Play all audios:
ABSTRACT B lymphoid development is initiated by the differentiation of hematopoietic stem cells into lineage committed progenitors, ultimately generating mature B cells. This highly
regulated process generates clonal immunological diversity via recombination of immunoglobulin V, D and J gene segments. While several transcription factors that control B cell development
and V(D)J recombination have been defined, how these processes are initiated and coordinated into a precise regulatory network remains poorly understood. Here, we show that the transcription
factor ETS Related Gene (_Erg_) is essential for early B lymphoid differentiation. Erg initiates a transcriptional network involving the B cell lineage defining genes, _Ebf1_ and _Pax5_,
which directly promotes expression of key genes involved in V(D)J recombination and formation of the B cell receptor. Complementation of Erg deficiency with a productively rearranged
immunoglobulin gene rescued B lineage development, demonstrating that Erg is an essential and stage-specific regulator of the gene regulatory network controlling B lymphopoiesis. SIMILAR
CONTENT BEING VIEWED BY OTHERS THE IMMUNOGLOBULIN HEAVY CHAIN SUPER ENHANCER CONTROLS CLASS SWITCH RECOMBINATION IN DEVELOPING B CELLS Article Open access 28 March 2024 A DOUBLE-NEGATIVE
THYMOCYTE-SPECIFIC ENHANCER AUGMENTS NOTCH1 SIGNALING TO DIRECT EARLY T CELL PROGENITOR EXPANSION, LINEAGE RESTRICTION AND Β-SELECTION Article 31 October 2022 THE TRANSCRIPTION FACTOR HHEX
COOPERATES WITH THE COREPRESSOR TLE3 TO PROMOTE MEMORY B CELL DEVELOPMENT Article 29 June 2020 INTRODUCTION Transcription factors are critical for controlling the expression of genes that
regulate B-cell development. The importance of specific B-lymphoid transcription factors is highlighted by the phenotype of gene knockout models. Failure of B-cell lineage specification from
multi-potential progenitors occurs with deletion of _Ikzf1_1 and _Spi1_ (_Pu.1)_2, while deletion of _Tcf3 (E2A)_3 and _Foxo1_4 results in failure of B-cell development from common lymphoid
progenitors (CLPs). Developmental arrest later in B lymphopoiesis is observed with deletion of _Ebf1_ and _Pax5_ at the pre–proB and proB stages, respectively5,6. This sequential pattern of
developmental arrest associated with the loss of gene function, along with ectopic gene complementation studies2, gene expression profiling7 and analysis of transcription factor binding to
target genes, support models in which transcription factors are organised into hierarchical gene regulatory networks that specify B-lymphoid lineage fate, commitment and function8. Two
transcription factors that have multiple roles during B-cell development are Ebf1, a member of the COE family, and Pax5, a member of the PAX family. While Ebf1 and Pax5 have been shown to
bind to gene regulatory elements of a common set of target genes in a co-dependent manner during later stages of B lineage commitment9, both manifest distinct roles during different
developmental stages. Ebf1 has been proposed to form a transcriptional network with E2A and Foxo1 in CLPs that appears important in early B-lymphoid fate determination10, while during later
stages of B lymphopoiesis, Ebf1 acts as a pioneer transcription factor that regulates chromatin accessibility at a subset of genes co-bound by Pax511 as well as at the _Pax5_ promoter
itself12. Pax5 in contrast, regulates B-cell genomic organisation13 including the _Immunoglobulin heavy chain (Igh)_ locus during V(D)J recombination, co-operating with factors such as
CTCF14, as well as transactivating15 and facilitating the activity of the recombinase activating gene (Rag) complex16. It is unclear, however, how these various functions of Ebf1 and Pax5
are co-ordinated during different stages of B-lymphoid development. In particular, it would be important to ensure co-ordinated _Ebf1_ and _Pax5_ co-expression before the pre-BCR checkpoint,
such that _Ebf1_ and _Pax5_ co-regulated target genes required for V(D)J recombination and pre-B-cell receptor complex formation are optimally expressed9. Here we show that the ETS-related
gene (_Erg_), a member of the ETS family of transcription factors, plays this vital role in B lymphopoiesis. Deletion of _Erg_ from early lymphoid progenitors resulted in developmental
arrest at the early pre–proB-cell stage and loss of VH-to-DJH recombination. Gene expression profiling, DNA-binding analysis and complementation studies demonstrated Erg to be a
transcriptional regulator that lies at the apex of an Erg-dependent Ebf1 and Pax5 gene regulatory network commencing in pre–proB cells. This co-dependent transcriptional network directly
controls expression of the _Rag1/Rag2_ recombinase activating genes and the _Lig4_ and _Xrcc6_ DNA repair genes required for V(D)J recombination, as well as expression of components of the
pre-BCR complex such as _CD19, Igll1, Vpreb1_ and _Vpreb2_. Taken together, we define an essential Erg-mediated transcription factor network required for regulation of _Ebf1_ and _Pax5_
expression that is exquisitely stage specific during early B-lymphoid development. RESULTS _ERG_ IS REQUIRED FOR B-CELL DEVELOPMENT To build on prior work defining the role of the
transcription factor _Erg_ in regulation of hematopoietic stem cells (HSCs)17 and megakaryocyte-erythroid specification18, we sought to identify whether _Erg_ played roles in other
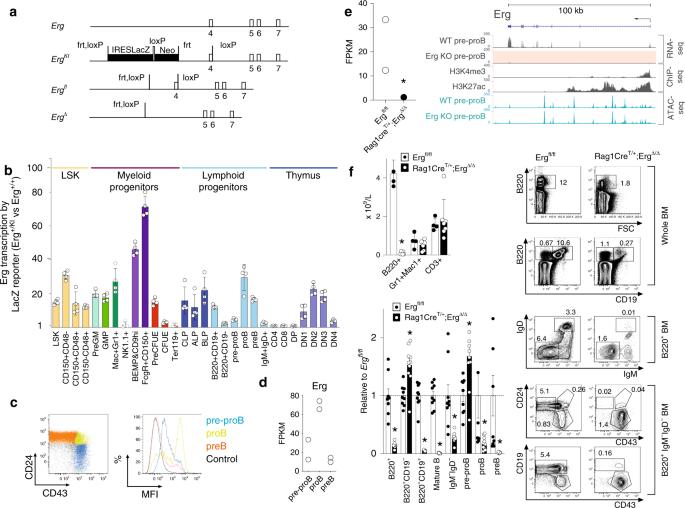
hematopoietic lineages. _Erg_ expression in adult hematopoiesis was first examined by generating mice carrying the _Erg_tm1a(KOMP)wtsi knock-in first reporter allele (_Erg__KI_) (Fig. 1a).
Consistent with the known role for _Erg_ in hematopoiesis17,18,19,20,21, significant _LacZ_ expression driven by the endogenous _Erg_ promoter was observed in HSCs and multi-potential
progenitor cells, as well as in granulocyte-macrophage and megakaryocyte-erythroid progenitor populations, with declining activity accompanying erythroid maturation (Fig. 1b with definitions
of cells examined provided in Supplementary Table 1 and representative flow cytometry plots in Supplementary Fig. 1). In other lineages, transcription from the _Erg_ locus was evident in
CLP, all lymphoid and B-cell-biased lymphoid progenitor cells, as well as in B lineage committed pre–proB, proB and preB cells and double-negative thymic T-lymphoid cell subsets, with a
reduction in transcription with later B- and T-cell maturation (Fig. 1b, c). We confirmed these findings with RNA-sequencing (RNA-seq) analysis that showed significant _Erg_ RNA in pre–proB,
proB and preB cells (Fig. 1d). This detailed characterisation of _Erg_ expression raised the possibility that _Erg_ plays a stage-specific function at early developmental stages of the
lymphoid lineages. To determine whether _Erg_ had a role in lymphoid development, mice carrying floxed _Erg_ alleles (_Erg__fl/fl_, Fig. 1a) were interbred with _Rag1Cre_ transgenic mice
that efficiently delete floxed alleles in CLPs and T- and B-committed progenitor cells22, but have normal lymphoid development (Supplementary Fig. 2a). The resulting _Rag1Cre_T/+;_Erg_Δ_/_Δ
mice specifically lack _Erg_ throughout lymphopoiesis (Fig. 1e_,_ Supplementary Fig. 2b). While numbers of red blood cells, platelets and other white cells were normal,
_Rag1Cre_T/+;_Erg_Δ_/_Δ mice displayed a deficit in circulating lymphocytes (Supplementary Table 2). This was due to a specific absence of B cells; the numbers of circulating T cells and
thymic progenitors were not decreased (Fig. 1f_,_ Supplementary Fig. 2c). B cells are produced from bone marrow progenitor cells that progress through regulated developmental stages.
B-lymphoid development was markedly compromised in _Rag1Cre_T/+;_Erg_Δ_/_Δ mice, with proB, preB, immature B and mature recirculating B cells (Hardy fractions C-F, defined in Supplementary
Table 1) markedly reduced in number or virtually absent (Fig. 1f). A B-lymphoid developmental block was clearly evident at the pre–proB (Hardy fraction A-to-B) stage, with excess numbers of
these cells present in the bone marrow. _ERG_ DEFICIENCY PERTURBS VH-TO-DJH RECOMBINATION To further characterise the developmental B lineage block in _Rag1Cre_T/+;_Erg_Δ_/_Δ mice, B220+
bone marrow cells were examined for _Igh_ somatic recombination. Unlike cells from control _Erg__fl/fl_ mice, B220+ cells from _Rag1Cre_T/+;_Erg_Δ_/_Δ mice had not undergone significant
VH-to-DJH immunoglobulin heavy chain gene rearrangement, although DH-to-JH recombination was relatively preserved (Fig. 2a). We next investigated the abnormalities underlying _Igh_
recombination in greater detail. We first undertook fluorescence in situ hybridisation (FISH) at the _Igh_ locus to measure the intra-chromosomal distance between distal VHJ558 and proximal
VH7183 VH family genes, as cell stage-specific contraction of the _Igh_ locus is essential for efficient V(D)J recombination23. This revealed that pre–proB cells from _Rag1Cre_T/+;_Erg_Δ_/_Δ
mice had reduced locus contraction compared with _Erg__fl/fl_ controls (Fig. 2b). To assess whether other structural perturbations across the _Igh_ locus were also present, high throughput
chromatin conformation capture (in situ Hi-C) was performed. We performed a differential analysis of the data and revealed a reduction of long-range interactions across the _Igh_ locus in
_Rag1Cre_T/+;_Erg_Δ_/_Δ pre–proB cells when compared with _Erg__fl/fl_ and C57BL/6 controls (Fig. 2c). As these findings were also observed in _Pax5_ deficient cells13,23 reflecting a direct
role for Pax5 in co-ordinating the structure of the _Igh_ locus14, we mapped _Erg_ binding sites across the _Igh_ locus by ChIP-seq. Unlike well-defined Pax5 binding to Pax5- and
CTCF-associated intergenic regions (PAIR domains)14,16, Erg binding to VH families was not identified across the locus (Fig. 2c, Supplementary Fig. 3a). Thus, a structural role for Erg in
maintaining the multiple long-range interactions and VH-to-DJH recombination in normal cells is unlikely and cannot account for the absence of these in _Rag1Cre_T/+;_Erg_Δ_/_Δ pre–proB
cells. Analysis of _Igh_ locus accessibility by ATAC-seq did not reveal any significant difference between Erg-deficient pre–proB, proB and preB cells and control cells (Supplementary Fig.
3a), suggesting that the loss of locus accessibility either by chromatin regulation24 or by peripheral nuclear positioning with lamina-associated domain silencing25 were not mechanisms that
could adequately explain reduced _Igh_ locus contraction, reduction of long range interactions, and loss of VH-to-DJH recombination in the absence of _Erg_. A potential role for ETS family
of transcription factors in regulation of immunoglobulin gene rearrangement was proposed from experiments investigating the iEμ enhancer: a complex _cis_-activating element located in the
intronic region between the _Igh_ joining region (JH) and constant region (Cμ) implicated in efficient VH-to-DJH recombination and _Igh_ chain transcription26. The iEμ enhancer is proposed
to nucleate a three-loop domain at the 3′ end of _Igh_ interacting with the VH region to juxtapose 5′ and 3′ ends of the heavy chain locus27. Erg and its closest related ETS family member,
Fli1, were shown to bind to the μA element and trans-activate iEμ co-operatively with a bHLH transcription factor in vitro28. We therefore sought to determine whether the lack of Erg, and
Erg binding in particular to the μA site of iEμ, could account for the loss of VH-to-DJH recombination observed in _Rag1Cre_T/+;_Erg_Δ_/_Δ mice in vivo. While ChIP-PCR demonstrated Erg
binding to the iEμ enhancer containing the μA element (Supplementary Fig. 3b), mice in which the μA region (μAΔ/Δ) was deleted had preserved numbers of circulating mature B cells compared
with cEμΔ/+ controls (Fig. 2d) and intact VH-to-DJH recombination (Supplementary Fig. 3c). This was in contrast to cEμΔ/Δ mice, in which a core 220 bp element of iEμ was deleted, in which a
marked reduction of circulating mature IgM+IgD+ B cells was evident in peripheral blood, in keeping with previous models29 (Fig. 2d). Importantly, ChIP-seq did not demonstrate Erg binding to
other iEμ enhancer regions in μAΔ/Δ proB cells (Supplementary Fig. 3d). Together these data show that while Erg can bind to the μA region of the iEμ in vivo, deletion of this region did not
result in significant perturbation of B lymphoid development. It is therefore unlikely that Erg binding to μA element of iEμ could account for the loss of VH-to-DJH recombination in
particular, or the _Rag1Cre_T/+;_Erg_Δ_/_Δ phenotype in general. REARRANGED _IGH_ ALLELE PERMITS _ERG_-DEFICIENT B LYMPHOPOIESIS Given the loss of VH-to-DJH recombination associated with
structural perturbation of the _Igh_ locus in Erg-deficient pre–proB cells, we sought to complement the loss of formation of a functional _Igh_ μ transcript and in doing so, determine
whether failure to form a pre-BCR complex was a principal reason for the developmental block in _Rag1Cre_T/+;_Erg_Δ_/_Δ mice30. Complementation with a functionally rearranged _Igh_ allele in
models of defective VH-to-DJH recombination such as deletion of _Rag1, Rag2_, or components of DNA-dependent protein kinase (DNA-PK) that mediate VH-to-DJH recombination, can overcome the
pre-BCR developmental block31,32,33,34. The _IgH__VH10tar_ knock-in allele that expresses productive _Igh_HEL transcripts under endogenous _Igh_ locus regulation32 was therefore used to
generate mice that lacked _Erg_ in B-cell progenitors but would undergo stage-appropriate expression of the rearranged _Igh_HEL chain (_Rag1Cre_T/+;_Erg_ Δ/Δ;_IgH_VH10tar/+). The presence of
the _IgH__VH10tar_ allele permitted B-cell development in the absence of _Erg_. The bone marrow of _Rag1Cre_T/+;_Erg_Δ/Δ;_IgH_VH10tar/+ mice contained significant numbers of B220+IgM+ B
cells and, notably, CD25+CD19+IgM− preB cells, a population coincident with successful pre-BCR formation35, that were virtually absent in _Rag1Cre_T/+;_Erg_Δ/Δ mice (Fig. 3a). The rescued
preB cells, however, were at lower numbers compared with _Erg__fl/fl_, _Erg__fl/fl_;_IgH_VH10tar/+ and _Rag1Cre_T/+ controls. This was likely due to restriction of the clonal repertoire
permitted by the _IgH__VH10tar_ allele as the predominant _Igh_ clone (Fig. 3c). Similarly, in the spleens of _Rag1Cre_T/+;_Erg_Δ/Δ;_IgH_VH10tar/+ mice, near normal numbers of all B-lymphoid
populations were observed, in contrast to the marked reduction in _Rag1Cre_T/+;_Erg_Δ/Δ mice (Fig. 3b). Notably, IgκL chain recombination had proceeded in
_Rag1Cre_T/+;_Erg_Δ/Δ;_IgH_VH10tar/+ cells (Fig. 3c). We next tested whether the rescued _Rag1Cre_T/+;_Erg_ Δ/Δ;_IgH_VH10tar/+ splenic B cells were functional in the absence of _Erg_.
_Rag1Cre_T/+;_Erg_Δ/Δ;_IgH_VH10tar/+ splenocytes were indistinguishable from wild-type controls in in vitro proliferative assays using anti-μ stimulation, T-cell dependent stimulation with
CD40 ligand, IL4 and IL5, or T-cell independent stimulation using lipopolysaccharide (Fig. 3d). _Rag1Cre_T/+;_Erg_ Δ/Δ;_IgH_VH10tar/+ splenic B cells were also able to differentiate normally
as measured by formation of CD138+ plasma cells and IgG1 class switch recombination (Fig. 3e, f). Circulating _Rag1Cre_T/+;_Erg_Δ/Δ;_IgH_VH10tar_/+_ B cells also expressed IgD, unlike their
_Rag1Cre_T/+;_Erg_Δ/Δ counterparts (Fig. 3g). These experiments demonstrated that loss of a functional _Igh_ μ transcript and failure to form a pre-BCR complex was a principal reason for
lack of B-cell development in _Rag1Cre_T/+;_Erg_Δ_/_Δ mice. _ERG_-DEFICIENT PRE–PROB CELLS DO NOT EXPRESS EBF1 AND PAX5 To define the mechanism by which _Erg_ regulates VH-to-DJH
recombination and pre-BCR formation, we undertook gene expression profiling of _Rag1Cre_T/+;_Erg_Δ/Δ pre–proB cells. Differential gene expression and gene-ontogeny analysis of differentially
expressed genes in _Rag1Cre_T/+;_Erg_Δ/Δ pre–proB compared with _Erg__fl/fl_ pre–proB cells demonstrated deregulated expression of multiple B lymphoid genes (Fig. 4a). These included genes
encoding cell surface or adhesion receptors and core components of the pre-BCR complex _CD19, CD22, Igll1, Vpreb1, Vpreb2, CD79a and CD79b_, genes required for _Igh_ recombination such as
_Rag1_ and _Rag2_ and components of non-homologous end-joining repair complex associated with V(D)J recombination: _Xrcc6_ (Ku70) and _Lig4_, and importantly, transcription factors
implicated in B-cell development (_Ebf1, Pax5, Tcf3, Bach2, Irf4, Myc, Pou2af1, Lef1, Myb_) (Fig. 4b). _Ebf1_ and _Pax5_ are critical for B lineage specification5 and maintenance36,37 and
act co-operatively to regulate a gene network in early B-cell fates9. Because we observed with the loss of _Erg,_ reduced expression of several critical B lineage genes previously identified
to be controlled by _Ebf1_ and/or _Pax5_, for example _CD19_, _Vpreb1_, and _Igll1_ (Fig. 4a), we speculated that Erg may play an important role in regulating the expression of these two
essential transcription factors and their targets. To determine if Erg bound _Ebf1_ and/or _Pax5_ gene regulatory regions and directly regulated their expression, we undertook ChIP-seq
analysis in wild-type proB cells and ATAC-seq to assess locus accessibility at the _Ebf1_ and _Pax5_ loci in the absence of _Erg_ in _Rag1Cre_T/+;_Erg_Δ/Δ pre–proB cells and proB and preB
cells rescued with the _IgH__VH10tar_ knock-in allele. This demonstrated direct Erg binding to the proximal (β) promoter region of _Ebf1_38 as well as to the _Pax5_ promoter and _Pax5_
lymphoid-specific intron 5 enhancer12 (Fig. 4c_,_ Supplementary Fig. 4b). Direct Erg binding to these regulatory regions together with the absence of _Ebf1_and _Pax5_ transcription in
_Rag1Cre_T/+;_Erg_Δ/Δ pre–proB cells and the loss of Ebf1 and Pax5 protein in _Rag1Cre_T/+;_Erg_Δ/Δ by western blot (Fig. 4d), demonstrated that Erg was a direct transcriptional regulator of
_Ebf1_ and _Pax5_. Importantly, the loss of _Ebf1_ and _Pax5_ expression occurred while expression of other known regulators of _Ebf1_ expression, namely, _Foxo1_, _Spi1, Tcf3_ and _Ikzf1_
were maintained (Supplementary Fig. 4a), and both _Ebf1_ and _Pax5_ loci remained accessible by ATAC-seq in _Rag1Cre_T/+;_Erg_Δ/Δ pre–proB cells (Fig. 4c). Reinforcing the observation that
Erg, Ebf1 and Pax5 may form a co-ordinated transcriptional network, the _Erg_ promoter region was directly bound by Pax5, and the _Erg_ enhancer region was bound by Pax5 and Ebf1 (Fig. 4c).
To better understand the roles of Erg, Ebf1 and Pax5 in the B-cell lineage trajectory, single-cell RNA-seq of CLP, pre–proB and CD19+ proB and preB populations was examined (GSE 114793, Fig.
4e). Consistent with our other analysis (Fig. 1b), an increase in _Erg_ expression in CLPs, pre–proB and proB cells was observed (Fig. 4e) with the identity of proB and preB populations
confirmed with analysis of additional B lineage genes (Supplementary Fig. 5). Importantly, _Erg_ expression preceded the expression of _Ebf1_ and _Pax5_ in the B lineage trajectory, with
_Ebf1_ and _Pax5_ expression increasing during the later proB and preB stages. Taken together, this data strongly supported an apical role for Erg in initiating _Ebf1_ and _Pax5_ expression
during early B-cell development. AN _ERG_, _EBF1_ AND _PAX5_ CO-DEPENDENT GENE REGULATORY NETWORK As we observed Ebf1 and Pax5 binding to _cis_-regulatory regions of the _Erg_ locus (Fig.
4c), we determined whether Ebf1 and Pax5 could regulate _Erg_ gene expression in B-cell progenitors by examining a publicly available dataset in which _Ebf1_ (_Ebf1_Δ/Δ) or _Pax5_
(_Pax5_Δ/Δ) had been deleted (Fig. 5a). Deletion of either _Ebf1_ or _Pax5_ resulted in reduced _Erg_ expression (Fig. 5b), with Ebf1 appearing to be the stronger influence. We next compared
gene expression changes in _Ebf1_Δ/Δ pre–proB cells and _Pax5_Δ/Δ proB cells to those genes regulated by Erg in pre–proB cells. As would be predicted if Erg, Ebf1 and Pax5 were components
of a co-dependent gene regulatory network, this analysis showed a highly significant correlation in gene expression changes observed with _Ebf1_ or _Pax5_ deletion in pre–proB and proB cells
and those observed with _Erg_ deletion in pre–proB cells. This was noted for downregulated genes in Erg, Ebf1 and Pax5 deficient cells in particular (Fig. 5c). Finally, to confirm that Ebf1
and Pax5 were transcriptional regulators downstream of Erg, transduction of _Rag1Cre_T/+;_Erg_Δ/Δ pre–proB cells with MSCV-driven constructs for constitutive overexpression of _Ebf1_ and
_Pax5_ was performed. This experiment demonstrated rescue of B220 expression with _Ebf1_ or _Pax5_ overexpression in Erg deficient cells (Fig. 5d). Notably, partial rescue of CD19 expression
and VH-to-DJH recombination was observed with _Ebf1_ overexpression, while no rescue was observed with _Pax5_ overexpression (Fig. 5d, e). RNA-seq analysis of _Erg_-deficient cells
transduced with _Ebf1_ or _Pax5_ expression vectors demonstrated _Ebf1_ overexpression could rescue the expression of several target genes of the transcriptional network including _Pax5_
itself, genes involved in pre-BCR signalling (such as _Vpreb1_, _Vpreb2, CD79a, CD79b, CD22 and CD19_), genes involved in V-to-DJH recombination (such as _Rag1_, _Rag2_), as well as
transcription from the _Igh_ locus (_Ighv1-5_, _Ighv1-7_, _Ighv1-4_). In the absence of _Ebf1_, _Pax5_ overexpression alone induced the expression of a much more limited set of these target
genes (Fig. 5f). Therefore these data suggest that _Pax5_ lies downstream of _Ebf1_ and supports the model where Ebf1 facilitates the role of Pax5 in B-cell development11. These findings
were also in keeping with a hierarchical model of Erg, Ebf1 and Pax5 forming a co-dependent transcriptional network that co-regulate critical target genes required for VH-to-DJH
recombination and pre-BCR signaling. ERG CO-BINDS COMMON EBF1 AND PAX5 TARGET GENES Because expression of multiple B-cell genes were deregulated in _Rag1Cre_T/+;_Erg_Δ/Δ pre–proB cells,
including those to which Ebf1 and Pax5 had been shown to directly bind and regulate, we investigated the possibility that Erg co-bound common target genes to reinforce the Ebf1 and Pax5 gene
network using a genome-wide motif analysis of Erg DNA-binding sites in proB cells. As expected, the most highly enriched motif underlying Erg binding was the ETS motif. However, significant
enrichment of _Ebf1-, E2A-, Pax5_- and _Foxo1_-binding motifs were also identified within 50 bp of Erg-binding sites (Fig. 6a), suggesting that Erg acts co-operatively with other
transcription factors to regulate target gene expression in a co-dependent gene network. Analysis of the binding of each of Erg, Ebf1 and Pax5 to regulatory regions of genes that were
differentially expressed in _Rag1Cre_T/+;_Erg_Δ/Δ pre–proB cells was then undertaken. This analysis identified significant overlap of Erg-, Ebf1- and Pax5-binding sites within 5 kb of the
transcriptional start site (TSS) of genes differentially expressed in _Rag1Cre_T/+;_Erg_Δ/Δ pre–proB cells compared with control pre–proB cells (Fig. 6b). Taken together, these data provided
further compelling evidence for a gene regulatory network in which Erg is required for initiating and maintaining expression of _Ebf1_ and _Pax5_ from the pre–proB cell stage of
development, as well as reinforcing expression of target genes within the network by co-operative binding and co-regulation of target genes with Ebf1 and Pax5. To further delineate the
directly regulated target genes in an Erg-dependent Ebf1 and Pax5 transcriptional network, we undertook mapping of ChIP-seq binding of Erg, Ebf1 and Pax5 to Erg-dependent genes at the
pre–proB cell stage of development. The majority of these target genes demonstrated direct combinatorial binding of Erg, Ebf1 and/or Pax5 to annotated promoter regions, gene body
enhancer/putative enhancer regions or putative distal enhancer regions of these genes (Fig. 6c). Detailed examination of several key target genes for which expression was completely
dependent on Erg in pre–proB cells identified direct binding of Erg to the promoter and enhancer regions for several pre-BCR components, including _CD19_, _Igll1, Vpreb1_ and _CD79a_. This
occurred with co-ordinate binding of Ebf1 and Pax5 to the regulatory regions of these genes15 (Fig. 6d). In addition, indirect regulation by Erg at the _Rag1/Rag2_ locus was also identified,
with downregulation of expression of transcription factors that bind and regulate the _Rag2_ promoter such _Pax5_, _Lef1_ and _c-Myb_ in _Rag1Cre_T/+;_Erg_Δ/Δ pre–proB cells (Fig. 4b)39, as
well as direct binding of Erg to the conserved B-cell specific _Erag_ enhancer40 (Supplementary Fig. 4a, b). Importantly, the loss of _Rag1_ and _Rag2_ expression in _Rag1Cre_T/+;_Erg_Δ/Δ
pre–proB cells occurred while expression of _Foxo1_, a positive regulator of the locus41 was relatively maintained (Supplementary Fig. 4a). An Erg-Ebf1-Pax5 mediated gene regulatory network
was then mapped using each target gene, expression of which was perturbed in _Rag1Cre_T/+;_Erg_Δ/Δ pre–proB cells, and that was directly bound by Erg, Ebf1 and/or Pax5 at promoter, proximal
or distal gene regions, to provide a comprehensive representation of this gene network (Fig. 6e). This highlights the interdependent roles of these transcription factors in multiple cellular
processes required for B lymphopoiesis. An important observation arising from our data was that the B-lymphoid developmental block arising in _Rag1Cre_T/+;_Erg_Δ/Δ pre–proB cells could be
overcome with the provision of a rearranged functional _Igh_ VH10tar allele. This suggested that once the pre-BCR checkpoint was bypassed, _Erg_ was no longer critical for further B-cell
development and function, including VLJL recombination of the _Igl_ and BCR formation (Fig. 3c, d). Indeed, beyond the pre-BCR checkpoint, re-emergence of _Ebf1_ and _Pax5_ expression
occurred (Fig. 4c) as well as expression of target genes of the _Ebf1_ and _Pax5_ network (Fig. 6d_,_ Supplementary Fig. 4a) in Erg-deficient _Rag1Cre_T/+;_Erg_Δ/Δ;_IgH_VH10tar/+ proB and
preB cells rescued with a VH10tar allele. This was in keeping with the expression pattern of _Erg_ in the B lineage trajectory (Figs. 1b–d and 4e) and defines the role of Erg as an
exquisitely stage-specific regulator of early B-cell development. DISCUSSION In this study we explored the role of the transcription factor Erg in B lymphopoiesis. Our studies suggest that
_Erg_ expression from the CLP stage of development initiates a transcriptional network comprised of Erg, Ebf1 and Pax5 in pre–proB and proB cells to regulate VH-to-DJH _Igh_ recombination
and pre-BCR signaling (Fig. 1b, Fig. 4e). This important role for Erg in B-cell development was demonstrated in mice in which _Erg_ had been deleted throughout lymphopoiesis, which exhibited
a developmental block at the pre–proB cell stage that was associated with profound defects in VH-to-DJH recombination, _Igh_ locus organisation and transcriptional changes in multiple
B-cell genes, including loss of expression of _Ebf1_, and _Pax5_. Combining RNA-seq, ChIP-seq and gene complementation studies, we were able to define a co-dependent transcriptional network
between Erg, Ebf1 and Pax5, with direct Erg binding to the proximal (β) _Ebf1_ promoter, to which Pax5, Ets1 and Pu.1 also co-operatively bind38, as well as Erg binding to the _Pax5_
promoter and potent intron 5 enhancer region, two critical regulatory elements required for correct transcriptional initiation of _Pax5_ in early B-cell development12. These data support a
model (Fig. 6f) in which increased _Erg_ expression from CLPs is required to initiate and maintain _Ebf1_ and _Pax5_ expression in pre–proB cells and proB cells, to establish an
inter-dependent B-lymphoid gene regulatory network. Together Erg, Ebf1 and Pax5 directly co-regulated the expression of multiple genes that had previously been identified as direct
transcriptional targets of Ebf1 and Pax5 (Fig. 6c–e). Direct Erg binding to promoters of the pre-BCR signalling complex genes such as _Igll1_, _VpreB_ and _CD79a_, establish Erg as a
transcriptional regulator of target genes in this network. In addition to _Rag1_ and _Rag2_, we also identified network regulation of expression of _Xrcc6_, the gene encoding the Ku70
subunit of DNA-dependent protein kinase holoenzyme (DNA-PK) that binds DNA double strand breaks during V(D)J recombination42, and _Lig4_, encoding the XRCC4-associated DNA-ligase that is
required for DNA-end joining during V(D)J recombination43 (Supplementary Fig. 4a, b). Along with direct Erg promotion of expression of _Pax5_, a structural regulator of the _Igh_ locus,
these findings are sufficient to explain the _Rag1Cre_T/+;_Erg_Δ/Δ phenotype in which VH-to-DJH recombination was lost. Together with loss of expression of components of the pre-BCR complex,
we can conclude B-cell development was blocked as a consequence of _Erg_ deletion due to the collapse of the Erg-mediated transcriptional network. Importantly, re-emergence of _Ebf1_ and
_Pax5_ expression beyond the pre-BCR checkpoint in _Igh_-rescued _Rag1Cre__T/+_;_Erg__Δ/Δ_;_IgH__VH10tar/+_ proB and preB cells was observed, along with expression of target genes of Ebf1
and Pax5. This demonstrates that Erg is a stage-specific regulator of early B-cell development, with emergence of an Erg-independent Ebf1 and Pax5 gene network during the later stages of
B-cell development, once clones have transitioned through the pre-BCR checkpoint. This would allow IgL chain VL to JL recombination and BCR formation to proceed in preB cells in which
endogenous _Erg_ expression is also reduced (Figs. 1b, c and 4e). The transcriptional regulators of _Ebf1_ and _Pax5_ expression during these later stages of B-cell development remain to be
defined. _Erg,_ however, is critical for initiating and maintaining _Ebf1_ and _Pax5_ expression in pre–proB and proB cells (Fig. 4e), orchestrating a transcriptional network required for
early B-cell development. In this role, Erg not only co-ordinates the transcriptional functions of Ebf1 and Pax5, but also directly binds and activates critical target genes required for
transition through the pre-BCR checkpoint. METHODS MICE Mice carrying the _Erg_tm1a(KOMP)wtsi knock-first reporter allele44 (_Erg__KI_, KOMP Knockout Mouse Project) were generated by gene
targeting in ES cells. Mice with a conditional _Erg_ knockout allele (_Erg__fl_) from which the IRES-LacZ cassette was excised were generated by interbreeding _Erg__KI_ mice with _Flpe_
transgenic mice45. _Rag1Cre_ mice46, in which Cre recombinase is expressed during lymphopoiesis from the CLP stage22, were interbred with _Erg__fl_ mice to generate mice lacking _Erg_ in
lymphopoiesis (_Rag1Cre_T/+;_Erg_Δ/Δ) and _Rag1Cre_+/+;_Erg_fl/fl (_Erg__fl/fl_) controls. Mice carrying the rearranged immunoglobulin heavy chain _IgH__VH10tar_ allele47 were a gift from
Professor Robert Brink. The cEμΔ/Δ and μΑΔ/Δ mice were generated by the MAGEC laboratory (Walter and Eliza Hall Institute of Medical Research)48 on a C57BL/6J background. To generate cEμΔ
mice, 20 ng/μl of Cas9 mRNA, 10 ng/μl of sgRNA (GTTGAGGATTCAGCCGAAAC and ATGTTGAGTTGGAGTCAAGA) and 40 ng/μl of oligo donor
(CAAGCTAAAATTAAAAGGTTGAACTCAATAAGTTAAAAGAGGACCTCTCCAGTTTCGGCTCAACTCAACATTGCTCAATTCATTTAAAAATATTTGAAACTTAATTTATTATTGTTAAAA) were injected into the cytoplasm of fertilised one-cell stage
embryos. To generate μΑΔ mice, 20 ng/μl of Cas9 mRNA, 10 ng/μl of sgRNA (GAACACCTGCAGCAGCTGGC) and 40 ng/μl of oligo donor
(GCTACAAGTTTACCTAGTGGTTTTATTTTCCCTTCCCCAAATAGCCTTGCCACATGACCTGCCAGCTGCTGCAGGTGTTCTGGTTCTGATCGGCCATCTTGACTCCAACTCAACATTGCT) were injected into the cytoplasm of fertilized one-cell stage
embryos. Twenty-four hours later, two-cell stage embryos were transferred into the oviducts of pseudo-pregnant female mice. Viable offspring were genotyped by next-generation sequencing.
Non-commercial unique materials are subject to Materials Transfer Agreements. Mice were co-housed in a barrier facility and analysed from 6 to 18 weeks of age. Male and female mice were
used. The primers and PCR conditions used for genotyping are provided in Supplementary Table 3. This study was performed in accordance with the Australian Code for the Care and Use of
Animals for Scientific Purposes, published by the Australian National Health and Medical Research Council. Euthanasia was performed by CO2 induction or cervical dislocation. Experimental
procedures were approved by the Walter and Eliza Hall Institute of Medical Research Animal Ethics Committee. PRIMARY CELL CULTURE B-cell progenitors were obtained from bone marrow that was
lineage depleted using biotinylated Ter119, Mac1, Gr1, CD3, CD4, and CD8 antibodies, anti-biotin microbeads and LS columns (Miltenyi Biotec) and cultured on OP9 stromal cells in Iscove’s
Modified Dulbecco’s Medium (Gibco, Invitrogen) supplemented with 10% (v/v) foetal calf serum (Gibco, Invitrogen), 50 μM β-mercaptoethanol as well as murine interleukin-7 (10 ng/mL) at 37 °C
in 10% CO2 for 7 days. Splenic B cells were purified by negative selection using a B-cell isolation kit (Miltenyi Biotec)49 and purity was confirmed by flow cytometry prior to labelling with
Cell Trace Violet (CTV; Life technologies) as per the manufacturer’s instructions. Labelled cells were seeded at 5 × 104 cells per well and cultured for 90 h. HAEMATOLOGY Blood was
collected into tubes containing EDTA (Sarstedt) and analysed on an Advia 2120 analyser (Bayer). FLOW CYTOMETRY Single-cell suspensions from bone marrow, lymph node or spleen were prepared in
balanced salt solution (BSS-CS: 0.15 M NaCl, 4 mM KCl, 2 mM CaCl2, 1 mM MgSO4, 1 mM KH2PO4, 0.8 mM K2HPO4, and 15 mM HEPES supplemented with 2% [vol/vol] bovine calf serum). Analysis of
blood was performed after erythrocyte lysis in buffered 156 mM NH4Cl. Staining was performed using biotinylated or fluorochrome-conjugated antibodies specific for murine antigens Ter119
(Ly-76), CD41 (MWReg30), Gr1 (Ly6G and Ly6C), Mac1 (CD11b), NK1.1, CD11c (N418), CD45R/B220 (RA3-6B2), CD19 (1D3), CD3 (17A2), CD4 (GK1.5), CD8a (53.6.7), Sca1 (Ly6A/E, D7), cKit (CD117,
ACK4 or 2B8), CD150 (TC15-12F12.2), CD105 (MJ7/18), CD16/32 (24G2), CD127 (A7R34), CD135 (A2F10), Ly6D (49-H4), CD21/CD35 (7G6), CD23 (B3B4), CD93 (AA4.1), CD24 (M1/69), CD43 (S7), CD45.2
(S450-15-2), CD45.1 (A20), IgMb (AF6-78), IgD (11-26 c.2a), CD138 (281.2), IgG1 (X56), CD25 (3C7), CD44 (IM7). Secondary staining used streptavidin PE-Texas-Red (Invitrogen). See
Supplementary Table 4 for antibody dilutions and catalogue numbers for commercial antibodies. FACS-Gal analysis was performed using warm hypotonic loading of fluorescein di
β-D-galactopyranoside (Molecular Probes) on single cells50 followed by immunophenotyping using relevant surface antigens as defined in Supplementary Table 1. Cells were analysed using a LSR
II or FACS Canto flow cytometer (Becton Dickinson) or sorted using a FACSAria II (Becton Dickinson) flow cytometer after antibody staining and lineage selection or depletion using
anti-biotin beads and LS columns (Miltenyi Biotec). Data were analysed using FlowJo software (Version 8.8.7, Tree Star). SPLENIC B-CELL CULTURE Purified and CTV labelled splenic B cells were
cultured with either AffiniPure F(ab’)2 Fragment Goat Anti-Mouse IgM µ Chain Specific (20 μg/ml; Jackson Immunoresearch), CD40L (produced in-house51) supplemented with IL4 (10 ng/ml;
R&D systems) and IL5 (5 ng/ml; R&D systems) to assess T-cell dependent responses, or LPS (25 ug/ml; Difco) to assess T-cell independent responses, and analysed by flow cytometry.
ANALYSIS OF PUBLICLY AVAILABLE RNA-SEQ DATASETS FASTQ files containing RNA-seq profiles of pre–proB cells from Ebf1Δ/Δ (GSM2879293, GSM2879294, GSM2879295), pro-B cells from Pax5Δ/Δ
(GSM2879296, GSM2879297, GSM2879298) and control populations from wild-type mice (GSM2879299, GSM2879300, GSM2879301). Reads were aligned to the mm10 genome using Rsubread’s align function
and read counts were summarised at the gene level as for the primary samples (See Supplementary Methods)52. Genes were filtered from downstream analysis using edgeR’s filterByExpr function
and library sizes were TMM normalised. Counts were transformed to log2-CPM and the mean-variance relationship estimated using the _voom_ function in limma53. Heatmaps were generated using
heatmap.2 function in gplots. Genes were tested for differential expression using linear modelling in limma 3.38.254. Gene set testing was performed using _camera_55 and barcode plots were
generated with limma. For single cell RNA-seq analysis raw counts corresponding to single-cell RNA-seq of wild-type mouse CLPs, pre–proB and CD19+ B lymphoid progenitor cells were downloaded
from Gene Expression Omnibus repository, accession GSE114793. Raw counts were filtered to remove low expressed genes and cells with low cell quality. Read counts were then L1 normalised
such that the sum of expression values for each cell sums to 1. Library sizes were then normalised by median counts per cell. Normalised read counts were then imputed using the MAGIC
algorithm56 (Rmagic v 1.4.0) with the settings _t_ = 11, _k_ = 30 with other parameters set at default values. The tSNE visualisation of the first 20 principal components of the imputed
values was obtained using Rtsne (v 0.15) package with the following parameters: perplexity parameter = 80, momentum of 0.5 for the first 250 iterations and a final momentum of 0.8. The
learning rate of the tSNE was set to 200 with an exaggeration factor of 12. PCA initialisation was disabled. All of the analysis was performed in R version 3.6.1. ATAC-SEQ ANALYSIS
ATAC-seq57 was performed on sorted pre–proB, proB and preB populations. Briefly, 5 × 104 nuclei were fragmented by sonication for 30 min at 37 °C and the DNA purified prior to amplification
with indexing primers (HiFi Ready Mix, Kapa Biosciences) for 13 PCR cycles followed by quality assessment by Bioanalyser. High quality libraries were size selected (150–700 base pairs) and
sequenced using a high output paired end 75 base pair kit on the NextSeq 500 (Illumina) to a minimum of 50 million reads. ATAC-seq reads were aligned to mm10 genome using Bowtie258
(http://bowtie-bio.sourceforge.net/bowtie2/index.shtml accessed 6th March 2017). Peak calling was performed using MACS259. Intersections of genetic coordinates were performed using Bedtools
(http://bedtools.readthedocs.io/en/latest/ accessed 6 March 2017). Heatmaps of unique peaks were generated using pHeatmap in R. These data have been deposited in Gene Expression Omnibus
database (accession number GSE132852). VISUALISATION OF RNA-SEQ, CHIP-SEQ AND ATASEQ DATA RNA-seq, ChIP-seq and ATAC-seq files were converted to BigWig files using deepTools (version 2)60
and uploaded to Cyverse (www.cyverse.org) for visualisation in UCSC Genome Browser61 (genome.ucsc.edu). GENE NETWORK ANALYSIS All Ebf1-, Pax5- and Erg-ChIP-seq peaks mapping to
differentially expressed genes in _Rag1Cre_T/+;_Erg__Δ/Δ_ pre–proB cells within 10 kb of the TSS were identified. Peaks inside the gene body were annotated as “proximal targets”, peaks
overlapping the TSS were labelled as promoter regulated targets, peaks less than 3 kb upstream or downstream of the TSS were labelled as putative promoter regulated targets, peaks more than
3 kb upstream or downstream TSS were labelled as putative distal targets. Gene Ontogeny (GO) annotation of differentially expressed genes was performed and underwent expert manual curation.
The network was constructed using62 CRAN package, and exported to Cytoscape63 for customisation using RCy364 R/Bioconductor package. HI-C ANALYSIS For in situ Hi-C analysis13,65, primary
immune cell libraries were generated in biological duplicates for each genotype. An Illumina NextSeq 500 was used to sequence libraries with 80 bp paired-end reads to produce libraries of
sizes between 42 million and 100 million valid read pairs. Each sample was aligned to the mm10 genome using the _diffHic_ package v1.14.066, which utilises cutadapt v0.9.567 and bowtie2
v2.2.558 for alignment. The resultant BAM file was sorted by read name, the FixMateInformation command from the Picard suite v1.117 (https://broadinstitute.github.io/picard/) was applied,
duplicate reads were marked and then re-sorted by name. Read pairs were determined to be dangling ends and removed if the pairs of inward-facing reads or outward-facing reads on the same
chromosome were separated by less than 1000 bp for inward-facing reads and 6000 bp for outward-facing reads. Read pairs with fragment sizes above 1000 bp were removed. An estimate of an
alignment error was obtained by comparing the mapping location of the 3′ segment of each chimeric read with that of the 5′ segment of its mate. A mapping error was determined to be present
if the two segments were not inward-facing and separated by less than 1000 bp, and around 1–2% were estimated to have errors. Differential interactions (DIs) between the three different
groups were detected using the _diffHic_ package66. Read pairs were counted into 100 kbp bin pairs. Bins were discarded if found on sex chromosomes, contained a count of less than 10,
contained blacklisted genomic regions as defined by ENCODE for mm1068 or were within a centromeric or telomeric region. Filtering of bin-pairs was performed using the filterDirect function,
where bin pairs were only retained if they had average interaction intensities more than 5-fold higher than the background ligation frequency. The ligation frequency was estimated from the
inter-chromosomal bin pairs from a 500 kbp bin-pair count matrix. The counts were normalised between libraries using a loess-based approach. Tests for DIs were performed using the
quasi-likelihood (QL) framework69 of the edgeR package. The design matrix was constructed using a layout that specified the cell group to which each library belonged and the mouse sex. A
mean-dependent trend was fitted to the negative binomial dispersions with the estimateDisp function. A generalised linear model (GLM) was fitted to the counts for each bin pair70, and the QL
dispersion was estimated from the GLM deviance with the glmQLFit function. The QL dispersions were then squeezed towards a second mean-dependent trend, using a robust empirical Bayes
strategy71. A _P_ value was computed against the null hypothesis for each bin pair using the QL _F_ test. _P_ values were adjusted for multiple testing using the Benjamini–Hochberg method. A
DI was defined as a bin pair with a false discovery rate (FDR) below 5%. DIs adjacent in the interaction space were aggregated into clusters using the diClusters function to produce
clustered DIs. DIs were merged into a cluster if they overlapped in the interaction space, to a maximum cluster size of 1 Mbp. The significance threshold for each bin pair was defined such
that the cluster-level FDR was controlled at 5%. Cluster statistics were computed using the _csaw_ package v1.16.072. Overlaps between unclustered bin pairs and genomic intervals were
performed using the InteractionSet package73. Plaid plots were constructed using the contact matrices and the plotHic function from the _Sushi R_ package74. The colour palette was inferno
from the _viridis_ package (https://github.com/sjmgarnier/viridis accessed 30 March 2018) and the range of colour intensities in each plot was scaled according to the library size of the
sample. The plotBedpe function of the _Sushi_ package was used to plot the unclustered DIs as arcs where the _z_-score shown on the vertical access was calculated as -log10(_p_-value). These
data have been deposited in Gene Expression Omnibus database (accession number GSE133246). FLUORESCENCE IN SITU HYBRIDISATION Cultured B-cell progenitors were resuspended in hypotonic 0.075
M KCl solution and warmed to 37 °C for 20 min. Cells were pelleted and resuspended in 3:1 (vol/vol) methanol:glacial acetic acid fixative. Fixed cells were dropped onto coated ShandonTM
polysine slides (ThermoFisher Scientific) and air dried. The cells were hybridised with FISH probes (Creative Bioarray) at 37 °C for 16 h beneath a coverslip sealed with Fixogum (Marabu)
after denaturation at 73 °C for 5 min. Cells were washed at 73 °C in 0.4× SSC/0.3%NP40 for 2 min followed by 2× SSC/0.1%NP40 for less than 1 min at room temperature and air dried in the dark
and cover slipped. Images of nuclei were captured on an inverted Zeiss LSM 880 confocal using a 63×/1.4 NA oil immersion objective. Z-stacks of images were then captured using the lambda
scan mode, a 405 and a multi-band pass beam splitter (488/561/633). The following laser lines were used: 405, 488, 561 and 633 nm. Spectral data were captured at 8 nm intervals. In all
cases, images were set up with a pixel size of 70 nm and an interval of 150 nm for z-stacks. Single dye controls using the same configuration were captured and spectra imported for spectral
unmixing using the Zen software (Zen 2.3, Zeiss Microscopy). Unmixed data were then deconvolved using the batch express tool in Huygens professional software (Scientific Volume Imaging).
Images were analysed using TANGO software75 after linear deconvolution. Nuclear boundaries were extracted in TANGO using the background nuclear signal in the Aqua channel. A 3D median filter
was applied and the 3D image projected with maximum 2D image projection for nuclei detection using the Triangle method for automated thresholding in ImageJ76. Binary image holes were filled
and a 2D procedure implemented to separate touching nuclei using ImageJ 2D watershed implementation. The 2D boundaries of the detected nuclei were expanded in 3D and inside each 3D
delimited region, Triangle thresholding was applied to detect the nuclear boundary in the 3D space. Acquired images from immunofluorescent probes were first filtered using 3D median and 3D
tophat filter to enhance spot-like structures followed by application of the “spotSegmenter” TANGO plugin with only the best four spots having the brightest intensity kept for analysis. The
spots identified by TANGO were manually verified against the original immunofluorescent image to identify and record the correct distance computed by TANGO between the aqua and 5-Rox
immunofluorescent probes for both _Igh_ alleles within a nucleus. STATISTICAL ANALYSIS Student’s unpaired two-tailed _t_ tests were used using GraphPad Prism (GraphPad Software) unless
otherwise specified. Unless otherwise stated, a _P_ value of <0.05 was considered significant. Details of reagents and software packages used are provided in Supplementary Table 4.
REPORTING SUMMARY Further information on research design is available in the Nature Research Reporting Summary linked to this article. DATA AVAILABILITY The following datasets analysed in
the current study are available at the NCBI Gene Expression Omnibus, accession GSE132852 (ATAC-seq), GSE132853 (ChIP-seq), GSE132854 (RNA-seq), GSE133246 (Hi-C). The source data underlying
Figs. 1b, d–f, 2a, b, d, 3a–d, f, g, 4d, 5b, d–f, Supplementary Figs. 2a–c, 3b, c are provided in the Source Data file. The data supporting this study are available in the Article,
Supplementary Information, Source Data or available from the authors upon reasonable requests. Source data are provided with this paper. REFERENCES * Reynaud, D. et al. Regulation of B cell
fate commitment and immunoglobulin heavy-chain gene rearrangements by Ikaros. _Nat. Immunol._ 9, 927–936 (2008). Article CAS PubMed PubMed Central Google Scholar * Medina, K. L. et al.
Assembling a gene regulatory network for specification of the B cell fate. _Dev. Cell_ 7, 607–617 (2004). Article CAS PubMed Google Scholar * Bain, G. et al. E2A proteins are required
for proper B cell development and initiation of immunoglobulin gene rearrangements. _Cell_ 79, 885–892 (1994). Article CAS PubMed Google Scholar * Mansson, R. et al. Positive intergenic
feedback circuitry, involving EBF1 and FOXO1, orchestrates B-cell fate. _Proc. Natl Acad. Sci. USA_ 109, 21028–21033 (2012). Article ADS CAS PubMed PubMed Central Google Scholar * Lin,
H. & Grosschedl, R. Failure of B-cell differentiation in mice lacking the transcription factor EBF. _Nature_ 376, 263–267 (1995). Article ADS CAS PubMed Google Scholar * Nutt, S.
L., Urbanek, P., Rolink, A. & Busslinger, M. Essential functions of Pax5 (BSAP) in pro-B cell development: difference between fetal and adult B lymphopoiesis and reduced V-to-DJ
recombination at the IgH locus. _Genes Dev._ 11, 476–491 (1997). Article CAS PubMed Google Scholar * Miyai, T. et al. Three-step transcriptional priming that drives the commitment of
multipotent progenitors toward B cells. _Genes Dev._ 32, 112–126 (2018). Article CAS PubMed PubMed Central Google Scholar * Nutt, S. L. & Kee, B. L. The transcriptional regulation
of B cell lineage commitment. _Immunity_ 26, 715–725 (2007). Article CAS PubMed Google Scholar * Li, R. et al. Dynamic EBF1 occupancy directs sequential epigenetic and transcriptional
events in B-cell programming. _Genes Dev._ 32, 96–111 (2018). Article CAS PubMed PubMed Central Google Scholar * Lin, Y. C. et al. A global network of transcription factors, involving
E2A, EBF1 and Foxo1, that orchestrates B cell fate. _Nat. Immunol._ 11, 635–643 (2010). Article CAS PubMed PubMed Central Google Scholar * Boller, S. et al. Pioneering activity of the
C-terminal domain of EBF1 shapes the chromatin landscape for B cell programming. _Immunity_ 44, 527–541 (2016). Article CAS PubMed Google Scholar * Decker, T. et al. Stepwise activation
of enhancer and promoter regions of the B cell commitment gene Pax5 in early lymphopoiesis. _Immunity_ 30, 508–520 (2009). Article CAS PubMed Google Scholar * Johanson, T. M. et al.
Transcription-factor-mediated supervision of global genome architecture maintains B cell identity. _Nat. Immunol._ 19, 1257–1264 (2018). Article CAS PubMed Google Scholar * Ebert, A. et
al. The distal V(H) gene cluster of the Igh locus contains distinct regulatory elements with Pax5 transcription factor-dependent activity in pro-B cells. _Immunity_ 34, 175–187 (2011).
Article MathSciNet CAS PubMed Google Scholar * Ochiai, K. et al. A self-reinforcing regulatory network triggered by limiting IL-7 activates pre-BCR signaling and differentiation. _Nat.
Immunol._ 13, 300–307 (2012). Article CAS PubMed PubMed Central Google Scholar * Zhang, Z. et al. Transcription factor Pax5 (BSAP) transactivates the RAG-mediated V(H)-to-DJ(H)
rearrangement of immunoglobulin genes. _Nat. Immunol._ 7, 616–624 (2006). Article CAS PubMed Google Scholar * Ng, A. P. et al. Erg is required for self-renewal of hematopoietic stem
cells during stress hematopoiesis in mice. _Blood_ 118, 2454–2461 (2011). Article CAS PubMed Google Scholar * Ng, A. P. et al. Early lineage priming by trisomy of erg leads to
myeloproliferation in a down syndrome model. _PLoS Genet._ 11, e1005211 (2015). Article PubMed PubMed Central CAS Google Scholar * Loughran, S. J. et al. The transcription factor Erg is
essential for definitive hematopoiesis and the function of adult hematopoietic stem cells. _Nat. Immunol._ 9, 810–819 (2008). Article CAS PubMed Google Scholar * Ng, A. P. et al.
Trisomy of Erg is required for myeloproliferation in a mouse model of Down syndrome. _Blood_ 115, 3966–3969 (2010). Article CAS PubMed Google Scholar * Knudsen, K. J. et al. ERG promotes
the maintenance of hematopoietic stem cells by restricting their differentiation. _Genes Dev._ 29, 1915–1929 (2015). Article CAS PubMed PubMed Central Google Scholar * Malin, S. et al.
Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro-B cell development. _Nat. Immunol._ 11, 171–179 (2010). Article CAS PubMed Google Scholar *
Fuxa, M. et al. Pax5 induces V-to-DJ rearrangements and locus contraction of the immunoglobulin heavy-chain gene. _Genes Dev._ 18, 411–422 (2004). Article CAS PubMed PubMed Central
Google Scholar * Kumari, G. & Sen, R. Chromatin Interactions in the control of immunoglobulin heavy chain gene assembly. _Adv. Immunol._ 128, 41–92 (2015). Article CAS PubMed Google
Scholar * Zullo, J. M. et al. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. _Cell_ 149, 1474–1487 (2012). Article CAS PubMed Google
Scholar * Perlot, T. & Alt, F. W. Cis-regulatory elements and epigenetic changes control genomic rearrangements of the IgH locus. _Adv. Immunol._ 99, 1–32 (2008). Article CAS PubMed
PubMed Central Google Scholar * Guo, C. et al. Two forms of loops generate the chromatin conformation of the immunoglobulin heavy-chain gene locus. _Cell_ 147, 332–343 (2011). Article CAS
PubMed PubMed Central Google Scholar * Rivera, R. R., Stuiver, M. H., Steenbergen, R. & Murre, C. Ets proteins: new factors that regulate immunoglobulin heavy-chain gene expression.
_Mol. Cell Biol._ 13, 7163–7169 (1993). Article CAS PubMed PubMed Central Google Scholar * Marquet, M. et al. The Emu enhancer region influences H chain expression and B cell fate
without impacting IgVH repertoire and immune response in vivo. _J. Immunol._ 193, 1171–1183 (2014). Article CAS PubMed Google Scholar * Rajewsky, K. Clonal selection and learning in the
antibody system. _Nature_ 381, 751–758 (1996). Article ADS CAS PubMed Google Scholar * Chang, Y., Bosma, G. C. & Bosma, M. J. Development of B cells in scid mice with immunoglobulin
transgenes: implications for the control of V(D)J recombination. _Immunity_ 2, 607–616 (1995). Article CAS PubMed Google Scholar * Cook, A. J. et al. Reduced switching in SCID B cells
is associated with altered somatic mutation of recombined S regions. _J. Immunol._ 171, 6556–6564 (2003). Article CAS PubMed Google Scholar * Spanopoulou, E. et al. Functional
immunoglobulin transgenes guide ordered B-cell differentiation in Rag-1-deficient mice. _Genes Dev._ 8, 1030–1042 (1994). Article CAS PubMed Google Scholar * Young, F. et al. Influence
of immunoglobulin heavy- and light-chain expression on B-cell differentiation. _Genes Dev._ 8, 1043–1057 (1994). Article CAS PubMed Google Scholar * Rolink, A., Grawunder, U., Winkler,
T. H., Karasuyama, H. & Melchers, F. IL-2 receptor alpha chain (CD25, TAC) expression defines a crucial stage in pre-B cell development. _Int Immunol._ 6, 1257–1264 (1994). Article CAS
PubMed Google Scholar * Nutt, S. L., Heavey, B., Rolink, A. G. & Busslinger, M. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. _Nature_ 401, 556–562
(1999). Article ADS CAS PubMed Google Scholar * Nechanitzky, R. et al. Transcription factor EBF1 is essential for the maintenance of B cell identity and prevention of alternative fates
in committed cells. _Nat. Immunol._ 14, 867–875 (2013). Article CAS PubMed Google Scholar * Roessler, S. et al. Distinct promoters mediate the regulation of Ebf1 gene expression by
interleukin-7 and Pax5. _Mol. Cell Biol._ 27, 579–594 (2007). Article CAS PubMed Google Scholar * Jin, Z. X. et al. Lymphoid enhancer-binding factor-1 binds and activates the
recombination-activating gene-2 promoter together with c-Myb and Pax-5 in immature B cells. _J. Immunol._ 169, 3783–3792 (2002). Article CAS PubMed Google Scholar * Hsu, L. Y. et al. A
conserved transcriptional enhancer regulates RAG gene expression in developing B cells. _Immunity_ 19, 105–117 (2003). Article CAS PubMed Google Scholar * Amin, R. H. & Schlissel, M.
S. Foxo1 directly regulates the transcription of recombination-activating genes during B cell development. _Nat. Immunol._ 9, 613–622 (2008). Article CAS PubMed PubMed Central Google
Scholar * Gu, Y. et al. Growth retardation and leaky SCID phenotype of Ku70-deficient mice. _Immunity_ 7, 653–665 (1997). Article CAS PubMed Google Scholar * Frank, K. M. et al. Late
embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. _Nature_ 396, 173–177 (1998). Article ADS CAS PubMed Google Scholar * Skarnes, W. C. et al. A
conditional knockout resource for the genome-wide study of mouse gene function. _Nature_ 474, 337–342 (2011). Article CAS PubMed PubMed Central Google Scholar * Farley, F. W., Soriano,
P., Steffen, L. S. & Dymecki, S. M. Widespread recombinase expression using FLPeR (flipper) mice. _Genesis_ 28, 106–110 (2000). Article CAS PubMed Google Scholar * McCormack, M. P.,
Forster, A., Drynan, L., Pannell, R. & Rabbitts, T. H. The LMO2 T-cell oncogene is activated via chromosomal translocations or retroviral insertion during gene therapy but has no
mandatory role in normal T-cell development. _Mol. Cell Biol._ 23, 9003–9013 (2003). Article CAS PubMed PubMed Central Google Scholar * Phan, T. G. et al. B cell receptor-independent
stimuli trigger immunoglobulin (Ig) class switch recombination and production of IgG autoantibodies by anergic self-reactive B cells. _J. Exp. Med._ 197, 845–860 (2003). Article CAS PubMed
PubMed Central Google Scholar * Kueh, A. J. et al. An update on using CRISPR/Cas9 in the one-cell stage mouse embryo for generating complex mutant alleles. _Cell Death Differ._ 24,
1821–1822 (2017). Article CAS PubMed PubMed Central Google Scholar * Hasbold, J., Corcoran, L. M., Tarlinton, D. M., Tangye, S. G. & Hodgkin, P. D. Evidence from the generation of
immunoglobulin G-secreting cells that stochastic mechanisms regulate lymphocyte differentiation. _Nat. Immunol._ 5, 55–63 (2004). Article CAS PubMed Google Scholar * Berger, C. N., Tan,
S. S. & Sturm, K. S. Simultaneous detection of beta-galactosidase activity and surface antigen expression in viable haematopoietic cells. _Cytometry_ 17, 216–223 (1994). Article CAS
PubMed Google Scholar * Kehry, M. R. & Castle, B. E. Regulation of CD40 ligand expression and use of recombinant CD40 ligand for studying B cell growth and differentiation. _Semin.
Immunol._ 6, 287–294 (1994). Article CAS PubMed Google Scholar * Liao, Y., Smyth, G. K. & Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and
quantification of RNA sequencing reads. _Nucleic Acids Res._ 47, e47 (2019). Article CAS PubMed PubMed Central Google Scholar * Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom:
precision weights unlock linear model analysis tools for RNA-seq read counts. _Genome Biol._ 15, R29 (2014). Article PubMed PubMed Central CAS Google Scholar * Ritchie, M. E. et al.
limma powers differential expression analyses for RNA-sequencing and microarray studies. _Nucleic Acids Res._ 43, e47 (2015). Article PubMed PubMed Central CAS Google Scholar * Wu, D.
& Smyth, G. K. Camera: a competitive gene set test accounting for inter-gene correlation. _Nucleic Acids Res_ 40, e133 (2012). Article CAS PubMed PubMed Central Google Scholar * van
Dijk, D. et al. Recovering gene interactions from single-cell data using data diffusion. _Cell_ 174, 716–729 e727 (2018). Article PubMed PubMed Central CAS Google Scholar * Buenrostro,
J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins
and nucleosome position. _Nat. Methods_ 10, 1213–1218 (2013). Article CAS PubMed PubMed Central Google Scholar * Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with
Bowtie 2. _Nat. Methods_ 9, 357–359 (2012). Article CAS PubMed PubMed Central Google Scholar * Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). _Genome Biol._ 9, R137 (2008).
Article PubMed PubMed Central CAS Google Scholar * Ramirez, F., Dundar, F., Diehl, S., Gruning, B. A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data.
_Nucleic Acids Res._ 42, W187–W191 (2014). Article CAS PubMed PubMed Central Google Scholar * Raney, B. J. et al. Track data hubs enable visualization of user-defined genome-wide
annotations on the UCSC Genome Browser. _Bioinformatics_ 30, 1003–1005 (2014). Article CAS PubMed Google Scholar * Csárdi, G. & Nepusz, T. The igraph software package for complex
network research. _Int. J. Complex Syst_. 1695 (2006). * Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. _Genome Res._ 13,
2498–2504 (2003). Article CAS PubMed PubMed Central Google Scholar * Ono, K., Muetze, T., Kolishovski, G., Shannon, P. & Demchak, B. CyREST: turbocharging cytoscape access for
external tools via a RESTful API. _F1000Res._ 4, 478 (2015). Article PubMed PubMed Central CAS Google Scholar * Rao, S. S. et al. A 3D map of the human genome at kilobase resolution
reveals principles of chromatin looping. _Cell_ 159, 1665–1680 (2014). Article CAS PubMed PubMed Central Google Scholar * Lun, A. T. & Smyth, G. K. diffHic: a bioconductor package
to detect differential genomic interactions in Hi-C data. _BMC Bioinform._ 16, 258 (2015). Article CAS Google Scholar * Martin, M. Cutadapt removes adapter sequences from high-throughput
sequencing reads. _EMBnet. J._ 17, 10–12 (2011). Article Google Scholar * Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. _Nature_ 489, 57–74 (2012).
Article ADS CAS Google Scholar * Chen, Y., Lun, A. T. & Smyth, G. K. From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the
edgeR quasi-likelihood pipeline. _F1000Res._ 5, 1438 (2016). PubMed PubMed Central Google Scholar * McCarthy, D. J., Chen, Y. & Smyth, G. K. Differential expression analysis of
multifactor RNA-Seq experiments with respect to biological variation. _Nucleic Acids Res._ 40, 4288–4297 (2012). Article CAS PubMed PubMed Central Google Scholar * Phipson, B., Lee, S.,
Majewski, I. J., Alexander, W. S. & Smyth, G. K. Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. _Ann. Appl.
Stat._ 10, 946–963 (2016). Article MathSciNet PubMed PubMed Central MATH Google Scholar * Lun, A. T. & Smyth, G. K. csaw: a Bioconductor package for differential binding analysis
of ChIP-seq data using sliding windows. _Nucleic Acids Res._ 44, e45 (2016). Article PubMed CAS Google Scholar * Lun, A. T., Perry, M. & Ing-Simmons, E. Infrastructure for genomic
interactions: bioconductor classes for Hi-C, ChIA-PET and related experiments. _F1000Res._ 5, 950 (2016). Article PubMed PubMed Central Google Scholar * Phanstiel, D. H., Boyle, A. P.,
Araya, C. L. & Snyder, M. P. Sushi.R: flexible, quantitative and integrative genomic visualizations for publication-quality multi-panel figures. _Bioinformatics_ 30, 2808–2810 (2014).
Article CAS PubMed PubMed Central Google Scholar * Ollion, J., Cochennec, J., Loll, F., Escude, C. & Boudier, T. TANGO: a generic tool for high-throughput 3D image analysis for
studying nuclear organization. _Bioinformatics_ 29, 1840–1841 (2013). Article CAS PubMed PubMed Central Google Scholar * Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image
to ImageJ: 25 years of image analysis. _Nat. Methods_ 9, 671–675 (2012). Article CAS PubMed PubMed Central Google Scholar * Zhang, J. A., Mortazavi, A., Williams, B. A., Wold, B. J.
& Rothenberg, E. V. Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. _Cell_ 149, 467–482 (2012). Article CAS PubMed
PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We thank Janelle Lochland, Jason Corbin, Jasmine McManus, Melanie Salzone, Carolina Alvarado, Keti Stoev, Nicole Lynch
and Shauna Ross for skilled assistance. We thank Professor Robert Brink for the VH10tar knock-in mouse line. This work was supported by Program Grants (1113577, 1016647, 1054618, 1054925),
Project Grant (A.P.N. 1060179, 1122783, M.J.H. 1186575, 1159658), Fellowship (D.M.T. 1060675, S.L.N. 1155342, WSA 1058344, T.M.J. 1124081, M.J.H. 1156095), C.R.B. Blackburn Scholarship
(M.S.Y.L., Australian National Health and Medical Research Council jointly with Royal Australasian College of Physicians) and Independent Research Institutes Infrastructure Support Scheme
Grant (361646) from the Australian National Health and Medical Research Council, the Australian Cancer Research Fund and Victorian State Government Operational Infrastructure Support. Y.C.C.
was supported through Maddie Riewoldt’s Vision. M.J.H. was supported through the Leukemia and Lymphoma Society of America (LLS SCOR 7001-13). The MAGEC laboratory was supported by the
Australian Phenomics Network and the Australian Government through the National Collaborative Research Infrastructure Strategy Program. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Blood
Cells and Blood Cancer Division, The Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, 3052, Australia Ashley P. Ng, Kira Behrens, Andrew J. Kueh, Ladina DiRago, Craig D.
Hyland, Helen Ierino, Sandra Mifsud, Elizabeth Viney, Tracy Willson, Marco J. Herold & Warren S. Alexander * Department of Medical Biology, The University of Melbourne, Parkville, VIC,
3010, Australia Ashley P. Ng, Hannah D. Coughlan, Timothy M. Johanson, Michael Sze Yuan Low, Andrew J. Kueh, Thomas Boudier, Rhys S. Allan, Marco J. Herold, Kelly Rogers, Gordon K. Smyth,
Melissa J. Davis, Stephen L. Nutt & Warren S. Alexander * Bioinformatics Division, The Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, 3052, Australia Hannah D.
Coughlan, Soroor Hediyeh-zadeh, Gordon K. Smyth & Melissa J. Davis * Immunology Division, The Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, 3052, Australia Timothy
M. Johanson, Michael Sze Yuan Low, Rhys S. Allan & Stephen L. Nutt * Monash Haematology, Monash Hospital, Clayton, VIC, 3004, Australia Michael Sze Yuan Low * Peter MacCallum Cancer
Centre, Parkville, VIC, 3000, Australia Charles C. Bell, Omer Gilan, Yih-Chih Chan & Mark A. Dawson * Sir Peter MacCallum Department of Oncology, The University of Melbourne, Parkville,
VIC, 3010, Australia Charles C. Bell, Omer Gilan, Yih-Chih Chan & Mark A. Dawson * Advanced Technology and Biology Division, The Walter and Eliza Hall Institute of Medical Research,
Parkville, VIC, 3052, Australia Thomas Boudier & Kelly Rogers * Inflammation Division, The Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, 3010, Australia Rebecca
Feltham & Anna Gabrielyan * Centre for Cancer Research, The University of Melbourne, Parkville, VIC, 3010, Australia Mark A. Dawson * Department of Immunology and Pathology, Monash
University, Melbourne, VIC, 3004, Australia David M. Tarlinton Authors * Ashley P. Ng View author publications You can also search for this author inPubMed Google Scholar * Hannah D.
Coughlan View author publications You can also search for this author inPubMed Google Scholar * Soroor Hediyeh-zadeh View author publications You can also search for this author inPubMed
Google Scholar * Kira Behrens View author publications You can also search for this author inPubMed Google Scholar * Timothy M. Johanson View author publications You can also search for this
author inPubMed Google Scholar * Michael Sze Yuan Low View author publications You can also search for this author inPubMed Google Scholar * Charles C. Bell View author publications You can
also search for this author inPubMed Google Scholar * Omer Gilan View author publications You can also search for this author inPubMed Google Scholar * Yih-Chih Chan View author
publications You can also search for this author inPubMed Google Scholar * Andrew J. Kueh View author publications You can also search for this author inPubMed Google Scholar * Thomas
Boudier View author publications You can also search for this author inPubMed Google Scholar * Rebecca Feltham View author publications You can also search for this author inPubMed Google
Scholar * Anna Gabrielyan View author publications You can also search for this author inPubMed Google Scholar * Ladina DiRago View author publications You can also search for this author
inPubMed Google Scholar * Craig D. Hyland View author publications You can also search for this author inPubMed Google Scholar * Helen Ierino View author publications You can also search for
this author inPubMed Google Scholar * Sandra Mifsud View author publications You can also search for this author inPubMed Google Scholar * Elizabeth Viney View author publications You can
also search for this author inPubMed Google Scholar * Tracy Willson View author publications You can also search for this author inPubMed Google Scholar * Mark A. Dawson View author
publications You can also search for this author inPubMed Google Scholar * Rhys S. Allan View author publications You can also search for this author inPubMed Google Scholar * Marco J.
Herold View author publications You can also search for this author inPubMed Google Scholar * Kelly Rogers View author publications You can also search for this author inPubMed Google
Scholar * David M. Tarlinton View author publications You can also search for this author inPubMed Google Scholar * Gordon K. Smyth View author publications You can also search for this
author inPubMed Google Scholar * Melissa J. Davis View author publications You can also search for this author inPubMed Google Scholar * Stephen L. Nutt View author publications You can also
search for this author inPubMed Google Scholar * Warren S. Alexander View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Conceptualisation,
A.P.N., M.S.Y.L, A.J.K., T.M.J., M.A.D., R.S.A., K.R., D.M.T., G.K.S., M.J.D., S.L.N. and W.S.A.; Methodology, A.P.N., M.S.Y.L, C.C.B., O.G., T.M.J., T.B., A.J.K., M.J.H., M.A.D., R.S.A.,
K.R., D.M.T., G.K.S., M.J.D., S.L.N. and W.S.A.; Investigation, A.P.N., H.D.C., S.H., K.B., T.M.J., M.S.Y.L., C.C.B., O.G., Y.C.C., T.B., L.D., C.D.H., H.I., S.M., E.V., A.J.K., R.F., A.G.,
T.W., K.R., G.K.S., M.J.D.; Formal analysis, A.P.N., H.D.C., S.H., M.S.Y.L., O.G., C.C.B., Y.C.C. T.B., K.R., M.J.D., S.L.N; Writing—Original Draft, A.P.N.; Writing—Review & Editing,
A.P.N., H.D.C., S.H., M.S.Y.L., A.J.K., G.K.S., S.L.N., and W.S.A.; Funding Acquisition, A.P.N. and W.S.A.; Supervision, A.P.N., M.A.D., D.M.T., G.K.S., M.J.D., S.L.N. and W.S.A.
CORRESPONDING AUTHOR Correspondence to Ashley P. Ng. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PEER REVIEW INFORMATION
_Nature Communications_ thanks John Pimanda and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. PUBLISHER’S
NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW
FILE REPORTING SUMMARY DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is
licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in
this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative
Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a
copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ng, A.P., Coughlan, H.D., Hediyeh-zadeh, S. _et al._ An
Erg-driven transcriptional program controls B cell lymphopoiesis. _Nat Commun_ 11, 3013 (2020). https://doi.org/10.1038/s41467-020-16828-y Download citation * Received: 02 October 2019 *
Accepted: 24 May 2020 * Published: 15 June 2020 * DOI: https://doi.org/10.1038/s41467-020-16828-y SHARE THIS ARTICLE Anyone you share the following link with will be able to read this
content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative