purchased from Vazyme
(Nanjing, China) to prevent detection of IP antibody heavy chains (55 kDa). STATISTICAL ANALYSIS In this study, all statistical analyses were performed using SPSS 19.0 software package (SPSS
Inc., Chicago, USA) and GraphPad Prism 8 software (GraphPad, USA). Clinical pathological data analysis employed chi-square analysis and Pearson correlation analysis. For two-group
comparisons, we employed the t-test to evaluate the significance of differences. When analyzing differences among multiple groups, we utilized analysis of variance (One-way ANOVA). All data
are presented as mean ± standard error of the mean (SEM) of independent replicates (_n_ ≥ 3). Differences with _p_ < 0.05 were considered statistically significant. RESULTS HIGH
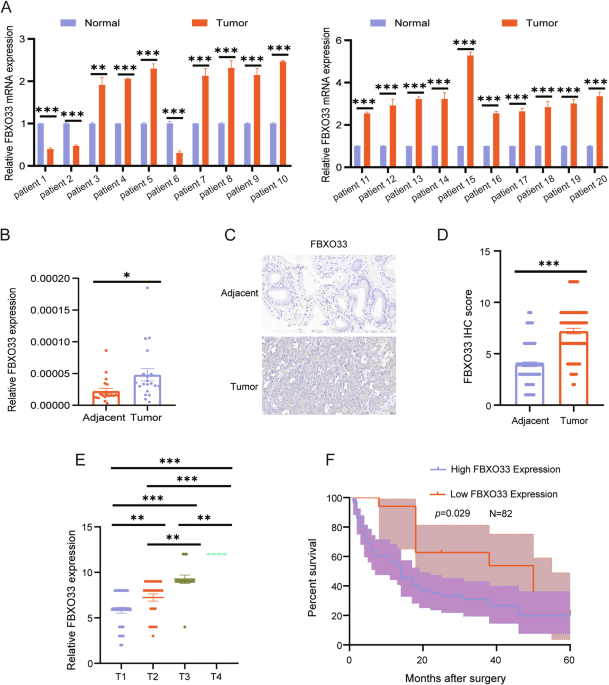
EXPRESSION OF FBXO33 IN GBC IS NEGATIVELY CORRELATED WITH OVERALL SURVIVAL IN GBC To investigate the role of FBXO33 in GBC, mRNA expression levels of FBXO33 were measured in 20 paired GBC
tumor and adjacent gallbladder tissues using RT-qPCR. The results revealed that FBXO33 expression was significantly higher in GBC tissues compared to adjacent tissues (Fig. 1A, B).
Additionally, IHC staining was performed to assess protein expression levels of FBXO33 in 82 paired GBC tumor and adjacent tissues. The findings confirmed that FBXO33 expression was elevated
in cancer tissues compared to adjacent tissues (Fig. 1C, D). FBXO33 expression was positively associated with distant metastasis and TNM staging but showed no significant correlation with
gender, age, or the presence of gallstones (Table 2). Moreover, FBXO33 expression progressively increased from T1 to T4 stages of GBC (Fig. 1E). Kaplan-Meier survival analysis demonstrated
that GBC patients with low FBXO33 expression had higher overall survival rates compared to those with high FBXO33 expression (Fig. 1F). These results indicate that FBXO33 acts as an oncogene
and is highly expressed in GBC. FBXO33 REGULATES THE PROGRESSION OF EMT IN GBC IN VIVO AND IN VITRO To clarify the role of FBXO33 in GBC invasion and metastasis, GBC cells with FBXO33
knockdown (sh-FBXO33) or overexpression (Flag-FBXO33) were constructed using lentiviral vectors (Fig. 2A, B). Scratch and Transwell assays revealed that cell migration was significantly
reduced in the sh-FBXO33 group compared to the sh-Scr control group (Fig. 2C, D), while migration was significantly enhanced in the Flag-FBXO33 group compared to the Vector control group
(Fig. 2E, F). Western blot analysis showed that in the sh-FBXO33 group, E-Cadherin expression was significantly upregulated, and Vimentin expression was significantly downregulated compared
to the sh-Scr group (Fig. 2G and Supplementary Fig. S1A). Conversely, in the Flag-FBXO33 group, E-Cadherin expression was significantly downregulated, and Vimentin expression was
significantly upregulated compared to the Vector group (Fig. 2H and Supplementary Fig. S1B). To further validate these findings, a subcutaneous xenograft model of GBC was established using
SGC-996 cells with either FBXO33 knockdown or overexpression and their respective control cells. Tumors in the sh-FBXO33 group were smaller and lighter compared to those in the sh-Scr group,
indicating that FBXO33 knockdown inhibited GBC tumor growth (Fig. 2I–K). In contrast, tumors in the Flag-FBXO33 group were larger and heavier, suggesting that FBXO33 overexpression promoted
tumor growth (Supplementary Fig. S1C–E). IHC analysis of subcutaneous xenografts confirmed that E-Cadherin expression was significantly increased and Vimentin expression significantly
decreased in the sh-FBXO33 group compared to the sh-Scr group (Fig. 2L). Conversely, E-Cadherin expression was significantly reduced and Vimentin expression significantly elevated in the
Flag-FBXO33 group compared to the Vector group (Supplementary Fig. S1F). In summary, these findings demonstrate that FBXO33 regulates the progression of EMT in GBC both in vivo and in vitro.
FBXO33 INTERACTS WITH P53 E3 ubiquitin ligases perform their biological functions by recognizing and regulating substrates [18], and FBXO33 is a member of this family. To understand the
mechanism by which FBXO33 functions in GBC, the online tool Ubibrowser was used to predict potential substrates of FBXO33 [19]. Among the predicted substrates, RELA (p65) had the highest
confidence score, followed by p53 (Fig. 3A and Supplementary Fig. S2A). Substrate ubiquitination is essential for understanding the function of E3 ubiquitin ligases, as well as their
substrate recognition and regulatory roles [20]. To investigate the mechanism of FBXO33, Co-IP experiments were performed to assess the effect of Flag-FBXO33 on the ubiquitination of p65 and
p53. The results showed that Flag-FBXO33 significantly increased the ubiquitination level of p53 compared to the Vector group, while it had minimal effect on p65 (Supplementary Fig. S2B,
C). Thus, p53 was selected as the substrate of FBXO33 for further investigation. Co-IP experiments confirmed that FBXO33 binds to p53 (Fig. 3B, C). Given that many E3 ubiquitin ligases can
target both wild-type p53 (wtp53) and mutant p53 (mutp53) [21], we investigated whether FBXO33 exhibits the same behavior. The p53 gene in GBC cell lines remained unmutated [22], indicating
that the p53 interacting with FBXO33 in GBC-SD and SGC-996 cells is wtp53. Using CRISPR/Cas9, the endogenous p53 gene was knocked out in GBC-SD and SGC-996 cells (Supplementary Fig. S2D, E),
followed by transfection with plasmids encoding Myc-tagged wtp53 and mutp53 variants. Co-IP experiments showed that Flag-FBXO33 significantly increased the ubiquitination level of wtp53 but
had minimal effect on common mutp53 variants (p53R175H, p53R248W, and p53R273H) [23] (Supplementary Fig. S2F, G). Therefore, wtp53 was selected as the substrate of FBXO33 for further
analysis. To identify the interaction domains of FBXO33 and p53, truncated forms of p53 (WT-p53, ▲1-100-p53, ▲100–293-p53, ▲293-393-p53) (Fig. 3D) and FBXO33 (WT-FBXO33, ▲1-111-FBXO33,
▲111-555-FBXO33) were constructed (Fig. 3E). Co-IP results demonstrated that His-WT-p53, His-▲1-100-p53, and His-▲293-393-p53 could bind to Flag-FBXO33, while His-▲100–293-p53 lost this
ability, indicating that the 100–293 domain of p53 is crucial for binding to FBXO33 (Fig. 3F, G). Similarly, Flag-WT-FBXO33 and Flag-▲1-111-FBXO33 could bind to His-p53, while
Flag-▲111-555-FBXO33 lost this ability, showing that the 111-555 domain of FBXO33 is essential for binding to p53 (Fig. 3H, I). These findings confirm that FBXO33 interacts with p53. FBXO33
REGULATES THE STABILITY OF THE P53 PROTEIN Western blot analysis revealed that sh-FBXO33 significantly increased p53 protein expression (Fig. 4A and Supplementary Fig. S3A), while
Flag-FBXO33 significantly decreased it (Fig. 4B and Supplementary Fig. S3B). RT-qPCR results indicated that neither sh-FBXO33 nor Flag-FBXO33 altered p53 mRNA expression (Fig. 4C, D),
suggesting that FBXO33 regulates p53 protein expression at the post-translational level. Proteasome inhibitor MG132 alleviated the inhibitory effect of Flag-FBXO33 on p53, indicating that
FBXO33 affects p53 protein expression through the ubiquitin-proteasome pathway (Fig. 4E and Supplementary Fig. S3C). To further evaluate the impact of FBXO33 on p53 protein stability,
cycloheximide (CHX) was used to block protein synthesis. The CHX assay showed that p53 protein had a longer half-life in sh-FBXO33 GBC cells (Fig. 4F, G) and a shorter half-life in
Flag-FBXO33 GBC cells (Fig. 4H, I). These results demonstrate that FBXO33 regulates p53 protein stability via the ubiquitin-proteasome pathway. REGULATION OF P53 UBIQUITINATION BY FBXO33
This study examined how FBXO33 regulates the stability of p53 protein. Co-IP assays were performed to assess endogenous p53 polyubiquitination levels. The results showed that sh-FBXO33
significantly reduced p53 polyubiquitination after MG132 treatment (Fig. 5A, B), whereas Flag-FBXO33 markedly increased p53 polyubiquitination (Supplementary Fig. S2B, C). Furthermore,
deletion of the 111-555 domain of FBXO33 abolished its ability to inhibit p53 (Fig. 5C, D), indicating that this domain is essential for promoting p53 polyubiquitination. Key ubiquitination
sites within the 100–293 domain of p53 protein, lysine-291 (K291) and lysine-292 (K292), were identified [24]. To evaluate the roles of these sites in FBXO33-mediated p53 polyubiquitination,
lysine-to-arginine mutations were introduced, resulting in His-K291R-p53 and His-K292R-p53 mutant plasmids. His-WT-p53, His-K291R-p53, and His-K292R-p53 were transfected into Flag-FBXO33
and Vector control cells. Western blot analysis revealed that K291R and K292R mutations reduced the inhibitory effect of Flag-FBXO33 on p53 protein (Fig. 5E). Co-IP experiments confirmed
that these mutations weakened the promotion of p53 polyubiquitination by Flag-FBXO33 (Fig. 5E). These findings demonstrate that K291 and K292 are critical for FBXO33-mediated regulation of
p53 protein. P53 REGULATES THE FUNCTION OF FBXO33 IN GBC AND IS NEGATIVELY CORRELATED WITH FBXO33 EXPRESSION To assess whether p53 contributes to FBXO33-mediated regulation of GBC cell
migration and EMT, His-WT-p53, His-K291R-p53, and His-K292R-p53 were transfected into Flag-FBXO33 group cells. Scratch assays revealed that cell migration was significantly reduced in the
Flag-FBXO33+His-WT-p53 group compared to the Flag-FBXO33 group. Further reductions in migration were observed in the Flag-FBXO33+His-K291R-p53 and Flag-FBXO33+His-K292R-p53 groups compared
to the Flag-FBXO33+His-WT-p53 group (Fig. 6A, B). Similar trends were observed in Transwell assays (Fig. 6C, D). Western blot analysis showed increased E-Cadherin expression and decreased
Vimentin expression in the Flag-FBXO33+His-WT-p53 group compared to the Flag-FBXO33 group. These changes were further amplified in the Flag-FBXO33+His-K291R-p53 and Flag-FBXO33+His-K292R-p53
groups compared to the Flag-FBXO33+His-WT-p53 group (Fig. 6E). These findings suggest that His-WT-p53 reduces the promotive effects of Flag-FBXO33 on GBC cell migration and EMT. Mutations
at K291 and K292 (His-K291R-p53 and His-K292R-p53) further attenuated the inhibitory effects of Flag-FBXO33 on p53, resulting in significantly enhanced anti-cancer activity of p53 in these
mutant groups compared to His-WT-p53. In conclusion, FBXO33 regulates GBC cell migration and EMT through p53, with K291 and K292 ubiquitination sites of p53 playing critical roles in this
process. The experimental results confirm that FBXO33 regulates GBC cell migration and EMT through p53. To further validate this conclusion, Western blot was used to analyze the protein
expression levels of FBXO33 and p53 in 15 GBC patients, regardless of the mutational status of p53. In 11 cases, FBXO33 was highly expressed in cancer tissues compared to adjacent tissues,
while p53 was more highly expressed in adjacent tissues than in cancer tissues. Conversely, in 3 cases, FBXO33 was more highly expressed in adjacent tissues, while p53 was more highly
expressed in cancer tissues (Fig. 6F). IHC analysis of 30 consecutive GBC pathological sections revealed that high expression of FBXO33 in GBC tissues corresponded to low p53 expression, and
vice versa (Fig. 6G). Correlation analysis indicated a negative relationship between the expression of FBXO33 and p53 (Fig. 6H). When examining the mutational status of p53 in 82 GBC
patients (Fig. 1C), Kaplan-Meier curves showed that patients with wtp53 had higher overall survival rates compared to those with mutp53 (Supplementary Fig. S4A). These findings align with
the results reported by Sunwang Xu et al. [22]. Correlation analysis revealed a negative association between FBXO33 and wtp53 expression (Supplementary Fig. S4B), but no correlation between
FBXO33 and mutp53 expression (Supplementary Fig. S4C). Moreover, high FBXO33 expression was significantly associated with poor prognosis in patients with wtp53 but showed no association with
prognosis in patients with mutp53 (Supplementary Fig. S4D, E). These results suggest that wtp53 regulates the function of FBXO33 in GBC and is negatively correlated with its expression.
TRANSCRIPTION FACTOR YY1 PROMOTES FBXO33 EXPRESSION IN GBC The results demonstrate that FBXO33 is significantly upregulated in GBC tissues compared to adjacent tissues and regulates EMT
progression, indicating its potential as a therapeutic target for GBC. To explore the regulatory mechanism of FBXO33 expression, the promoter sequence of the human FBXO33 gene (2000 bp
upstream of the transcription start site) was obtained from the NCBI database. Potential transcription factors for FBXO33 were predicted using the PROMO, Harmonizome, and GTRD databases.
When the PROMO database was set to 0% Maximum matrix dissimilarity rate, YY1 was identified as the only intersecting transcription factor across all three databases (Fig. 7A). To evaluate
whether YY1 regulates FBXO33 expression in GBC, GBC cells with YY1 knockdown (sh-YY1) and overexpression (OE-YY1) were constructed (Fig. 7B, C). RT-qPCR analysis revealed that sh-YY1
significantly reduced FBXO33 mRNA expression (Fig. 7D), while OE-YY1 significantly increased it (Fig. 7E). Western blot results confirmed similar effects at the protein level (Fig. 7F, G).
To further investigate, a wild-type plasmid (WT-PRO) containing the potential YY1 binding sequence in the FBXO33 promoter and a mutant plasmid (MUT-PRO) with a mutated binding sequence were
constructed (Fig. 7H). Dual-luciferase reporter assays demonstrated that fluorescence intensity in the WT-PRO group was higher than in the MUT-PRO group (Fig. 7I, J). Furthermore, sh-YY1
reduced fluorescence intensity in the WT-PRO group but had no significant effect on the MUT-PRO group (Fig. 7I). Conversely, OE-YY1 increased fluorescence intensity in the WT-PRO group but
had no significant effect on the MUT-PRO group (Fig. 7J). ChIP-qPCR analysis showed that YY1 was enriched in the FBXO33 promoter region. Knockdown of YY1 significantly reduced enrichment,
while overexpression of YY1 significantly increased it (Fig. 7K, L). These results confirm that in GBC, YY1 promotes the transcriptional activation of FBXO33. TRANSCRIPTIONAL ACTIVATION OF
FBXO33 BY YY1 IN GBC DEPENDS ON YY1 LACTYLATION MODIFICATION Protein lactylation modification (Kla) is a recently identified post-translational modification that plays a vital role in
regulating gene expression [25]. Notably, YY1 is one of the proteins susceptible to lactylation modification [26], with the lactylation site K183 playing a critical role in its
transcriptional function [27]. Based on this, it was hypothesized that the transcriptional activation of FBXO33 by YY1 is also regulated by lactylation modification. To test this hypothesis,
experiments were conducted. RT-qPCR and Western blot analysis showed that exogenous lactate did not alter the protein expression of YY1 (Fig. 8A) but significantly increased the mRNA and
protein expression of FBXO33 (Fig. 8B, C). Co-IP experiments demonstrated that lactate enhanced the lactylation level of YY1 (Fig. 8D, E). Since YY1 has only one lactylation site (K183)
[27], lysine K183 was mutated to arginine, creating a wild-type YY1 plasmid (Flag-WT-YY1) and a K183 mutant plasmid (Flag-K183R-YY1). Co-IP results indicated that mutation of the K183
lactylation site reduced the lactylation level of YY1 (Fig. 8F, G). Further, Western blot analysis revealed that in the Flag-K183R-YY1 group, exogenous lactate failed to increase FBXO33 mRNA
and protein expression, unlike in the Flag-WT-YY1 group (Fig. 8H, I). Dual-luciferase reporter assays confirmed that lactate significantly enhanced fluorescence intensity in the WT-PRO
group but had no significant effect on the MUT-PRO group (Fig. 8J). After K183 mutation, lactate could no longer increase fluorescence intensity in the WT-PRO group (Fig. 8J). ChIP-qPCR
analysis showed that lactate significantly elevated the enrichment level of YY1 at the FBXO33 promoter region (Fig. 8K), but this effect was lost after K183 mutation (Fig. 8K). In
conclusion, the K183 lactylation modification of YY1 enhances its ability to bind to the FBXO33 promoter region, thereby regulating the transcriptional activation of FBXO33. DISCUSSION In
recent years, studies have increasingly highlighted the critical role of protein ubiquitination in tumor development and progression [28, 29]. Targeting ubiquitin signaling pathways presents
significant potential for cancer therapy [20, 30]. A detailed understanding of ubiquitination targets and their specific mechanisms in GBC could serve as a foundation for developing novel
therapeutic strategies. Among E3 ubiquitin ligases, the F-box family has emerged as a key regulator of EMT progression and holds promise for cancer treatment [31, 32]. For instance, FBXO43,
which is highly expressed in various malignancies, promotes oncogenic activity by stabilizing SKP2, a cell cycle regulator [33]. Similarly, FBXO45 facilitates pancreatic cancer progression
by regulating the stability of the tumor suppressor USP49 [34]. FBXO33, an E3 ubiquitin ligase associated with Cullin 1 [35], is a member of the F-box protein family, characterized by
uncharacterized domains [31, 36]. Experimental results in this study confirmed that FBXO33 is upregulated in GBC and positively correlates with distant metastasis and TNM staging. Knockdown
of FBXO33 inhibits EMT progression in GBC, while its overexpression promotes EMT. These findings demonstrate that FBXO33 functions as an oncogene in GBC. E3 ubiquitin ligases link target
proteins with specific E2 ubiquitin-conjugating enzymes [8, 37], modulating the biological functions of their substrates [38]. However, limited studies have explored the target proteins of
FBXO33, with only EIF3F [39],YBX1 [40], and MYC [15] confirmed as its ubiquitination targets. p53, one of the first identified tumor suppressors [41], has been extensively studied in cancer
research [42, 43]. When mutated, p53 loses its tumor-suppressive functions, and mutp53 gains oncogenic properties, promoting cancer cell survival, proliferation, and metastasis [44].
Mutations in p53 are common in GBC [45] and are associated with poorer patient prognoses [22]. The ubiquitination of p53 has been extensively studied in recent years. MDM2, a well-known
ubiquitin ligase, promotes p53 ubiquitination and represents a promising target for cancer therapy through the MDM2/p53 axis [46, 47]. Additionally, p53 has been identified as a substrate
for FBXL8 [48], VPRBP [49], TRIM31 [50], and SCML2 [51]-mediated ubiquitination. Interestingly, both wtp53 and mutp53 can undergo ubiquitination [52], with some E3 ubiquitin ligases
targeting both forms [53, 54], while others exhibit specificity for either wtp53 or mutp53 [55,56,57]. In this study, p53 was predicted to be a ubiquitination target of FBXO33 through the
Ubibrowser platform and subsequently confirmed by Co-IP experiments. Further analysis revealed that FBXO33’s effect on wtp53 is significantly stronger than on mutp53. Mechanistic studies
identified the DNA binding domain (DBD) of p53 as the critical region for FBXO33-mediated ubiquitination, with K291 and K292 in the DBD playing essential roles in regulating GBC EMT
progression. Analysis of tissue samples from GBC patients further confirmed the correlation between FBXO33 and wtp53, but not mutp53. It was also observed that high expression of FBXO33 is
negatively correlated with prognosis in patients with wtp53, whereas no such correlation was observed in patients with mutp53. The transcription factor YY1 regulates the expression of
various genes in malignant tumors by mediating enhancer-promoter interactions [58, 59]. Using predictions from multiple databases, potential binding sequences of YY1 were identified in the
promoter region of FBXO33. Experimental verification confirmed that YY1 acts as a transcription factor for the FBXO33 gene, partially explaining the high expression of FBXO33 in GBC. In the
context of protein post-translational modification, protein lactylation induced by tumor metabolite lactate plays a critical role in regulating protein function [60]. The K183 site on YY1 is
a key lactylation site [26, 27]. Dual-luciferase reporter assays and ChIP-qPCR analysis demonstrated that lactate enhances the enrichment of YY1 in the promoter region of FBXO33. Further
experiments confirmed that the binding ability of YY1 to the FBXO33 promoter region is strengthened following lactylation modification at K183, revealing a novel mechanism for the
transcriptional activation of FBXO33 in GBC. In conclusion, this study highlights the positive correlation between FBXO33 expression and the prognosis of GBC. As an oncogene, FBXO33 promotes
EMT progression by enhancing p53 ubiquitination at K291 and K292, leading to decreased p53 expression. Furthermore, YY1 activates FBXO33 transcription in GBC through K183 lactylation
modification, sustaining high FBXO33 expression in GBC. These findings elucidate the mechanism of action of FBXO33 in GBC and provide potential strategies for targeted ubiquitin signaling
therapy in GBC. DATA AVAILABILITY The datasets used and analyzed during the current study are available from the corresponding author on reasonable request. REFERENCES * Roa JC, Garcia P,
Kapoor VK, Maithel SK, Javle M, Koshiol J. Gallbladder cancer. Nat Rev Dis Primers. 2022;8:69. Article PubMed Google Scholar * Wang J, Ren M, Yu J, Hu M, Wang X, Ma W, et al. Single-cell
RNA sequencing highlights the functional role of human endogenous retroviruses in gallbladder cancer. EBioMedicine. 2022;85:104319. Article CAS PubMed PubMed Central Google Scholar * Li
Y, Yang B, Miao H, Liu L, Wang Z, Jiang C, et al. Nicotinamide N -methyltransferase promotes M2 macrophage polarization by IL6 and MDSC conversion by GM-CSF in gallbladder carcinoma.
Hepatology. 2023;78:1352–67. PubMed Google Scholar * Feo CF, Ginesu GC, Fancellu A, Perra T, Ninniri C, Deiana G, et al. Current management of incidental gallbladder cancer: A review. Int
J Surg. 2022;98:106234. Article PubMed Google Scholar * Saitoh M. Transcriptional regulation of EMT transcription factors in cancer. Semin Cancer Biol. 2023;97:21–9. Article CAS PubMed
Google Scholar * Cockram PE, Kist M, Prakash S, Chen SH, Wertz IE, Vucic D. Ubiquitination in the regulation of inflammatory cell death and cancer. Cell Death Differ. 2021;28:591–605.
Article CAS PubMed PubMed Central Google Scholar * Han D, Wang L, Jiang S, Yang Q. The ubiquitin-proteasome system in breast cancer. Trends Mol Med. 2023;29:599–621. Article CAS
PubMed Google Scholar * Dewson G, Eichhorn PJA, Komander D. Deubiquitinases in cancer. Nat Rev Cancer. 2023;23:842–62. Article CAS PubMed Google Scholar * Cruz Walma DA, Chen Z,
Bullock AN, Yamada KM. Ubiquitin ligases: guardians of mammalian development. Nat Rev Mol Cell Biol. 2022;23:350–67. Article CAS PubMed Google Scholar * Song Y, Lin M, Liu Y, Wang ZW,
Zhu X. Emerging role of F-box proteins in the regulation of epithelial-mesenchymal transition and stem cells in human cancers. Stem Cell Res Ther. 2019;10:124. Article PubMed PubMed
Central Google Scholar * Zheng H, Shen M, Zha YL, Li W, Wei Y, Blanco MA, et al. PKD1 phosphorylation-dependent degradation of SNAIL by SCF-FBXO11 regulates epithelial-mesenchymal
transition and metastasis. Cancer Cell. 2014;26:358–73. Article CAS PubMed PubMed Central Google Scholar * Simon C, Brunke ID, Stielow B, Forné I, Steitz AM, Geller M, et al. SAMD1
suppresses epithelial-mesenchymal transition pathways in pancreatic ductal adenocarcinoma. PLoS Biol. 2024;22:e3002739. Article CAS PubMed PubMed Central Google Scholar * Sahu SK,
Tiwari N, Pataskar A, Zhuang Y, Borisova M, Diken M, et al. FBXO32 promotes microenvironment underlying epithelial-mesenchymal transition via CtBP1 during tumour metastasis and brain
development. Nat Commun. 2017;8:1523. Article PubMed PubMed Central Google Scholar * Wu J, Wen T, Marzio A, Song D, Chen S, Yang C, et al. FBXO32-mediated degradation of PTEN promotes
lung adenocarcinoma progression. Cell Death Dis. 2024;15:282. Article CAS PubMed PubMed Central Google Scholar * Wei Q, Liu Z, Zhu J, Jiang W, Xie H, Feng G, et al. The Ubiquitin E3
Ligase FBXO33 Suppresses Stem Cell-Like Properties and Metastasis in Non-Small-Cell Lung Cancer by Promoting Ubiquitination and Degradation of Myc. Front Biosci. 2024;29:296. * Landrum MJ,
Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–d7. Article CAS
PubMed Google Scholar * Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019;47:D941–d7. Article
CAS PubMed Google Scholar * Berndsen CE, Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol. 2014;21:301–7. Article CAS PubMed Google Scholar * Li Y,
Xie P, Lu L, Wang J, Diao L, Liu Z, et al. An integrated bioinformatics platform for investigating the human E3 ubiquitin ligase-substrate interaction network. Nat Commun. 2017;8:347.
Article PubMed PubMed Central Google Scholar * Sampson C, Wang Q, Otkur W, Zhao H, Lu Y, Liu X, et al. The roles of E3 ubiquitin ligases in cancer progression and targeted therapy. Clin
Transl Med. 2023;13:e1204. Article CAS PubMed PubMed Central Google Scholar * Hao Q, Chen Y, Zhou X. The Janus Face of p53-Targeting Ubiquitin Ligases. Cells. 2020;9:1656. * Xu S, Zhan
M, Jiang C, He M, Yang L, Shen H, et al. Genome-wide CRISPR screen identifies ELP5 as a determinant of gemcitabine sensitivity in gallbladder cancer. Nat Commun. 2019;10:5492. Article CAS
PubMed PubMed Central Google Scholar * Efe G, Rustgi AK, Prives C. p53 at the crossroads of tumor immunity. Nat Cancer. 2024;5:983–95. Article PubMed Google Scholar * Li Y, Cui K,
Zhang Q, Li X, Lin X, Tang Y, et al. FBXL6 degrades phosphorylated p53 to promote tumor growth. Cell Death Differ. 2021;28:2112–25. Article CAS PubMed PubMed Central Google Scholar *
Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575–80. Article CAS PubMed PubMed Central Google
Scholar * Huang J, Wang X, Li N, Fan W, Li X, Zhou Q, et al. YY1 Lactylation Aggravates Autoimmune Uveitis by Enhancing Microglial Functions via Inflammatory Genes. Adv Sci.
2024;11:e2308031. * Wang X, Fan W, Li N, Ma Y, Yao M, Wang G, et al. YY1 lactylation in microglia promotes angiogenesis through transcription activation-mediated upregulation of FGF2. Genome
Biol. 2023;24:87. Article PubMed PubMed Central Google Scholar * Dang F, Nie L, Wei W. Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ. 2021;28:427–38.
Article CAS PubMed Google Scholar * Trulsson F, Akimov V, Robu M, van Overbeek N, Berrocal DAP, Shah RG, et al. Deubiquitinating enzymes and the proteasome regulate preferential sets of
ubiquitin substrates. Nat Commun. 2022;13:2736. Article CAS PubMed PubMed Central Google Scholar * Zhou X, Sun SC. Targeting ubiquitin signaling for cancer immunotherapy. Signal
Transduct Target Ther. 2021;6:16. Article CAS PubMed PubMed Central Google Scholar * Tekcham DS, Chen D, Liu Y, Ling T, Zhang Y, Chen H, et al. F-box proteins and cancer: an update from
functional and regulatory mechanism to therapeutic clinical prospects. Theranostics. 2020;10:4150–67. Article CAS PubMed PubMed Central Google Scholar * Díaz VM, de Herreros AG. F-box
proteins: Keeping the epithelial-to-mesenchymal transition (EMT) in check. Semin Cancer Biol. 2016;36:71–9. Article PubMed Google Scholar * Zheng L, Shen J, Chen Y, Lin J, Li P, Zhao X,
et al. FBXO43 promotes cell cycle progression in cancer cells through stabilizing SKP2. Cancer Lett. 2024;591:216848. Article CAS PubMed Google Scholar * Wu L, Yu K, Chen K, Zhu X, Yang
Z, Wang Q, et al. Fbxo45 facilitates pancreatic carcinoma progression by targeting USP49 for ubiquitination and degradation. Cell Death Dis. 2022;13:231. Article CAS PubMed PubMed Central
Google Scholar * Chen ZS, Wong AKY, Cheng TC, Koon AC, Chan HYE. FipoQ/FBXO33, a Cullin-1-based ubiquitin ligase complex component modulates ubiquitination and solubility of polyglutamine
disease protein. J Neurochem. 2019;149:781–98. Article CAS PubMed Google Scholar * Zhang C, Pan G, Qin JJ. Role of F-box proteins in human upper gastrointestinal tumors. Biochim Biophys
Acta Rev Cancer. 2024;1879:189035. Article CAS PubMed Google Scholar * Loix M, Zelcer N, Bogie JFJ, Hendriks JJA. The ubiquitous role of ubiquitination in lipid metabolism. Trends Cell
Biol. 2024;34:416–29. * Kaushik A, Parashar S, Ambasta RK, Kumar P. Ubiquitin E3 ligases assisted technologies in protein degradation: Sharing pathways in neurodegenerative disorders and
cancer. Ageing Res Rev. 2024;96:102279. Article CAS PubMed Google Scholar * Lagirand-Cantaloube J, Offner N, Csibi A, Leibovitch MP, Batonnet-Pichon S, Tintignac LA, et al. The
initiation factor eIF3-f is a major target for atrogin1/MAFbx function in skeletal muscle atrophy. EMBO J. 2008;27:1266–76. Article CAS PubMed PubMed Central Google Scholar * Xiao Y,
Cai GP, Feng X, Li YJ, Guo WH, Guo Q, et al. Splicing factor YBX1 regulates bone marrow stromal cell fate during aging. EMBO J. 2023;42:e111762. Article CAS PubMed PubMed Central Google
Scholar * Gallagher WM, Brown R. p53-oriented cancer therapies: current progress. Ann Oncol. 1999;10:139–50. Article CAS PubMed Google Scholar * Selivanova G. Therapeutic targeting of
p53 by small molecules. Semin Cancer Biol. 2010;20:46–56. Article CAS PubMed Google Scholar * Levine AJ. p53: 800 million years of evolution and 40 years of discovery. Nat Rev Cancer.
2020;20:471–80. Article CAS PubMed Google Scholar * Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9:701–13. Article CAS PubMed
Google Scholar * Nepal C, Zhu B, O’Rourke CJ, Bhatt DK, Lee D, Song L, et al. Integrative molecular characterisation of gallbladder cancer reveals micro-environment-associated subtypes. J
Hepatol. 2021;74:1132–44. Article CAS PubMed Google Scholar * Soussi T, Kroemer G. MDM2-TP53 Crossregulation: An Underestimated Target to Promote Loss of TP53 Function and Cell Survival.
Trends Cancer. 2018;4:602–5. Article CAS PubMed Google Scholar * Ning Y, Hui N, Qing B, Zhuo Y, Sun W, Du Y, et al. ZCCHC10 suppresses lung cancer progression and cisplatin resistance
by attenuating MDM2-mediated p53 ubiquitination and degradation. Cell Death Dis. 2019;10:414. Article PubMed PubMed Central Google Scholar * Yao J, Wang XP, Yang J, Yang Z, Zhang ZY.
SCF-FBXL8 contributes to liver metastasis and stem-cell-like features in colorectal cancer cells by mediating ubiquitination and degradation of TP53. Clin Transl Med. 2023;13:e1208. Article
CAS PubMed PubMed Central Google Scholar * Yi J, Tavana O, Li H, Wang D, Baer RJ, Gu W. Targeting USP2 regulation of VPRBP-mediated degradation of p53 and PD-L1 for cancer therapy. Nat
Commun. 2023;14:1941. Article CAS PubMed PubMed Central Google Scholar * Guo Y, Li Q, Zhao G, Zhang J, Yuan H, Feng T, et al. Loss of TRIM31 promotes breast cancer progression through
regulating K48- and K63-linked ubiquitination of p53. Cell Death Dis. 2021;12:945. Article CAS PubMed PubMed Central Google Scholar * Peng Q, Shi X, Li D, Guo J, Zhang X, Zhang X, et
al. SCML2 contributes to tumor cell resistance to DNA damage through regulating p53 and CHK1 stability. Cell Death Differ. 2023;30:1849–67. Article CAS PubMed PubMed Central Google
Scholar * Wang J, Liu W, Zhang L, Zhang J. Targeting mutant p53 stabilization for cancer therapy. Front Pharmacol. 2023;14:1215995. Article CAS PubMed PubMed Central Google Scholar *
Yue X, Zhao Y, Huang G, Li J, Zhu J, Feng Z, et al. A novel mutant p53 binding partner BAG5 stabilizes mutant p53 and promotes mutant p53 GOFs in tumorigenesis. Cell Discov. 2016;2:16039.
Article CAS PubMed PubMed Central Google Scholar * Wang S, Hao Q, Li J, Chen Y, Lu H, Wu X, et al. Ubiquitin ligase DTX3 empowers mutant p53 to promote ovarian cancer development. Genes
Dis. 2022;9:705–16. Article CAS PubMed Google Scholar * Liu J, Zhang C, Xu D, Zhang T, Chang CY, Wang J, et al. The ubiquitin ligase TRIM21 regulates mutant p53 accumulation and gain of
function in cancer. J Clin Invest. 2023;133:e164354. * Li D, Marchenko ND, Moll UM. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through
inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011;18:1904–13. Article CAS PubMed PubMed Central Google Scholar * Su X, Feng C, Wang S, Shi L, Gu Q, Zhang H, et al.
The noncoding RNAs SNORD50A and SNORD50B-mediated TRIM21-GMPS interaction promotes the growth of p53 wild-type breast cancers by degrading p53. Cell Death Differ. 2021;28:2450–64. Article
CAS PubMed PubMed Central Google Scholar * Hays E, Bonavida B. YY1 regulates cancer cell immune resistance by modulating PD-L1 expression. Drug Resist Updat. 2019;43:10–28. Article
PubMed Google Scholar * Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, et al. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell. 2017;171:1573–88.e28. Article
CAS PubMed PubMed Central Google Scholar * Chen Y, Wu J, Zhai L, Zhang T, Yin H, Gao H, et al. Metabolic regulation of homologous recombination repair by MRE11 lactylation. Cell.
2024;187:294–311.e21. Article CAS PubMed Google Scholar Download references FUNDING This study was funded by the Natural Science Foundation of Fujian Province (Grant No. 2022J011300).
AUTHOR INFORMATION Author notes * These authors contributed equally: Zhenheng Wu, You Peng, Wen Chen. AUTHORS AND AFFILIATIONS * Department of Hepatobiliary Surgery and Fujian Institute of
Hepatobiliary Surgery, Fujian Medical University Union Hospital, Fuzhou, 350001, Fujian, China Zhenheng Wu * The Third Affiliated Hospital of Sun Yat-sen University, Zhaoqing Hospital,
Health Management Center, Zhaoqing, 526070, Guangdong, China You Peng * Department of Hepatobiliary Surgery, Fuzhou First Hospital Affiliated with Fujian Medical University, Fuzhou, 350009,
Fujian, China Wen Chen * Department of Hepatic Surgery Center, Tongji Hospital of Tongji Medical College of Huazhong University of Science and Technology, Wuhan, China Feng Xia * The Basic
Medical School, Hubei University of Science and Technology, Xianning, 437100, Hubei, China Tieshan Song & Qiming Ke * Department of General Surgery, Tianjin Medical University General
Hospital, Tianjin, 300052, China Qiming Ke Authors * Zhenheng Wu View author publications You can also search for this author inPubMed Google Scholar * You Peng View author publications You
can also search for this author inPubMed Google Scholar * Wen Chen View author publications You can also search for this author inPubMed Google Scholar * Feng Xia View author publications
You can also search for this author inPubMed Google Scholar * Tieshan Song View author publications You can also search for this author inPubMed Google Scholar * Qiming Ke View author
publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Study design (Qiming Ke, Zhenheng Wu), immunohistochemistry analysis (Zhenheng Wu, Wen Chen, Feng Xia),
cell and animal experiments (Zhenheng Wu, Qiming Ke, Wen Chen), qPCR and WB experiments (Qiming Ke, You Peng, Tieshan Song), data analysis (Qiming Ke, Zhenheng Wu, You Peng, Wen Chen),
drafting of the manuscript (Qiming Ke), approval of the final version of the manuscript (all authors). CORRESPONDING AUTHOR Correspondence to Qiming Ke. ETHICS DECLARATIONS COMPETING
INTERESTS The authors declare no competing interests. ETHICS APPROVAL Research involving human subjects is reviewed and approved by the Ethics Committee of Fujian Medical University Union
Hospital. Patients/participants provide written informed consent to participate in this study. The animal study has been approved by the Ethics Committee of the Medical Faculty of the Fujian
Medical University. CONSENT TO PARTICIPATE Informed consent was obtained from all individual participants included in the study. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature
remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Edited by Francesca Bernassola SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION
ORIGINAL WESTERN BLOT RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation,
distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and
indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to
the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will
need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE
CITE THIS ARTICLE Wu, Z., Peng, Y., Chen, W. _et al._ Lactylation-driven transcriptional activation of FBXO33 promotes gallbladder cancer metastasis by regulating p53 polyubiquitination.
_Cell Death Dis_ 16, 144 (2025). https://doi.org/10.1038/s41419-025-07372-y Download citation * Received: 01 June 2024 * Revised: 20 December 2024 * Accepted: 21 January 2025 * Published: 28
February 2025 * DOI: https://doi.org/10.1038/s41419-025-07372-y SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative