Play all audios:
ABSTRACT Metastasis is a major challenge for colorectal cancer (CRC) treatment. In this study, we identified autophagy activation as a prognostic indicator in CRC and observed that the
expression of key autophagy proteins is elevated in metastatic and recurrent cases. Our subsequent goal was to identify potential genes associated with the autophagy panel and assess their
prognostic significance, biological roles, and mechanisms in CRC metastasis. Among the candidates, CENPF emerged as the top gene in our screening process. We found that CENPF expression was
preferentially elevated in CRC tissues compared to adjacent normal tissues, with significantly higher levels in CRC patients with tumor recurrence. Furthermore, a multicenter cohort study
demonstrated that upregulated CENPF expression was strongly associated with poorer disease-free survival in CRC. Functional experiments showed that CENPF knockdown inhibited CRC cell
invasion and metastasis both in vitro and in vivo. Intriguingly, we found CENPF undergoes degradation in CRC via the ubiquitination-proteasome pathway. Mechanistically, we observed that USP4
interacted with and stabilized CENPF via deubiquitination. Furthermore, USP4-mediated CENPF upregulation was critical regulators of metastasis of CRC. Examination of clinical samples
confirmed that USP4 expression positively correlates with CENPF protein expression, but not mRNA transcript levels. Taken together, this study describes a novel USP4-CENPF signaling axis
which is crucial for CRC metastasis, potentially serving as a therapeutic target and a promising prognostic biomarker for CRC. SIMILAR CONTENT BEING VIEWED BY OTHERS USP21-EGFR SIGNALING
AXIS IS FUNCTIONALLY IMPLICATED IN METASTATIC COLORECTAL CANCER Article Open access 18 December 2024 THE NR3C2-SIRT1 SIGNALING AXIS PROMOTES AUTOPHAGY AND INHIBITS EPITHELIAL MESENCHYMAL
TRANSITION IN COLORECTAL CANCER Article Open access 14 April 2025 USP36 PROMOTES COLORECTAL CANCER PROGRESSION THROUGH INHIBITION OF P53 SIGNALING PATHWAY VIA STABILIZING RBM28 Article Open
access 29 September 2024 INTRODUCTION Colorectal cancer (CRC) is the second most common malignancy worldwide and ranks as the third leading cause of cancer-related deaths [1]. Despite the
progress in treatment modalities, CRC recurrence remains a significant concern due to various factors such as tumor heterogeneity, microenvironmental influences, and acquired drug
resistance. The recurrence of CRC often signifies a more aggressive disease phenotype, posing a considerable threat to patient survival. A thorough understanding of the molecular processes
underlying CRC metastasis has the potential to reveal a plethora of new novel biomarkers and therapeutic targets. The major type of genomic instability was chromosomal instability (CIN),
which was observed in both pre-cancerous lesions and malignant growth [2]. CIN was characterized by frequent chromosomal abnormalities, including whole chromosome or large segmental
gains/losses (non-diploidy), structural rearrangements, and focal aberrations (e.g., amplifications and deletions), resulting in tumor heterogeneity and other malignant features. The
centromeres and their associated kinetochores were necessary for proper spindle attachment, chromosome alignment, mitotic checkpoint activation, and separation of sister chromatids during
mitosis, which were considered major causes of CIN when abnormally expressed. Centromere proteins (CENPs) family participates in centromere formation and organization during mitosis.
Centromere protein F (CENPF), one of the CENPs, is the largest member of the centromere protein family (approximately 350 kDa in molecular weight) and plays critical roles in protein
complexes, participating in microtubule functions such as attachment and dynamics, centromere assembly, and mitotic checkpoints [3]. Recently, CENPF has been confirmed to potentially induce
CIN in primary breast cancer and participate in the progression of various malignant tumors, making it a key factor in tumor progression and a promising indicator for prognosis [4,5,6,7].
However, the biological roles and prognostic value of CENPF in CRC remained unknown. Ubiquitination, a vital posttranslational modification, orchestrates numerous cellular processes, such as
cell-cycle advancement, transcriptional modulation, and signal transduction [8,9,10]. This process involves the attachment of ubiquitin molecules to target proteins, marking them for
various fates within the cell. However, alongside ubiquitination, the reversal process known as deubiquitination, managed by deubiquitinating enzymes (DUBs), has emerged as a pivotal
regulatory mechanism governing protein turnover [11, 12]. Deubiquitination, in essence, acts as a molecular undo button, removing ubiquitin molecules from target proteins and thereby
influencing their stability, activity, and localization within the cell. In recent years, targeting DUBs to regulate ubiquitination modification of substrate proteins has become a hot
research direction, and targeting DUBs is emerging as a promising agent for anticancer therapy [13, 14]. Previous studies have shown that several members of the CENP family [15,16,17] are
regulated via the ubiquitin-proteasome pathway. Based on this, we investigated specific deubiquitinating enzymes (DUBs) that regulate CENPF protein expression and further explored the role
and potential mechanisms of targeting the DUB-CENPF axis for CRC treatment. In this research, we employed bioinformatics approaches and conducted siRNA functional screening to identify CENPF
as a pivotal gene involved in CRC metastasis. We utilized both in vitro and in vivo CRC cell models to investigate the functional role of CENPF. Additionally, we also evaluated the
prognostic relevance of CENPF in CRC by analyzing data from multiple patient cohorts across three different medical centers. Furthermore, our study revealed that CENPF serves as a novel
substrate for the Ub-specific protease (USP) family member USP4, a deubiquitinating enzyme closely associated with numerous oncogenic proteins [18,19,20,21,22,23,24,25]. Notably, the
interaction between CENPF and USP4 is intricately linked to CRC metastasis. MATERIALS AND METHODS ETHICS STATEMENT Ethical approval was obtained from the institutional review boards at each
participating center (KY2021-R019, 2023KY251) for the retrospective analysis of anonymized data. All procedures involving animals were conducted in accordance with institutional ethical
standards for animal experimentation and were approved by the Ethics Committee of Fujian Medical University/Laboratory Animal Center (IACUC FJMU 2022-0488). STUDY PATIENTS The study enrolled
a total of 519 patients diagnosed with typical colorectal adenocarcinoma histology. Formalin-fixed, paraffin-embedded (FFPE) specimens were collected from 393 patients who underwent
curative surgery at the First Affiliated Hospital, Wenzhou Medical University (Wenzhou, China) from April 2014 to December 2016, constituting Cohort I for the training dataset. Additionally,
126 patients who underwent curative surgery at Union Hospital, Fujian Medical University (Fuzhou, China) from January 2010 to December 2011 formed Cohort II for external validation.
Detailed information on the inclusion and exclusion criteria, along with details of the recruitment process, can be found in Supplemental Fig. 10. The baseline characteristics of these
patients are summarized in Supplemental Table 1. The retrospective study was approved by the ethics committees of the two centers involved, and the informed consent requirement was waived.
FOLLOW-UP AND SURVIVAL ANALYSIS Patients included in the study underwent follow-up examinations every 3 months during the initial 2 years post-surgery, followed by 6-month intervals for the
subsequent 3 years. The last recorded follow-up data were available until September 6, 2021, for patients from Cohort I and until September 1, 2017, for patients from Cohort II. Survival
analysis was conducted on patients with complete immunohistochemistry (IHC) data. To determine the optimal cutoff point for epithelial CENPF IHC scores, X-tile software was utilized to
establish this cutoff, which correlates CENPF protein expression with DFS. The same threshold values (CENPF IHC score = 170) were then applied to the independent external validation cohorts.
Survival analyses were conducted to evaluate the potential relationship between CENPF protein expression and OS and DFS. Additionally, the optimal cutoff values (IHC Score=100) for USP4
were also determined using X-tile software, followed by subsequent survival analysis. IMMUNOHISTOCHEMISTRY Immunohistochemistry (IHC) was performed at the Pathology Laboratory of Union
Hospital using rabbit polyclonal antibodies to USP4 (1:1000, ab236987, Abcam) and rabbit polyclonal antibodies to CENPF (1:500, ab5, Abcam), Beclin-1 (1:500, ab62557, Abcam), LC3B (1:400,
3868, CST), ATG7 (1:500, ab52472, Abcam) following the manufacturer’s instructions. IHC scores were calculated by multiplying two components: the intensity score and the extent score. The
intensity score is assigned based on a scale (0 for no staining, 1 for weak, 2 for moderate, and 3 for strong), while the extent score represents the percentage of cells showing positive
staining. Before evaluating the staining intensity of each protein, a reference standard was set according to the baseline staining levels of each protein. Two experienced pathologists then
assess the target protein scores in each patient’s CRC tissue using the established grading system [26], with both pathologists blinded to the sample information. The reliability of the
scoring system was evaluated by analyzing the agreement between the scoring results obtained by the two independent investigators using contingency tables (Supplemental Tables 2,3). Cohen’s
Kappa Indices were calculated to measure inter-rater agreement, with a result considered to indicate excellent concordance if the Cohen’s Kappa index exceeded 0.8 [27]. GENOMIC DATA MINING
Data from GSE14333, GSE41258, and TCGA datasets were obtained from the Gene Expression Ominous (GEO) and TCGA. Then, we extracted corresponding autophagy genes (Supplemental Table 4)
expression profile and protein-coding genes from 3 dataset, respectively. In the R 3.2.0 environment, we iteratively explored the correlation between them to identify a candidate set of
autophagy-related genes. In the R 3.2.0 environment, we employed the plyr, reshape2, and ggpubr packages to process the raw data from TCGA dataset and display the expression pattern of CENPF
mRNA across various cancers. The limma package was employed to explore differentially expressed genes between colorectal cancer (CRC) and adjacent normal tissues in GSE41258 and GSE49355.
Additionally, two datasets (GSE8671, GSE22598) were used to investigate the differential expression of CENPF mRNA between colorectal cancer and adjacent normal tissues. Survival data
including disease-free survival (DFS) were annotated from the GSE14333 dataset. The prognostic relevance of CENPF mRNA expression level was investigated in GSE14333 cohort. The X-tile plot
curve was utilized to present the relationship between CENPF mRNA expression and DFS, determining the optimal cut-off value to stratify patients. Patients with CENPF mRNA expression above
the cut-off value were categorized into the CENPF-high group, while those below were categorized into the CENPF-low group. The Kaplan-Meier method was employed to examine the relationship
between different patient groups and DFS. CELL MODELS Cell lines including HCT116, SW480, DLD1 (human colorectal cancer cell lines), and HEK293T (human embryonic kidney epithelial cell line)
were obtained from Punuo Sai Life Sciences & Technology Co., Ltd. in Wuhan, China. Authentication of these cell lines was performed via STR profiling. Culture conditions were as
follows: HCT116 and DLD1 cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (Sigma, Missouri, USA) and penicillin/streptomycin, maintained in a 5% CO2
humidified incubator. SW480 and HEK293T cells were cultured in DMEM supplemented with 10% fetal bovine serum and penicillin/streptomycin, also within a 5% CO2 humidified incubator. All cell
lines used in this study were confirmed to be free of mycoplasma contamination. IMMUNOPRECIPITATION AND UBIQUITINATION ASSAYS In both experiments, cell lysis was performed using a homemade
lysis buffer kept on ice. The lysis buffer contained 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, 2.5 mM Sodium pyrophosphate, 1 mM β-Glycerophosphate, and 1
mM Na3VO4, supplemented with a protease inhibitor cocktail. After centrifugation at 12,000 × _g_ for 30 min at 4 °C, 5% of the supernatant lysates were retained as input for future use. For
the immunoprecipitation of endogenous proteins, protein A/G agarose (obtained from Santa Cruz Biotechnology, Santa Cruz, USA) was washed thrice with lysis buffer and then incubated with the
specified antibody along with the remaining cell lysate supernatant at 4 °C overnight. In the case of immunoprecipitation for exogenously overexpressed proteins, anti-Flag (GNI4510-FG), HA
(GNI4510-HA), or Myc (GNI4510-MC) affinity gel (obtained from GNI, Tokyo, Japan) was washed three times with lysis buffer and directly incubated with the remaining cell lysate supernatant at
4 °C overnight. Subsequent Western blot analysis was conducted the following day after washing three times with lysis buffer. TRANSWELL MIGRATION ASSAY Tumor cell migration assays were
conducted following the manufacturer’s guidelines. Initially, cells were harvested and suspended in serum-free medium. Subsequently, they were seeded onto Transwell inserts at a
concentration of 200,000 cells per well. These inserts were then positioned in a lower chamber containing 600 μl of culture media supplemented with 20% FBS. The Transwells were then
incubated for 24 h at 37 °C. Following incubation, cells on the interior of the Transwell inserts were eliminated using a cotton swab. The cells that had migrated to the lower surface of the
membrane were then fixed using 4% paraformaldehyde and stained with 0.1% crystal violet. Photographs were captured from five randomly selected fields, and the cells were counted to
determine the average number of cells that had migrated. TUMOR GROWTH AND LIVER METASTASIS ASSAY IN NUDE MICE Five-week-old female athymic nude mice were obtained from the GemPharmatech Co.,
Ltd (Nanjing, China). After the purchased experimental mice have adapted to the SPF-level barrier for one week, their body weight will be measured. Based on the body weight, the mice will
be randomly assigned into the experimental group and the control group. The mice (_n_ = 5 per group) were subcutaneously injected with either 3 × 106 DLD-1 cells or SW480 cells or HCT116
cells transduced with lenti-shCtrl or lenti-shCENPF1 or lenti-shCENPF2. Tumor volume (mm3) was calculated using the formula: Volume = 0.5 × length × (width)2. The investigator was blinded to
group assignments throughout the experiment and when evaluating the outcomes. To assess the metastatic potential of colorectal cancer (CRC) cells in the liver, athymic nude mice (_n_ = 6
per group) were utilized, following established protocols. Briefly, a small incision was made in the left abdomen, and the spleen was isolated and exposed. Viable cancer cells (3 × 106
cells/50 μl PBS) were injected into the spleen using a sterile tuberculin syringe and a 27-gauge needle. Then, the abdominal cavity was closed using nylon sutures. Mice were euthanized after
eight weeks (for DLD-1 and SW480 cells), and liver metastases were subsequently evaluated. CCK-8 ASSAY, COLONY FORMATION ASSAY The viability of cells was assessed using the Cell Counting
Kit-8 (CCK-8) from Dojindo Molecular Technologies, Inc. Cells were seeded into triplicate wells of 96-well plates at a density of 2000 cells per well for the CCK-8 assay. For the colony
formation assay, cells were cultured in triplicate wells of 6-well plates at a density of 500 cells per well. Following a two-week incubation period under standard growth conditions, the
colonies were fixed with ice-cold 4% paraformaldehyde, stained with crystal violet solution, and examined using an inverted microscope. WESTERN BLOT ANALYSIS Western blot analysis for
conventional protein was performed as previously described [28]. For the analysis of high molecular weight proteins in this study, we utilized an extended-range prestained protein marker
(Proteintech, Wuhan, China) and a specialized gel kit (Epizyme, Shanghai, China). Electrophoresis was conducted at 130 V for 3 h, with the running buffer replaced 1–2 times during the
process. For protein transfer, the transfer buffer was supplemented with 0.1% SDS and 10% methanol, and the transfer was carried out on ice at 300 mA for 3 h, with the buffer replaced 1–2
times. All other steps were performed according to the standard Western blot protocol. Antibodies used in Western blot were USP4 (1:1000, ab236987, Abcam), CENPF (1:1000, ab5, Abcam), HA
(1:3000, ab236632, Abcam), Flag (1:2000, 14793, CST), Myc (1:3000, AE070, Abclonal), GAPDH (1:5000, 60004-1-Ig, Proteintech), β-actin (1:3000, ab8226, Abcam). IMMUNOFLUORESCENCE (IF)
Immunofluorescence staining was carried out as described previously [28]. In brief, Cells were cultured on 35 mm glass-bottom dishes (Thermo Scientific™, Massachusetts, USA), fixed in 4%
paraformaldehyde for 15 min, and blocked with 5% goat serum in PBS with 0.3% Triton X-100 (Sigma, Missouri, USA). They were incubated overnight with CENPF (1:400, ab5, Abcam) and USP4
(1:200, 66822-1-Ig, Proteintech) primary antibodies, followed by Alexa Fluor 488 anti-Rabbit and Alexa Fluor 594 anti-Mouse secondary antibodies. Nuclei were stained with DAPI (Roche, Basel,
Switzerland) and observed using Leica SP5 fluorescence microscopy. STATISTICAL ANALYSIS Statistical analyses were performed using R (version 3.5.0) and SPSS (version 16.0.2). Significance
was set at _p_ < 0.05 for all two-tailed tests. Experiments included 3 to 10 samples per group, with results reported as mean ± SE from at least 3 independent experiments. The
Shapiro-Wilk test assessed data normality. For normally distributed data, independent sample _t_-tests compared two groups, and one-way ANOVA compared three or more groups. Pearson’s
correlation tested relationships between variables. For non-normally distributed data, the Mann–Whitney test compared two groups, Kruskal-Wallis test compared three or more groups, and
Spearman’s rank correlation tested relationships. Two-way ANOVA analyzed repeated measurements. Clinical characteristic comparisons used Mann–Whitney U, Fisher’s exact, or chi-square tests.
X-tile software optimized cutoff values for marker expression subgroups. Kaplan-Meier analysis and log-rank tests estimated overall and disease-free survival, while multivariate Cox
regression assessed marker contributions to survival outcomes. RESULTS IDENTIFICATION OF AUTOPHAGY-RELATED CENPF AS A KEY PARTICIPANT IN CRC METASTASIS Autophagy is considered a
self-degradative and conservative process [29], playing a crucial role in controlling the quality of cellular components and maintaining cellular homeostasis. Recently, it has been found
that autophagy not only supports tumor growth in harsh environments but also plays a critical role in tumor metastasis [30,31,32,33,34,35,36,37]. To explore the involvement of autophagy in
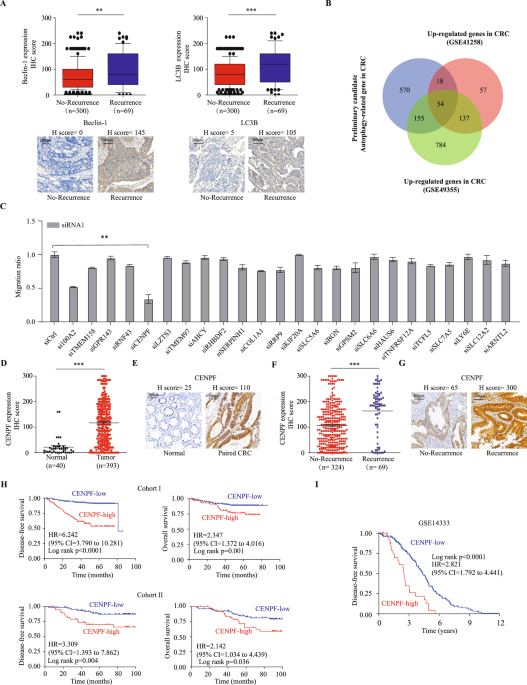
CRC metastasis, we analyzed the expression patterns and clinical relevance of key autophagy regulators in colorectal cancer tissues. Our findings revealed that the expression levels of
Beclin-1, LC3B, and ATG7 proteins in epithelial cells progressively increased from adjacent normal tissues to primary tumors, and were further elevated in metastatic cancers (all _P_ <
0.001; Supplemental Fig. 1A). Moreover, significant upregulation of these autophagy proteins was observed in CRC cases with recurrence compared to those without recurrence (all _P_ <
0.01; Fig. 1A, Supplemental Fig. 1B). Survival analysis of CRC patients from cohort I with follow-up data indicated that high expression of Beclin-1, LC3B, and ATG7 was associated with
shorter disease-free survival (_P_ < 0.0001; Supplemental Fig. 1C). These results suggest that autophagy activation may contribute to CRC metastasis. Building on these observations, we
further explored gene expression data from biobank databases to identify genes potentially involved in CRC metastasis and correlated with the autophagy gene set. We initially obtained raw
data from GSE14333, GSE41258, and TCGA gene sets, extracted the expression profile of autophagy genes and the remaining protein-coding genes, and performed correlation analysis on both sets.
The potential autophagy-related gene sets (correlation > 0.3 and _p_ < 0.001) were obtained for GSE14333, GSE41258, and TCGA gene sets, and their intersection was taken as the initial
candidate set of 797 autophagy-related genes (Supplemental Fig. 1D). We then compared those candidates with a group of genes that were highly expressed (logFC > 1 and fdr < 0.0001) in
GSE41258 and GSE49355 datasets, resulting in 54 potential important CRC-related genes (Fig. 1B). We further narrowed down the candidate genes based on single-cell databases, literature
searches to exclude stromal or immune cell localization, and previous studies on genes in CRC, focusing on 24 candidate genes that may play a role in CRC. We obtained two small interfering
RNAs (siRNAs) targeting each of the 24 genes and a control siRNA (siCtrl), then transfected into HCT116 CRC cells separately, and evaluated the impact on the migration ability of HCT116
cells using migration assays. It was found that knocking down CENPF most significantly inhibited the migration of HCT116 cells (Fig. 1C and Supplementary Fig. 1E). Also, we analyzed
published CRC mRNA microarray datasets from GSE8671, GSE22598 and TCGA. In TCGA, we observed that the expression of CENPF mRNA is significantly elevated in colorectal cancer and rectal
cancer, similar to many other broad-spectrum tumors, compared to adjacent normal tissues (Supplemental Fig. 2A). This finding is further validated by two additional datasets, GSE8671 and
GSE22598, confirming the upregulation of CENPF expression in CRC (Supplemental Fig. 2B, C). Additionally, we have collected an independent cohort of tissues microarrays (TMAs) composed of
colorectal cancer and adjacent normal tissues (Cohort I). We examine the expression of CENPF protein using immunohistochemistry staining. We observed CENPF protein was mainly distributed in
both cytoplasm and nucleus of colorectal epithelial cell as representative immunostaining presented (Fig. 1E), in which the expression pattern of CENPF protein mirrors that of mRNA
expression (Fig. 1D). Likewise, CENPF protein expression was significantly higher in CRC patients with tumor recurrence (Fig. 1F and Supplementary Fig. 2D) as demonstrated by representative
immunostaining (Fig. 1G). Thus, these findings suggest that CENPF may play a critical role in CRC metastasis. To assess the potential prognostic significance of dysregulated CENPF expression
in CRC, we then conducted time-to-event analyses using 3 cohorts with follow-up records. In Cohort I, high CENPF protein expression was associated with more advanced TNM stage (_p_ = 0.002)
(Supplementary Table 1). Importantly, high CENPF protein expression was correlated with poorer DFS and OS when compared to the low-expression group (HR 6.242, 95% CI 3.790 to 10.281,
Log-rank p < 0.0001; HR 2.347, 95% CI 1.372 to 4.016, Log-rank _p_ = 0.001, respectively) (Fig. 1H). Furthermore, multivariate Cox regression analysis demonstrated that high CENPF protein
expression was an independent predictor for both DFS and OS in CRC patients (HR 5.282, 95% CI 3.162 to 8.822, _p_ < 0.001; HR 2.074, 95% CI 1.192 to 3.607, _p_ = 0.01, respectively)
(Table 1). To ensure the reliability and applicability of these results, we conducted analogous examinations within an independent external validation cohort. Consistently, high CENPF
protein expression was linked to poorer DFS and OS compared to the low-expression group in Cohort II (HR 3.309, 95% CI 1.393 to 7.862, Log-rank _p_ = 0.004; HR 2.142, 95% CI 1.034 to 4.439,
Log-rank _p_ = 0.036, respectively) (Fig. 1H). Further validation through multivariate Cox regression analyses consistently demonstrated that elevated CENPF protein expression remained a
strong and independent prognostic factor for both DFS and OS among CRC patients within the external validation cohort II (Table 2). In GSE14333, it was found that higher CENPF expression in
tumors was significantly associated with decreased DFS (HR 2.821, 95% CI 1.792 to 4.441, Log-rank _p_ < 0.0001) (Fig. 1I). Collectively, these findings indicate a tumor-promoting role of
CENPF in CRC. KNOCKDOWN OF CENPF INHIBITS MIGRATION, INVASION OF COLORECTAL CANCER CELLS IN VITRO AND IN VIVO The prevalence of CENPF upregulation raises an intriguing possibility that CENPF
overexpression may be a cancer-promoting event in CRC. To examine this possibility, CENPF expression was stably knock down using lentiviral vectors carrying two shRNAs targeting CENPF
(shCENPF1 and shCENPF2) in DLD-1, HCT116, and SW480 cells. The knockdown efficiency was confirmed by Western blot assays (Supplementary Fig. 3A). Colorectal cancer cell migration and
invasion ability was assessed in vitro. The migration and invasion assays showed that knocking down CENPF significantly weakened the ability of colorectal cancer cell migration and invasion
(Fig. 2A). Similarly, scratch-wound-healing assays revealed that a lower migration ability in shCENPF cells than in shCtrl cells (Fig. 2B). To further exclude off-target effects of CENPF
shRNA in CRC, CENPF-HA overexpression plasmids was transiently transfected into stably CENPF-knockdown DLD1, HCT116, and SW480 cells, examined by Western blot assays (Supplementary Fig. 4A).
The migration and invasion assays revealed that CENPF overexpression significantly reversed the reduced migration and invasion compared to the vector group (Supplementary Fig. 4B).
Moreover, scratch wound healing assays demonstrated a noticeable restoration of migration capacity in CENPF-overexpressing DLD1 and SW480 cells compared to vectors (Supplementary Fig. 4C).
However, CCK-8 assays and Colony assays were used to investigate the role of CENPF in cell proliferation. The results revealed that knockdown of CENPF had no significant effect on CRC cell
growth in vitro (Supplementary Fig. 5A, B). Overall, these findings showed that CENPF can enhance the migration, and invasion of colorectal cancer cells in vitro. We further examine whether
the same result can be observed in vivo. CRC cells with or without CENPF knockdown were injected into spleen of the nude BALB/C mice (Supplementary Fig. 3B). Eight weeks post-injection, the
ability of SW480 and DLD1 cells to develop liver micrometastases were significantly impaired when cells lacked CENPF expression (Fig. 2C, D). Similarly, histological examination showed that
the shCENPF group developed less liver micrometastases (Supplementary Fig. 3C, D). Consistently, CENPF knockdown prolonged mouse survival (Fig. 2E). However, no significant difference in
tumor size was observed between CENPF knock-down group and shCtrl group (Supplementary Fig. 5C). Together, these results indicated that CENPF functioned as a tumor promoter in CRC
metastasis. USP4 INTERACTS WITH CENPF AND IS IDENTIFIED AS A CANDIDATE DUB FOR CENPF Previous studies have revealed that several members of the CENP family, such as CENPA [15], CENPH [16],
and CENPN [17], are degraded through the ubiquitin-proteasome pathway. Given this, we further investigated whether the CENPF protein follows a similar degradation pathway. Initially, we
utilized protein modification site prediction tools and identified several potential ubiquitination sites on the CENPF protein structure (Supplemental Fig. 6A). Western blotting showed CENPF
protein increased with MG132 treatment, proteasome inhibitor, in a dose-dependent manner (Fig. 3A). And we performed co-immunoprecipitation (Co-IP) assays by transfecting HCT116 cells with
CENPF-HA and Flag-Ubiquitin, and anti-HA immunoprecipitates were probed for the level of ubiquitination using Flag-antibody (Fig. 3B). It was discovered that CENPF undergoes ubiquitination,
suggesting its degradation in CRC via the ubiquitination-proteasome pathway. The proteasome identifies proteins tagged with ubiquitin and breaks them down into smaller peptides and amino
acids. Conversely, deubiquitinating enzymes (DUBs) work to remove or cleave ubiquitin molecules from protein substrates, playing a crucial role in numerous cellular processes as a key
regulatory mechanism [38]. To identify potential DUBs for CENPF, we screened DUBs expression library consisting of 53 human DUBs in HEK293T cells. HA-tagged CENPF was co-transfected with 53
DUBs-Flag plasmids separately into HEK293T cells, and expression of CENPF was detected using WB 72 h after transfection. After the initial round of screening (Supplementary Fig. 6B),
eighteen candidate DUBs that stabilized CENPF level significantly, were tested in the second round of screening in HCT116 CRC cells (Fig. 3C). Among the candidates, USP4 could interact with
CENPF. Immunoprecipitation with anti-Myc antibody demonstrated that Myc-USP4 interacted with overexpressed CENPF-HA (Fig. 3D). To determine whether the interaction between USP4 and CENPF
also occurs endogenously in CRC cells, immunoprecipitation was performed using anti-USP4 or anti-CENPF antibodies on lysates from HCT116 cells. IP with either antibody resulted in the
co-detection of both USP4 and CENPF, indicating that these endogenous proteins interact in CRC cells (Fig. 3E, F). Furthermore, immunofluorescence staining revealed that CENPF (green) and
USP4 (red) were colocalized with the nucleus, as indicated by the DAPI stain, in multiple CRC cell lines (Fig. 3G). We then identified which USP4 regions are critically required for its
interaction with CENPF. We generated four USP4 truncations (Fig. 3H), and through Co-IP assays, we found that the USP domain mediated association with CENPF (Fig. 3I). Therefore, USP4 could
interact with CENPF and markedly stabilized CENPF protein expression levels, which was the most promising candidate based on the overlap of these two screenings. USP4 DEUBIQUITINATES AND
STABILIZES CENPF To further elucidate the role of USP4 in its interaction with CENPF, further investigations demonstrated that USP4 could stabilize endogenously and exogenously expressed
CENPF (Fig. 4A, B). USP4 knockdown significantly reduced CENPF protein expression (Fig. 4C). However, neither the overexpression (Supplementary Fig. 7A) nor the knockdown (Supplementary Fig.
7B) of USP4 affected CENPF mRNA levels, indicating that USP4 stabilized CENPF protein levels post-transcriptionally. USP4 is categorized as a cysteine protease, with bioinformatic analyses
pinpointing C311 as the crucial catalytic cysteine. Consequently, USP4 mutant variant was created, wherein a substitution of cysteine to serine at position 311 (C311S) was introduced,
rendering the enzyme catalytically deficient. To whether USP4 can indeed deubiquitinate CENPF, HCT116 cells were transfected with CENPF-HA, Flag-UB, USP4-Myc or USP4 C311S-Myc, and then
anti-HA immunoprecipitates were probed for the level of ubiquitination using Flag-antibody. Co-expression of USP4 and CENPF led to a notable effect on diminishing ubiquitination of CENPF
(Fig. 4D). However, enzyme-inactive mutant USP4 C311S was not able to reduce CENPF ubiquitination (Fig. 4D). Moreover, overexpression of WT USP4, but not the enzyme-inactive mutant USP4
C311S, could stabilize CENPF protein (Fig. 4E). These results suggested that deubiquitylation and stabilization of CENPF was dependent on the catalytic activity of USP4. We next tested
whether USP4 indeed extended CENPF’s protein half-life by cycloheximide chase assay. Similarly, overexpression of WT USP4, but not USP4 C311S, prolonged the half-life of CENPF (Fig. 4F).
Conversely, when USP4 was depleted from HCT116 cells, the half-life of CENPF was markedly reduced (Fig. 4F). These data demonstrated that USP4 interacts with CENPF and facilitates
ubiquitination of CENPF, stabilizing it and prolonging its lifespan when overexpressed. Conversely, depletion of USP4 reduces CENPF’s lifespan. To confirm the relevance of the USP4-CENPF
interaction, we first analyzed protein expression of USP4 and CENPF in our clinical samples from two different Cohort. We found that USP4 and CENPF expression were positively correlated
(Fig. 4G). Representative immunostaining results were presented in Fig. 4H. However, public dataset analysis of USP4 and CENPF mRNA levels did not mirror these results (Supplementary Fig.
7C). Notably, a high expression level of USP4 correlated with poor prognosis of CRC patients from two different cohorts (Supplementary Fig. 8A). Taken together, these findings suggested that
USP4 was a strong DUB for CENPF, which at least partially, contributed to the upregulated CENPF protein expression. CENPF IS POSITIVELY REGULATED BY USP4 AND AFFECTS THE METASTATIC ABILITY
OF CRC CELLS After identifying the interaction between USP4 and CENPF, we further examined the effectiveness of USP4 on the CENPF mediated enhanced CRC migration, invasion, and metastasis
capacity. In HCT116 and DLD1 cell lines, we knocked down CENPF using shCENPF and stably overexpressed USP4 for subsequent research. Western blot analysis was utilized to detect the levels of
USP4 and CENPF in cell lines from various groups, as illustrated in Fig. 5A. The transwell assays and wound healing assays were employed to assess the invasion and migration capabilities.
The results showed that overexpressing USP4 significantly enhanced CRC’s migratory ability under CENPF-knocking down conditions (Fig. 5B, C and Supplementary Fig. 9). Additionally, we
investigated whether similar results could be observed in vivo. These findings were reinforced by conducting an animal study, where DLD1 cells were injected into the spleen of nude mice
using established methods. Compared to the shCtrl, tumor lesions in the shCENPF group were scarcely visible in the liver (Fig. 5D). However, upregulating USP4 expression could greatly
enhance the the ability of CENPF-knockdown CRC cell to form liver micrometastases (Fig. 5D). The same result was also observed in HE stains result (Fig. 5E). Thus, we proved that CENPF can
positively be regulated by USP4 and affects the metastatic ability of CRC cells in vitro and in vivo. THE USP4-CENPF AXIS WAS CORRELATED WITH CLINICAL OUTCOMES OF CRC PATIENTS Expanding on
previous discoveries linking CENPF and USP4 to CRC patient prognosis and their involvement in CRC metastasis regulation, we explored the prognostic relevance of USP4-CENPF association. We
categorized samples from cohort I into four groups according to their USP4 and CENPF protein levels determined by IHC analysis. These categories comprised low USP4/low CENPF, low USP4/high
CENPF, high USP4/low CENPF, and high USP4/high CENPF. Subsequently, we conducted comparisons of clinical outcomes among these groups. Kaplan–Meier analysis suggested that patients with high
USP4 and high CENPF expression tended to have the poorest DFS and OS compared with the other groups (Fig. 6A). These data imply that the USP4–CENPF axis plays a role in CRC development.
DISCUSSION Tumor recurrence is identified as a significant adverse prognostic indicator in patients with colorectal cancer (CRC) following curative resection [39]. In our study, we’re
looking into new key genes for CRC using bioinformatics analysis, especially those linked to autophagy-related genes. We firstly identified CENPF as a top candidate gene in CRC progression,
examined its functional role in metastatic progression and demonstrated the therapeutic value of targeting CENPF in inhibiting CRC liver metastasis. Mechanically, we found CENPF protein
could undergo ubiquitination, leading to subsequent proteasomal degradation. And USP4 as a deubiquitinating enzyme (DUB), interacted with and deubiquitinated CENPF, thereby stabilizing it.
Taken together, our data showed that a novel USP4-CENPF axis played an important regulatory role in CRC metastasis and may serve as a potential target for CRC treatment. Due to the
complexity and variability of CRC, effective targeted therapies for CRC and the availability of effective signatures that can accurately predict recurrence remain limited. Autophagy, as a
mechanism supporting cell survival, has emerged as a pivotal factor in cancer metastasis. Its impact can be twofold: either promoting or inhibiting metastasis, contingent upon factors such
as cancer cell subtype, the tumor microenvironment, and the stage of tumor progression [40, 41]. We conducted correlation analysis using multiple transcriptome sequencing datasets in CRC and
an autophagy gene list to identify candidate gene sets. We then intersected the differences between cancer and adjacent tissues to identify potential important targets for CRC treatment, in
which is a candidate set comprising 54 genes. Among them, fifteen candidate genes, including DACH1, GALNT6, IFITM1, EGFL6, WNT5A, CDK1, GDF15, SOX9, KIF23, CPNE1, BHLHE40, NEK2, ASPM,
PLA2G16, and PMEPA1, have been proven to play important roles in CRC [42,43,44,45,46,47,48,49,50,51,52,53,54,55,56], which indicated a certain degree of reliability in our screening process.
Using two different siRNAs to target candidate genes, we examined their effects on the migratory capacity of HCT116 colorectal cancer cells. Interestingly, knocking down CENPF with siRNA
showed the most pronounced inhibition of HCT116 migration, which has not been previously reported. Therefore, the role of CENPF in CRC has become the focus of our research. Moreover,
leveraging data from GEO, TCGA databases, and our own colorectal cancer tissue microarrays from two different centers, we explored CENPF expression patterns and prognostic significance in
CRC. Consistently, findings indicated upregulation of both CENPF mRNA and protein in colorectal tumors compared to adjacent normal tissues. Similarly, we observed that CRC with recurrence
exhibited significantly higher levels of CENPF than CRC without recurrence. In addition, CENPF expression correlated with CRC prognosis, showing more significant predictive efficacy in
disease-free survival, indicating its potential oncogenic role in CRC metastasis. Previous results have demonstrated that CENPF was highly expressed in the lung adenocarcinoma (LUAD), and
CENPF expression correlated with T stage and poor prognosis [57]. Moreover, CENPF knockout significantly inhibited LUAD cell growth, the tumor growth of mice [57]. Also, CENPF is markedly
elevated in pancreatic cancer (PC) and linked to poor patient outcomes [5]. Knocking down CENPF inhibits PC cell proliferation, migration, and EMT, inducing G2/M phase cell cycle arrest and
restraining in vivo pancreatic cell growth [5]. Importantly, knocking down CENPF expression significantly altered invasive and migratory capacity of CRC cells, as evidenced from a series of
in vitro experiments and in xenograft nude mice models of liver metastasis in vivo. Therefore, the consistent results from our comprehensive study verified that CENPF functions as a novel
metastasis-promoting gene in CRC. Another major finding of our study is that we’ve uncovered the role of ubiquitination in controlling CENPF protein expression and its functions. Until now,
there have been few reports confirming the factors that regulate CENPF expression. Previous research has shown that several members of the CENP family, including CENPA, CENPH, and CENPN, are
degraded via the ubiquitin-proteasome pathway [15,16,17]. First, we discovered that MG132 effectively blocked the degradation of CENPF protein and CENPF underwent ubiquitination
modifications, suggesting that CENPF could be regulated by ubiquitin-proteasome axis. To further identify the DUBs that can potentially deubiquitinate and stabilize CENPF, we screened a
human DUB expression library consisting of 53 DUBs-Flag plasmids in HEK293T cells. The top 18 knockdowns of DUB genes that stabilized CENPF level most significantly were selected for a
second-round screening genes. Each of those DUBs was then co-overexpressed together with HA-CENPF in HCT116 cells. Coimmunoprecipitation (co-IP) experiments showed the interaction exists
exclusively between USP4 and CENPF. Based on two rounds of screening, we propose that USP4 is the most likely deubiquitinase regulating CENPF stability. Mechanically, USP4 interacts with
CENPF and decreases CENPF ubiquitination levels, thus stabilizing it. Accordingly, USP4 expression was significantly positively correlated with CENPF in human CRC samples from two different
tertiary hospitals in China, as confirmed by immunohistochemistry. Importantly, the interaction of CENPF and USP4 then controlled the invasion and migration of colorectal cancer. Previous
evidence has confirmed a critical role for USP4 in regulating p53, TGFβ, Wnt/β-catenin, and NF-κB signaling, implicating dysregulation of USP4 expression in the development of cancer [58].
In CRC, USP4 has been shown to promote colorectal cancer cell metastasis in vitro and in vivo by regulating the stability and activity of β-catenin and PRL-3 [23, 25]. Previous studies
showed that mutating the catalytic residue Cys311 to Ala abolishes USP4’s deubiquitination and stabilization of β-catenin. In our study, the C311S mutation in USP4 also lost its ability to
stabilize and deubiquitinate CENPF. Thus, we speculate that CENPF is a key downstream target of USP4 in regulating colorectal cancer metastasis. Furthermore, high USP4 and high CENPF are
significant determinants of poor survival in patients with CRC. This study had some limitations as follows. First, we explained how post-translational modifications regulate CENPF’s abnormal
expression and function. However, we also found that CENPF transcription levels are abnormally elevated in colorectal cancer. Understanding the mechanisms behind this upregulation is
essential for effectively targeting CENPF abnormalities. Second, the role of the CENPF-USP4 axis in CRC metastasis has been identified, but the specific downstream molecular mechanisms
remain unclear and require further investigation, In particular, the role of autophagy activation in mediating CRC metastasis through CENPF requires more in-depth exploration. In summary,
our groundbreaking research, for the first time, has unveiled CENPF as a novel promoter of CRC metastasis and elucidate the molecular mechanism of the interaction between CENPF and USP4 in
inducing migration and invasion of colorectal cancer cells, evidenced from molecular, cellular, animal models, and clinical specimens. Thus, our findings provide a theoretical foundation and
innovative insights for the potential development of a possible treatment strategy targeting the USP4-CENPF for CRC metastasis. DATA AVAILABILITY Data and materials supporting the findings
of this study are available from the corresponding author (P Chi) upon reasonable request. REFERENCES * Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer
statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. Article PubMed Google Scholar * Pihan GA,
Wallace J, Zhou Y, Doxsey SJ. Centrosome abnormalities and chromosome instability occur together in pre-invasive carcinomas. Cancer Res. 2003;63:1398–404. CAS PubMed Google Scholar * Chen
EB, Qin X, Peng K, Li Q, Tang C, Wei YC, et al. HnRNPR-CCNB1/CENPF axis contributes to gastric cancer proliferation and metastasis. Aging. 2019;11:7473–91. Article CAS PubMed PubMed
Central Google Scholar * Li M, Zhao J, Yang R, Cai R, Liu X, Xie J, et al. CENPF as an independent prognostic and metastasis biomarker corresponding to CD4+ memory T cells in cutaneous
melanoma. Cancer Sci. 2022;113:1220–34. Article CAS PubMed PubMed Central Google Scholar * Chen H, Wang X, Wu F, Mo X, Hu C, Wang M, et al. Centromere protein F is identified as a novel
therapeutic target by genomics profile and contributing to the progression of pancreatic cancer. Genomics. 2021;113:1087–95. Article CAS PubMed Google Scholar * Aytes A, Mitrofanova A,
Lefebvre C, Alvarez MJ, Castillo-Martin M, Zheng T, et al. Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer
malignancy. Cancer Cell. 2014;25:638–51. Article CAS PubMed PubMed Central Google Scholar * O’Brien SL, Fagan A, Fox EJ, Millikan RC, Culhane AC, Brennan DJ, et al. CENP-F expression is
associated with poor prognosis and chromosomal instability in patients with primary breast cancer. Int J Cancer. 2007;120:1434–43. Article PubMed PubMed Central Google Scholar *
Wilkinson KD. Protein ubiquitination: a regulatory post-translational modification. Anticancer Drug Des. 1987;2:211–29. CAS PubMed Google Scholar * Cardozo T, Pagano M. The SCF ubiquitin
ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5:739–51. Article CAS PubMed Google Scholar * Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis
and treatment. Nat Med. 2014;20:1242–53. Article CAS PubMed Google Scholar * Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating
enzymes. Annu Rev Biochem. 2009;78:363–97. Article CAS PubMed Google Scholar * Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape.
Cell. 2009;138:389–403. Article CAS PubMed PubMed Central Google Scholar * Mofers A, Pellegrini P, Linder S, D’Arcy P. Proteasome-associated deubiquitinases and cancer. Cancer
Metastasis Rev. 2017;36:635–53. Article CAS PubMed PubMed Central Google Scholar * Clague MJ, Urbé S, Komander D. Breaking the chains: deubiquitylating enzyme specificity begets
function. Nat Rev Mol Cell Biol. 2019;20:338–52. Article CAS PubMed Google Scholar * Wang K, Liu Y, Yu Z, Gu B, Hu J, Huang L, et al. Phosphorylation at Ser68 facilitates DCAF11-mediated
ubiquitination and degradation of CENP-A during the cell cycle. Cell Rep. 2021;37:109987. Article CAS PubMed Google Scholar * Miyajima N, Maruyama S, Nonomura K, Hatakeyama S. TRIM36
interacts with the kinetochore protein CENP-H and delays cell cycle progression. Biochem Biophys Res Commun. 2009;381:383–7. Article CAS PubMed Google Scholar * Li B, Li Z, Lu C, Chang
L, Zhao D, Shen G, et al. Heat Shock Cognate 70 Functions as A Chaperone for the Stability of Kinetochore Protein CENP-N in Holocentric Insect Silkworms. Int J Mol Sci. 2019;20:5823. Article
CAS PubMed PubMed Central Google Scholar * Li T, Yan B, Ma Y, Weng J, Yang S, Zhao N, et al. Ubiquitin-specific protease 4 promotes hepatocellular carcinoma progression via cyclophilin
A stabilization and deubiquitination. Cell Death Dis. 2018;9:148. Article PubMed PubMed Central Google Scholar * Zhang H, Han Y, Xiao W, Gao Y, Sui Z, Ren P, et al. USP4 promotes the
proliferation, migration, and invasion of esophageal squamous cell carcinoma by targeting TAK1. Cell Death Dis. 2023;14:730. Article PubMed PubMed Central Google Scholar * Zhang X,
Berger FG, Yang J, Lu X. USP4 inhibits p53 through deubiquitinating and stabilizing ARF-BP1. EMBO J. 2011;30:2177–89. Article CAS PubMed PubMed Central Google Scholar * Zhao B,
Schlesiger C, Masucci MG, Lindsten K. The ubiquitin specific protease 4 (USP4) is a new player in the Wnt signalling pathway. J Cell Mol Med. 2009;13:1886–95. Article PubMed PubMed Central
Google Scholar * Li Z, Hao Q, Luo J, Xiong J, Zhang S, Wang T, et al. USP4 inhibits p53 and NF-κB through deubiquitinating and stabilizing HDAC2. Oncogene. 2016;35:2902–12. Article CAS
PubMed Google Scholar * Yun SI, Kim HH, Yoon JH, Park WS, Hahn MJ, Kim HC, et al. Ubiquitin specific protease 4 positively regulates the WNT/β-catenin signaling in colorectal cancer. Mol
Oncol. 2015;9:1834–51. Article CAS PubMed PubMed Central Google Scholar * Zhang L, Zhou F, Drabsch Y, Gao R, Snaar-Jagalska BE, Mickanin C, et al. USP4 is regulated by AKT
phosphorylation and directly deubiquitylates TGF-β type I receptor. Nat Cell Biol. 2012;14:717–26. Article CAS PubMed Google Scholar * Xing C, Lu XX, Guo PD, Shen T, Zhang S, He XS, et
al. Ubiquitin-Specific Protease 4-Mediated Deubiquitination and Stabilization of PRL-3 is required for potentiating colorectal oncogenesis. Cancer Res. 2016;76:83–95. Article CAS PubMed
Google Scholar * Chang W, Gao X, Han Y, Du Y, Liu Q, Wang L, et al. Gene expression profiling-derived immunohistochemistry signature with high prognostic value in colorectal carcinoma. Gut.
2014;63:1457–67. Article CAS PubMed Google Scholar * Xu X, Zhu L, Yang Y, Pan Y, Feng Z, Li Y, et al. Low tumour PPM1H indicates poor prognosis in colorectal cancer via activation of
cancer-associated fibroblasts. Br J Cancer. 2019;120:987–95. Article CAS PubMed PubMed Central Google Scholar * Xie Z, Lin H, Huang Y, Wang X, Lin H, Xu M, et al. BAP1-mediated MAFF
deubiquitylation regulates tumor growth and is associated with adverse outcomes in colorectal cancer. Eur J Cancer. 2024;210:114278. Article CAS PubMed Google Scholar * Mathew R,
Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–7. Article CAS PubMed PubMed Central Google Scholar * Farhan M, Silva M, Li S, Yan F, Fang J, Peng
T, et al. The role of FOXOs and autophagy in cancer and metastasis-Implications in therapeutic development. Med Res Rev. 2020;40:2089–113. Article CAS PubMed PubMed Central Google
Scholar * Garg M. Epithelial plasticity, autophagy and metastasis: potential modifiers of the crosstalk to overcome therapeutic resistance. Stem Cell Rev Rep. 2020;16:503–10. Article CAS
PubMed Google Scholar * Gundamaraju R, Lu W, Paul MK, Jha NK, Gupta PK, Ojha S, et al. Autophagy and EMT in cancer and metastasis: Who controls whom? Biochim Biophys Acta Mol Basis Dis.
2022;1868:166431. Article CAS PubMed Google Scholar * Dong Y, Jin Q, Sun M, Qi D, Qu H, Wang X, et al. CLDN6 inhibits breast cancer metastasis through WIP-dependent actin
cytoskeleton-mediated autophagy. J Exp Clin Cancer Res. 2023;42:68. Article CAS PubMed PubMed Central Google Scholar * Fan T, Wang X, Zhang S, Deng P, Jiang Y, Liang Y, et al. NUPR1
promotes the proliferation and metastasis of oral squamous cell carcinoma cells by activating TFE3-dependent autophagy. Signal Transduct Targeted Ther. 2022;7:130. Article CAS Google
Scholar * Han JH, Kim YK, Kim H, Lee J, Oh MJ, Kim SB, et al. Snail acetylation by autophagy-derived acetyl-coenzyme A promotes invasion and metastasis of KRAS-LKB1 co-mutated lung cancer
cells. Cancer Commun. 2022;42:716–49. Article Google Scholar * Marsh T, Debnath J. Autophagy suppresses breast cancer metastasis by degrading NBR1. Autophagy. 2020;16:1164–5. Article CAS
PubMed PubMed Central Google Scholar * Zhu Q, Zhang Q, Gu M, Zhang K, Xia T, Zhang S, et al. MIR106A-5p upregulation suppresses autophagy and accelerates malignant phenotype in
nasopharyngeal carcinoma. Autophagy. 2021;17:1667–83. Article CAS PubMed Google Scholar * Mennerich D, Kubaichuk K, Kietzmann T. DUBs, Hypoxia, and Cancer. Trends Cancer. 2019;5:632–53.
Article CAS PubMed Google Scholar * Mahar AL, Compton C, Halabi S, Hess KR, Weiser MR, Groome PA. Personalizing prognosis in colorectal cancer: a systematic review of the quality and
nature of clinical prognostic tools for survival outcomes. J Surg Oncol. 2017;116:969–82. Article PubMed PubMed Central Google Scholar * Dikic I, Elazar Z. Mechanism and medical
implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–64. Article CAS PubMed Google Scholar * Dower CM, Wills CA, Frisch SM, Wang HG. Mechanisms and context underlying
the role of autophagy in cancer metastasis. Autophagy. 2018;14:1110–28. Article CAS PubMed PubMed Central Google Scholar * Hu X, Zhang L, Li Y, Ma X, Dai W, Gao X, et al. Organoid
modelling identifies that DACH1 functions as a tumour promoter in colorectal cancer by modulating BMP signalling. EBioMedicine. 2020;56:102800. Article PubMed PubMed Central Google
Scholar * Duan J, Chen L, Gao H, Zhen T, Li H, Liang J, et al. GALNT6 suppresses progression of colorectal cancer. Am J Cancer Res. 2018;8:2419–35. CAS PubMed PubMed Central Google
Scholar * Yu F, Xie D, Ng SS, Lum CT, Cai MY, Cheung WK, et al. IFITM1 promotes the metastasis of human colorectal cancer via CAV-1. Cancer Lett. 2015;368:135–43. Article CAS PubMed
Google Scholar * Sung TY, Huang HL, Cheng CC, Chang FL, Wei PL, Cheng YW, et al. EGFL6 promotes colorectal cancer cell growth and mobility and the anti-cancer property of anti-EGFL6
antibody. Cell Biosci. 2021;11:53. Article CAS PubMed PubMed Central Google Scholar * Tufail M, Wu C. WNT5A: a double-edged sword in colorectal cancer progression. Mutat Res Rev Mutat
Res. 2023;792:108465. Article CAS PubMed Google Scholar * Zeng K, Li W, Wang Y, Zhang Z, Zhang L, Zhang W, et al. Inhibition of CDK1 Overcomes oxaliplatin resistance by regulating
ACSL4-mediated ferroptosis in colorectal cancer. Adv Sci. 2023;10:e2301088. Article Google Scholar * Lin H, Luo Y, Gong T, Fang H, Li H, Ye G, et al. GDF15 induces chemoresistance to
oxaliplatin by forming a reciprocal feedback loop with Nrf2 to maintain redox homeostasis in colorectal cancer. Cell Oncol. 2024;47:1149–65. Article CAS Google Scholar * Liang X, Duronio
GN, Yang Y, Bala P, Hebbar P, Spisak S, et al. An Enhancer-driven stem cell-like program mediated by SOX9 blocks intestinal differentiation in colorectal cancer. Gastroenterology.
2022;162:209–22. Article CAS PubMed Google Scholar * Ji Z, Mi A, Li M, Li Q, Qin C. Aberrant KIF23 expression is associated with adverse clinical outcome and promotes cellular malignant
behavior through the Wnt/β-catenin signaling pathway in Colorectal Cancer. J Cancer. 2021;12:2030–40. Article CAS PubMed PubMed Central Google Scholar * Wang Y, Pan S, He X, Wang Y,
Huang H, Chen J, et al. CPNE1 enhances colorectal cancer cell growth, glycolysis, and drug resistance through regulating the AKT-GLUT1/HK2 pathway. Onco Targets Ther. 2021;14:699–710.
Article PubMed PubMed Central Google Scholar * Shan E, Huo Y, Wang H, Zhang Z, Hu J, Wang G, et al. Differentiated embryonic chondrocyte expressed gene-1 (DEC1) enhances the development
of colorectal cancer with an involvement of the STAT3 signaling. Neoplasia. 2022;27:100783. Article CAS PubMed PubMed Central Google Scholar * Ko MJ, Seo YR, Seo D, Park SY, Seo JH,
Jeon EH, et al. RPL17 promotes colorectal cancer proliferation and stemness through ERK and NEK2/β-catenin signaling pathways. J Cancer. 2022;13:2570–83. Article CAS PubMed PubMed Central
Google Scholar * Wang L, Hu XD, Li SY, Liang XY, Ren L, Lv SX. ASPM facilitates colorectal cancer cells migration and invasion by enhancing β-catenin expression and nuclear translocation.
Kaohsiung J Med Sci. 2022;38:129–38. Article CAS PubMed Google Scholar * Yang L, Zheng L, Xie X, Luo J, Yu J, Zhang L, et al. Targeting PLA2G16, a lipid metabolism gene, by Ginsenoside
Compound K to suppress the malignant progression of colorectal cancer. J Adv Res. 2022;36:265–76. Article CAS PubMed Google Scholar * Zhang L, Wang X, Lai C, Zhang H, Lai M. PMEPA1
induces EMT via a non-canonical TGF-β signalling in colorectal cancer. J Cell Mol Med. 2019;23:3603–15. Article CAS PubMed PubMed Central Google Scholar * Hexiao T, Yuquan B, Lecai X,
Yanhong W, Li S, Weidong H, et al. Knockdown of CENPF inhibits the progression of lung adenocarcinoma mediated by ERβ2/5 pathway. Aging. 2021;13:2604–25. Article PubMed PubMed Central
Google Scholar * Wang Y, Zhou L, Lu J, Jiang B, Liu C, Guo J. USP4 function and multifaceted roles in cancer: a possible and potential therapeutic target. Cancer Cell Int. 2020;20:298.
Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by the Construction Project of Fujian Province Minimally Invasive Medical
Center ([2017]171), Joint Funds for innovation of Science and Technology, Fujian province (2020Y9071), Fujian provincial health technology project (2020CXA025). AUTHOR INFORMATION Author
notes * These authors contributed equally: Zhongdong Xie, Hanbin Lin, Yuecheng Wu, Yanan Yu, Xintong Liu. AUTHORS AND AFFILIATIONS * Department of Colorectal Surgery, Union Hospital, Fujian
Medical University, Fuzhou, China Zhongdong Xie, Xiaojie Wang, Yu Deng, Yan Linzhu, Li Qingyun, Ying Huang & Pan Chi * Department of Colorectal Surgery, The First Affiliated Hospital of
Wenzhou Medical University, Wenzhou, China Zhongdong Xie & Yating Zheng * Central Laboratory, Affiliated Hospital of Putian University, Putian, China Hanbin Lin * Key Laboratory of
Gastrointestinal Cancer (Fujian Medical University), Ministry of Education, Fuzhou, China Hanbin Lin, Yuecheng Wu, Yuting Han & Xinjian Lin * Guilin Medical University, Guilin, China
Yanan Yu * Central Laboratory, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, China Xintong Liu * Department of Science and Technology, The First Affiliated Hospital
of Wenzhou Medical University, Wenzhou, China Jiashu Wu * Department of Pathology, Union Hospital, Fujian Medical University, Fuzhou, China Meifang Xu & Lin Lin * Department of
Pathology, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, China Qiongying Zhang Authors * Zhongdong Xie View author publications You can also search for this author
inPubMed Google Scholar * Hanbin Lin View author publications You can also search for this author inPubMed Google Scholar * Yuecheng Wu View author publications You can also search for this
author inPubMed Google Scholar * Yanan Yu View author publications You can also search for this author inPubMed Google Scholar * Xintong Liu View author publications You can also search for
this author inPubMed Google Scholar * Yating Zheng View author publications You can also search for this author inPubMed Google Scholar * Xiaojie Wang View author publications You can also
search for this author inPubMed Google Scholar * Jiashu Wu View author publications You can also search for this author inPubMed Google Scholar * Meifang Xu View author publications You can
also search for this author inPubMed Google Scholar * Yuting Han View author publications You can also search for this author inPubMed Google Scholar * Qiongying Zhang View author
publications You can also search for this author inPubMed Google Scholar * Yu Deng View author publications You can also search for this author inPubMed Google Scholar * Lin Lin View author
publications You can also search for this author inPubMed Google Scholar * Yan Linzhu View author publications You can also search for this author inPubMed Google Scholar * Li Qingyun View
author publications You can also search for this author inPubMed Google Scholar * Xinjian Lin View author publications You can also search for this author inPubMed Google Scholar * Ying
Huang View author publications You can also search for this author inPubMed Google Scholar * Pan Chi View author publications You can also search for this author inPubMed Google Scholar
CONTRIBUTIONS ZX: writing–original draft, project administration, formal analysis, investigation and validation. HL, YW, YY, XL: Investigation, project administration, formal analysis and
validation. YZ, XW, JW, MX, YH, QZ, YD, LL, LY, QL: Investigation and validation. XL: writing–original draft, supervision, project administration, writing–review and editing. YH:
writing–original draft, supervision, project administration. PC: Supervision, funding acquisition, project administration, writing–review and editing. CORRESPONDING AUTHORS Correspondence to
Xinjian Lin, Ying Huang or Pan Chi. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains
neutral with regard to jurisdictional claims in published maps and institutional affiliations. Edited By Francesca Bernassola SUPPLEMENTARY INFORMATION SUPPLEMENTARY MATERIAL RIGHTS AND
PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any
medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The
images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not
included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly
from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Xie, Z., Lin, H.,
Wu, Y. _et al._ USP4-mediated CENPF deubiquitylation regulated tumor metastasis in colorectal cancer. _Cell Death Dis_ 16, 81 (2025). https://doi.org/10.1038/s41419-025-07424-3 Download
citation * Received: 03 July 2024 * Revised: 14 January 2025 * Accepted: 03 February 2025 * Published: 08 February 2025 * DOI: https://doi.org/10.1038/s41419-025-07424-3 SHARE THIS ARTICLE
Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided
by the Springer Nature SharedIt content-sharing initiative