Play all audios:
ABSTRACT In plants, epigenetic regulation is critical for silencing transposons and maintaining proper gene expression. However, its impact on the genome-wide transcription initiation
landscape remains elusive. By conducting a genome-wide analysis of transcription start sites (TSSs) using cap analysis of gene expression (CAGE) sequencing, we show that thousands of TSSs
are exclusively activated in various epigenetic mutants of _Arabidopsis thaliana_ and referred to as cryptic TSSs. Many have not been identified in previous studies, of which up to 65% are
contributed by transposons. They possess similar genetic features to regular TSSs and their activation is strongly associated with the ectopic recruitment of RNAPII machinery. The activation
of cryptic TSSs significantly alters transcription of nearby TSSs, including those of genes important for development and stress responses. Our study, therefore, sheds light on the role of
epigenetic regulation in maintaining proper gene functions in plants by suppressing transcription from cryptic TSSs. SIMILAR CONTENT BEING VIEWED BY OTHERS RNA INTERFERENCE-INDEPENDENT
REPROGRAMMING OF DNA METHYLATION IN _ARABIDOPSIS_ Article 30 November 2020 LONG-READ DIRECT RNA SEQUENCING REVEALS EPIGENETIC REGULATION OF CHIMERIC GENE-TRANSPOSON TRANSCRIPTS IN
_ARABIDOPSIS THALIANA_ Article Open access 05 June 2023 H3K36 METHYLATION STAMPS TRANSCRIPTION RESISTIVE TO PRESERVE DEVELOPMENT IN PLANTS Article 31 March 2025 INTRODUCTION Eukaryotic
genomes are comprised a large part of mobile genetic sequences, so-called transposable elements (TEs)1. Due to their mobility, TEs induce various alterations to the host genome, ranging from
genetic mutations to large-scale genomic rearrangements, such as inversions and translocations2,3. Genetic variations caused by TEs can introduce novel regulatory elements and therefore be
a major driving force underlying genome evolution1,2. On the other hand, uncontrolled activities of TEs can severely damage gene expression and the integrity of the host genomes3. To
suppress negative impacts without losing potential benefits brought in by TEs, both plants and animals have evolved numerous epigenetic mechanisms involving DNA methylation, histone
modifications, and small non-coding RNAs, allowing TEs remain silenced in their genomes4,5. Compared to mammals, plants are equipped with a different set of epigenetic mechanisms for greater
adaptability to dynamic environmental changes, partly due to their sessile nature. For example, in mammalian genomes, DNA sequences are mainly methylated at the cytosine in the CG
dinucleotides, while in plants cytosine methylation exists in both CG and non-CG contexts, which has different functional impacts on gene and TE regulation6. In the plant model _Arabidopsis
thaliana_ (_A. thaliana_), DNA methylation is established de novo by the RNA-directed DNA methylation (RdDM) pathway, which requires the functional activity of PolIV and PolV, two
plant-specific RNA polymerases5. After establishment, methylation patterns can be maintained by different factors depending on cytosine contexts. CG methylation is maintained by
METHYLTRANSFERASE 1 (MET1), a plant homologue of the mammalian DNA (cytosine-5)-methyltransferase 1 (DNMT1). Maintenance of DNA methylation in CHG context, on the other hand, is facilitated
by CHROMOMETHYLASE 3 (CMT3) in a positive feedback loop with the histone H3K9 methylase KRYPTONITE (KYP) (or SUPPRESSOR OF VARIEGATION 3-9 HOMOLOGUE 4 (SUVH4))7. Together with two of its
paralogues, SUVH5 and SUVH6, KYP regulates the genome-wide accumulation of H3K9me2 and consequently, CHG methylation6. CHH methylation can be maintained by either CMT2 or DOMAINS REARRANGED
METHYLASE 2 (DRM2) depending on the features of their targets, in which DRM2 often methylates short, euchromatic TEs, while CMT2 targets long TEs located in histone H1-containing
heterochromatic regions with the help of chromatin remodeler DECREASED DNA METHYLATION 1 (DDM1)8. These epigenetic pathways in plants, however, are highly interwoven. For example, MET1 and
CMT3 are involved in maintaining asymmetric methylation, while DMR2 and CMT2 may also affect DNA methylation in other contexts9. Epigenetic silencing of TEs inevitably confers regulatory
impacts on gene expression, especially when TEs are located close to transcription units2,4. In plants, repressive modifications triggered by TE insertions within introns or promoter regions
can attenuate or even turn off the expression of the associated genes10,11,12. At a global scale, genes harboring, or located close to, silenced TEs exhibit lower expression than their
counterparts13,14. Due to such unfavorable impacts, plants have evolved specific pathways to keep transcription units clear of repressive modifications, or to tolerate the presence of such
modifications when necessary. For example, in _A. thaliana_ the Jumonji C (jmjC) domain-containing histone demethylase INCREASE IN BONSAI METHYLATION 1 (IBM1) prevents repressive H3K9
methylation and consequently, CHG methylation, from accumulating at actively transcribed genes15. On the other hand, host factors, such as INCREASE IN BONSAI METHYLATION 2 (IBM2) and
Enhanced Downy Mildew 2 (EDM2) are required for proper transcription of genes containing heterochromatic domains16,17, likely due to the functional importance of these domains14. The
development of high resolution \({5}^{\prime}\) end-centered expression profiling techniques, such as oligo-capping methods18 or cap analysis of gene expression (CAGE)19, has greatly
advanced our understanding of gene regulation at a transcription initiation level. Studies employing these techniques have revealed both common and distinct features of the core promoters
and their origin and regulation, in many organisms20,21,22. In mammals, for example, CAGE sequencing (CAGE-seq) analyses revealed that a large fraction of cell-type specific transcripts in
stem and cancer cells originate from long terminal repeats (LTRs) of retroelements23,24. The loss of DNA methylation also causes spurious transcription within thousands of genes in mouse
embryonic stem cells25. In addition, modulating DNA methylation and histone deacetylation pathways pervasively activates cryptic transcription start sites (TSSs) normally silenced in human
cells26. These examples demonstrate the importance of epigenetic mechanisms in regulating transcription initiation in mammalian genomes. In plants, large-scale analyses have determined
thousands of TSSs, providing fundamental information about genetic structure and regulatory elements important for transcription in plant genomes27,28. Previous studies have also revealed
core promoter structures and sequence elements associated with plant TSSs29,30,31. However, these studies mainly focus on active TSSs in the wild-type background. The contribution of
epigenetic regulation to shaping the genome-wide transcription initiation landscape and its functional significance in plants, therefore, remains largely unexplored. To dissect the
functional impacts of epigenetic regulation in shaping the plant transcription initiation landscape, we employ CAGE-seq to generate the genome-wide profiles of TSSs at a high resolution for
various mutants of _A. thaliana_ that compromise epigenetic control. Our analysis identifies thousands of TSSs exclusively activated in the mutant backgrounds, demonstrating that epigenetic
regulation profoundly affects transcription initiation in _Arabidopsis_. These so-called cryptic TSSs are mainly located at heterochromatic regions, which hinder their accessibility to RNA
Polymerase II (RNAPII) transcription machinery. The alteration of DNA methylation maintenance in _met1_ activates the largest number of cryptic TSSs, which significantly overlap with the
targets regulated by other epigenetic pathways. A large fraction of cryptic TSSs originate from TEs of both retro and DNA-transposon families, suggesting that TEs are reservoirs of putative
TSSs in the _A. thaliana_ genome. Strikingly, the activation of cryptic TSSs significantly alters the regular transcription of nearby TSSs, which includes those of genes important for
development and stress responses in _Arabidopsis_. This study, therefore, sheds light on the role of epigenetic regulation in maintaining proper gene functions in plants by suppressing
transcription initiated from cryptic TSSs. In addition, the accompanying data are a valuable resource for studying the epigenetic control of the transcription of genes and TEs in plants.
RESULTS MAPPING TSSS IN EPIGENETIC MUTANTS OF _A_. _T__H__A__L__I__A__N__A_ BY CAGE-SEQ To gain a comprehensive view regarding the epigenetic regulation of transcription initiation in
plants, we performed CAGE-seq analyses of various _A. thaliana_ mutants, where epigenetic control is compromised, including mutants of maintenance DNA methyltransferase _met1_, the chromatin
remodeler _ddm1_, RdDM pathway components _nrpd1_ and _nrpe1_, histone H3K9 methyltransferases _suvh456_, histone H3K9 demethylase _ibm1_, and intragenic heterochromatin regulatory factors,
_ibm2_ and _edm2_. A total of 1,250,203,294 CAGE-seq reads were mapped to the _A. thaliana_ Col reference genome, achieving an average mapping efficiency of 97.53%. Of which, 402,814,394
reads were mapped uniquely, compiling a large collection of CAGE-seq data for this model plant (Supplementary Data 1). The expression of individual CAGE-based TSSs (CTSSs) was highly
correlated between replicates (the median of Pearson correlation coefficients was 0.95) (Supplementary Fig. 1a, b), confirming the reproducibility of our data. In total, 37,726 consensus tag
clusters representing single TSSs were identified across all samples (hereafter TSSs is used to refer to consensus tag clusters identified in this study, to distinguish from the
TAIR-annotated TSSs), of which about 30% were exclusively expressed in the mutant backgrounds (Supplementary Data 2). To confirm the relevance of our data, we analyzed the genome
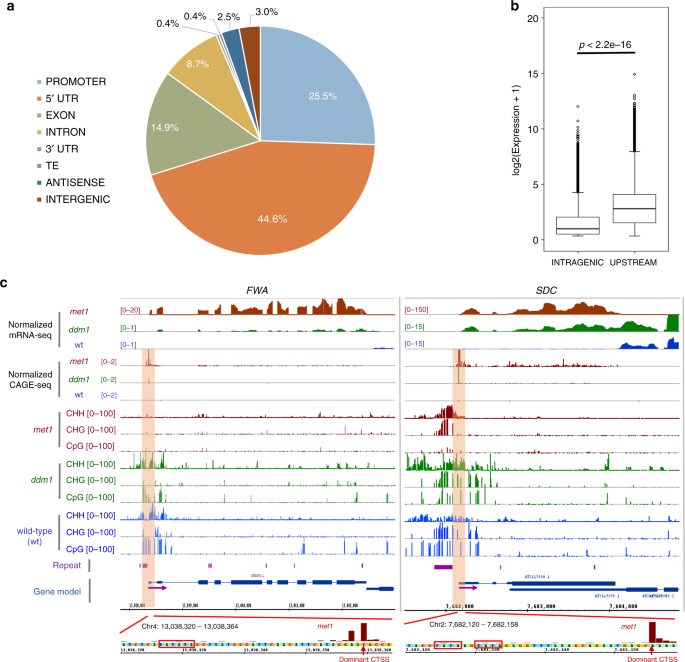
distribution of 26,561 TSSs identified in wild-type sample. A majority of them (18,634 or ~70%) were located in promoters and \({5}^{\prime}\) UTRs of 17,722 (~64%) annotated genes (Fig.
1a), and about one-fourth (~24%) were located in intragenic regions, of which exonic TSSs were more prevalent than the intronic counterparts. Although the mechanisms leading to the
prevalence of exonic TSSs in the plant genomes have yet been clear21, a part of them may represent \({5}^{\prime}\)-end capped products of post-transcriptional processing of mature mRNAs, as
described in human and vertebrate genomes32,33. Alternatively, some may correspond to cryptic promoters that trigger spurious transcription from gene bodies25,34, or to mis-annotated
TSSs22. Nevertheless, consistent with a previous study21, the expression of intragenic TSSs was significantly lower than that of their counterparts located in promoters and \({5}^{\prime}\)
UTRs (Fig. 1b). Moreover, the TSSs in promoters and \({5}^{\prime}\) UTRs were found in close proximity to the TAIR10-annotated TSSs (Supplementary Fig. 2a, b). A similar result was obtained
using the Araport11 genome annotations, (Supplementary Fig. 3a–c), with a shift in the numbers of TSSs assigned to each genome feature (Fig. 1a, Supplementary Fig. 3a). Because of the
higher consistency with the TSSs identified by our CAGE-seq (Supplementary Figs. 2a, b, 3b, c), TAIR10 annotations were used in further downstream analysis. On the other hand, active genes
supported by CAGE and mRNA-seq were largely overlapped (Supplementary Fig. 2c), suggesting that active transcription events in _A. thaliana_ can be efficiently captured by our CAGE-seq data.
We then compared wild-type TSSs identified by CAGE-seq with those reported by the paired end analysis of transcription start site (PEAT) method31. They were indeed consistent even though
the samples were prepared from different tissues (Supplementary Fig. 3d–f). At a local scale, the promoter architecture of two well-studied genes, _ALMT1_ (_AT1G08430_) and _sAPX_
(_AT4G08390_), was also reexamined. The former has three functional TSSs within its promoter and the latter has one upstream and one intragenic TSS21. Our data recapitulated these structures
(Supplementary Fig. 4a, b), confirming its consistency with previous studies21,31. It has been found that the loss of CG methylation at a SINE-related repeat in the promoter region
triggered the ectopic expression of the homeobox gene _FLOWERING WAGENINGEN_ (_FWA_), causing a late flowering phenotype of _Arabidopsis_11,35,36. CAGE-seq analysis identified a TSS encoded
within the SINE repeat, which was highly activated in _met1_ and _ddm1_ backgrounds (Fig. 1c). In addition, the ectopic activation of the TSS of the F-box gene _SUPPRESSOR OF drm1 drm2 cmt3_
(_SDC_), whose promoter contains a tandem repeat co-regulated by H3K9 methylation and the RdDM pathway8, was also detected by our data (Fig. 1c). Taken together, these results demonstrate
that our CAGE-seq data can be effectively exploited for the detection and analysis of both regular and cryptic TSSs under epigenetic control. MODULATING EPIGENETIC REGULATION ACTIVATES MANY
CRYPTIC TSSS Next, we investigated the impact of epigenetic regulation on the transcription initiation landscape in the _A. thaliana_ genome in greater details. Compromising epigenetic
controls significantly affected the transcription initiated from hundreds to thousands of TSSs, in which the defect of the maintenance DNA methylation pathway in _met1_ induced changes at
the largest number of targets (Fig. 2a), followed by _ibm1_, _ddm1_, _suvh456_, and _pol4_. To our surprise, _ibm2_ and _edm2_, which cause the transcriptional defect of _IBM1_16,17, had a
lower number of affected TSSs than _ibm1_, suggesting that the IBM1 function is partially maintained in these mutants. Of the altered TSSs, many were activated de novo in the mutant
backgrounds and were not associated with any TAIR10-annotated TSSs (Fig. 2a, Supplementary Fig. 2b, Supplementary Data 3). They were also largely distinct from the TSSs reported by
PEAT-seq31 and the TSSs identified in multiple tissues and light stress conditions in _A. thaliana_21 (Supplementary Fig. 5a, b), suggesting that they are cryptic TSSs suppressed by
epigenetic mechanisms (referred herein as EPICATs, for EPigenetically Induced Consensus tAg clusTers). Our data showed that the EPICATs activated in _met1_ largely overlapped with the
EPICATs regulated by other mutants, confirming the profound regulatory impact of MET1 on the genome-wide transcription initiation in _A. thaliana_ (Fig. 2b). On the other hand, _ddm1_ and
RdDM-associated mutants (_pol4_ and _pol5_) induced stronger activation of the EPICATs than _met1_ (Fig. 2c). Due to the minor numbers of instances, targets of _ibm2_ and _edm2_ were
excluded from further analysis. Similar results were obtained using the Araport11 annotations (Supplementary Figs. 3c, 5c), confirming the robustness of our analysis. As the transcription
orientation at regulatory regions of eukaryotes can be either unidirectional20 or bidirectional37, we examined the directionality of transcription initiated at EPICATs. Our data showed that
transcription at the EPICATs in _met1_ was mainly uni-directional, similar to that of the TAIR10-annotated TSSs in _A. thaliana_ (Supplementary Fig. 6a20). Moreover, the expression levels of
EPICATs were not significantly different from those of the annotated TSSs activated de novo in epigenetic mutants (Supplementary Fig. 6b). We also found that, tag clusters corresponding to
the EPICATs mainly had narrow peaks (NPs), especially those activated in _ddm1_, _met1_, and _pol5_ (Supplementary Fig. 6c), suggesting that they may have a well-defined underlying genetic
architecture31,38. To elucidate putative mechanisms regulating the activity of EPICATs, we first examined the genomic regions where they reside. EPICATs were mainly located at intergenic
regions, except the EPICATs in _ibm1_, of which a majority were intragenic (Fig. 3a, Supplementary Fig. 6d). These intragenic EPICATs, however, may not be directly regulated by the activity
of IBM1, because they were not associated with increased CHG methylation in the _ibm1_ background (Fig. 3b). In contrast, the EPICATs in other mutants were located in genomic regions
decorated with repressive chromatin modifications, such as DNA methylation, H3K9me2, and H3K27me1 (Supplementary Fig. 7a, b). Compared to the EPICATs in other mutants, those activated in
_pol4_ and _pol5_ were also associated with a higher level of CHH methylation and 24 nt siRNAs, the hallmarks of the RdDM pathway (Supplementary Fig. 7a, c). Moreover, DNA methylation at the
EPICATs in all mutants, except in _ibm1_, was significantly reduced, in concomitant with their activation (Fig. 3b, Supplementary Fig. 7d), suggesting that in wild-type plants transcription
initiation at EPICATs is directly suppressed by repressive epigenetic modifications. Since heterochromatic modifications, such as DNA methylation and H3K9me2, are often associated with
closed chromatin in plant genomes39, their loss may alter the access to genomic regions harboring EPICATs. We therefore examined how the accessibility of these loci changes in the mutant
backgrounds. For this purpose, the EPICATs activated in _ddm1_ were used as a proxy due to the large number of instances and the availability of public data characterizing chromatin openness
in _ddm1_40. Indeed, chromatin around the EPICATs became highly accessible in _ddm1_, compared to wild-type plants, as measured by the sensitivity to DNaseI (Fig. 3c). Furthermore, ChIP-seq
analysis showed that RNAPII phosphorylated at Ser5 (Ser5P) and Ser2 (Ser2P) in the C-terminal domain (CTD), the hallmarks of transcription initiation and elongation41 respectively, were
also highly accumulated at the EPICATs in most mutant backgrounds (Fig. 3d, Supplementary Fig. 7e). These data demonstrate that repressive chromatin suppresses the activity of EPICATs by
preventing the access of transcription machinery to genomic regions encompassing potential TSSs. Ectopic transcription initiation in mutants and the convergence of various epigenetic
pathways on a large number of EPICATs (Fig. 2b), together with the narrow shapes of tag clusters corresponding to most of the EPICATs (Supplementary Fig. 6c), suggest that these loci harbor
functional genetic features, such as promoter structure and/or regulatory sequences21, in addition to repressive chromatin modifications. Therefore, genetic sequences surrounding EPICATs
were analyzed. Interestingly, DNA elements and motifs enriched around EPICATs exhibited spatial architecture similar to that of regular plant promoters20,30, with a sharp accumulation of
TATA-box at 36 nt upstream and CA-rich/CT-rich (Y-patch) motifs around the TSSs (Fig. 3e, Supplementary Fig. 8). TATA-box, a core promoter motif conserved in both plants and animals30,38,
was especially enriched at the EPICATs in _met1_ and _ddm1_. The enrichment of the Telobox motif (AAACCCTA), which is known to recruit development-associated repressive modification H3K27me3
in _A. thaliana_42, was also found at the EPICATs in _met1_, _ddm1_, and _suvh456_. The presence of the Telobox sequence around EPICATs may partially explain the accumulation of H3K27me3 at
the heterochromatic regions upon the loss of DNA methylation and H3K9 methylation43. Taken together, we conclude that the _A. thaliana_ genome harbors hundreds of potential TSSs equipped
with functional core promoter architecture similar to that of regular TSSs. Their activities, however, are suppressed by repressive chromatin restricting their accessibility to transcription
machinery. GENE BODY METHYLATION AND THE SUPPRESSION OF INTRAGENIC TSSS In _A. thaliana_, about 20% of protein coding genes accumulate CG methylation in their bodies44. Moreover, gene body
methylation (gbM) is largely conserved across plant species, especially in angiosperms45, suggesting its functional importance. Although many hypotheses have been proposed regarding the
biological functions of gbM, such as suppressing spurious intragenic transcription25, impeding transcriptional elongation46, or reducing transcription noise47, so far its role in plants has
been largely elusive48. By exploiting the high resolution CAGE-seq data of genome-wide TSSs, we reexamined the relationship between gbM and intragenic transcription initiation in _A.
thaliana_. Our data showed that, in wild-type plants, a similar fraction of both body methylated (BM) and non body methylated (non-BM) genes harbored intragenic TSSs, suggesting that the
methylation state of gene body is not significantly associated with the occurrence of intragenic TSSs (Fig. 4a). Moreover, only a few BM genes activated intragenic EPICATs when gbM was
strongly lost in _met1_ background (Fig. 4b, Supplementary Fig. 9a), meanwhile intragenic EPICATs could be activated at some loci without gbM (Fig. 4d). These evidences, which are consistent
with the conclusions of a previous study48, suggested that gbM alone is dispensable for suppressing intragenic transcription at a global scale in _A. thaliana_ (Supplementary Fig. 9b).
Although some BM genes harbored intragenic EPICATs in _met1_ (Fig. 4c, d), at this time, we do not know if this is a direct or indirect effect of _met1_ mutant. Future testing using targeted
demethylation could help resolve if BM is causal at these loci. The intragenic EPICATs in _met1_ may correspond to \({5}^{\prime}\)-end capped products of post-transcriptional processing of
mature mRNAs generated at the associated gene loci, a mechanism well-described in mammals32,33. Although we did not rule out this possibility, our data provided evidences supporting that
some of these EPICATs are genuine TSSs. First, these loci exhibited a stronger accumulation of RNAPII in _met1_ (Supplementary Fig. 9c). Second, only 1/124 genes harboring intragenic EPICATs
also had upstream EPICATs (Supplementary Fig. 9d), suggesting that these intragenic EPICATs correspond to independent, de novo transcribed mRNAs. Third, promoter-associated DNA sequences
were also present at some of these intragenic loci (Fig. 4d). Besides _met1_, _ibm1_ also activated a comparable number of intragenic EPICATs (Supplementary Fig. 6d, Supplementary Data 4).
However, it is unlikely that they are directly regulated by the activity of IBM1 (Fig. 3b, Supplementary Fig. 10a). On the other hand, although the expression of _IBM1_ is significantly
reduced in _met1_ background49, the intragenic EPICATs activated in _ibm1_ and _met1_ were largely un-overlapped (Supplementary Fig. 10b). Moreover, the accumulation of RNAPII at these loci
was not significantly affected in _ibm1_ background (Supplementary Fig. 10c), suggesting that intragenic EPICATs in _ibm1_ and _met1_ are regulated differently. Given that none of the
associated genes simultaneously harbored upstream EPICATs, and that promoter-associated DNA sequences were present at some of these intragenic targets (Fig. 4e), we speculate that some of
them are genuine TSSs, while some others could be derived from post-transcriptionally processed mRNAs. RNAPII AND POLIV EXCLUSIVELY BIND TO RDDM-REGULATED EPICATS It has been reported that,
although PolIV-dependent RNAs (P4RNAs) feature PolII-like TSSs, PolIV and PolII target distinct genomic territories50. Our data, however, showed that 24 nt siRNAs were highly enriched at
genomic loci harboring the EPICATs activated in the mutants of the RdDM pathway’s components, such as _pol4_ and _pol5_ (Supplementary Fig. 7c). The biogenesis of these 24 nt siRNAs was
indeed dependent on PolIV, which is responsible for the transcription of P4RNAs initiated from the corresponding EPICATs (Supplementary Fig. 11a–c). Moreover, in _pol4_ and _pol5_
backgrounds, RNAPII was highly recruited to these loci (Supplementary Fig. 7e). These evidences suggest that, genomic regions harboring the EPICATs regulated by the RdDM pathway likely
possess distinct features compared to those of its regular targets, which allow PolII and PolIV exclusively function at these loci (Supplementary Fig. 11d). TES ARE A MAJOR SUPPLIER OF
CRYPTIC TSSS IN _ARABIDOPSIS_ The existence of a large number of cryptic TSSs within a small and compact genome, like that of _A. thaliana_, has raised important questions regarding their
origin. Investigations involving mammalian genomes have shown that TEs are a major genetic element that can be exapted as TSSs in the host genomes51,52. Although less prevalent, several
lines of study have demonstrated a similar function of TEs in plant genomes53,54. Together with the evidence that EPICATs are mainly located at intergenic regions decorated with repressive
chromatin modifications (Fig. 3a, Supplementary Fig. 7a, b), we speculated that many cryptic TSSs in the _A. thaliana_ genome may have originated from TEs. The data indicated that TEs
contribute to up to 65% of the EPICATs activated in the mutant backgrounds (Supplementary Fig. 12a). Additionally, hundreds of TEs harboring active TSSs were identified in wild-type
background (Fig. 5a, Supplementary Data 5). TEs, therefore, may serve as a reservoir of potential functional TSSs in _A. thaliana_, similar to their role in animal genomes. There are
numerous types of TEs with different origins and mobility strategies1,2 which greatly affect their abilities to induce genetic variations to the host genomes. Therefore, the TSS-encoding
potential of each TE family in the _A. thaliana_ genome was examined. Although EPICATs were associated with various TE families (Fig. 5a), compared to the genome-wide average, LTR/Gypsy
members were enriched among TEs harboring the EPICATs in _ddm1_ and _met1_ (_p_ = 2.0e-52 and 6.0e-49, respectively, Hypergeometric test), while members of the LTR/Copia family were highly
represented among the TE targets of _ddm1_ and _suvh456_ (_p_ = 8.0e-10 and 2.0e-31, respectively, Hypergeometric test). In addition, the DNA/En-Spm family was highly associated with the
EPICATs in _met1_, _ddm1_, and _suvh456_ (Fig. 5a, _p_ < 1.6e-16 for all, Hypergeometric test). Due to the minor numbers of TE instances associated with the EPICATs in _ibm1_, _pol4_, and
_pol5_, they were skipped from enrichment analysis. The data suggest that both retro- and DNA transposons are genetic suppliers of cryptic TSSs in the _A. thaliana_ genome. Since _ddm1_
affected the largest number of TEs harboring EPICATs, and these elements largely overlapped with TEs activated in other mutants (Fig. 5a, Supplementary Fig. 12b), we examined if they possess
any specific features that facilitate their ectopic activation in _ddm1_ background. Compared to their counterparts, which either contain active TSSs in wild-type plants or do not harbor
any EPICATs, TEs harboring EPICATs were more highly methylated in both CG and non-CG contexts (Fig. 5b). They were also substantially longer (Fig. 5b), suggesting that these TEs are likely
younger insertions that still maintain intact structures with transcription and transposition capacities, that may be a trigger for greater accumulation of DNA methylation and other
repressive modifications at the associated loci. Analysis of the core promoter motifs identified at the _ddm1_-activated EPICATs (Supplementary Fig. 8) showed that they were more prevalent
among EPICAT-harboring TEs (Fig. 5c). However, there were still hundreds to thousands of inactive TEs associated with these motifs (Supplementary Fig. 12c). As a case study, the genetic
structure associated with the EPICATs located in the LTR regions of the Gypsy TEs was investigated in a more detail. This was because the LTR/Gypsy family contributed a large number of
elements harboring the EPICATs in _ddm1_ and _met1_ (Fig. 5a), and its members still maintain transcription/transposition potential in the _Arabidopsis_ genome55. Although LTR sequences
surrounding the CAGE-seq peaks were largely diverged between and within Gypsy sub-families, they commonly shared putative TATA-box and TSS-associated YR motifs (Fig. 5d, Supplementary Fig.
12d). However, the conservation of sequences/motifs surrounding the LTR-encoded TSSs could not fully explain their activation in the mutant backgrounds. Moreover, although a significant loss
of repressive modifications (e.g., DNA methylation) was observed at many TEs regardless of their association with the EPICATs in _ddm1_ (Fig. 5e), only EPICAT-harboring elements became
highly accessible in the mutant, especially at their two ends (Fig. 5f). Concomitantly, RNAPII was highly recruited to these loci, together with an increased production of the associated
transcripts (Fig. 5g, Supplementary Fig. 12e). These data suggest that, in addition to the presence of core promoter sequences, factors regulating chromatin environment are required for
RNAPII recruitment and the ectopic activation of TE-encoded EPICATs. REGULATORY IMPACT OF TRANSCRIPTION FROM CRYPTIC TSSS In mammals, TE sequences frequently act as alternative promoters to
regulate development-associated gene expression programs51,52. While the contribution of TEs to plant transcriptomes has been much less clear56, this evidence suggests that regulatory
elements supplied by TEs can be co-opted for transcriptional regulation in plant genomes28. Using the EPICATs activated in _met1_ as a proxy, we therefore investigated the potential
alteration in the _A. thaliana_ transcriptome induced by cryptic TSSs. About ~80% of the EPICATs in _met1_ were associated with the transcripts assembled from mRNA-seq data (Supplementary
Fig. 13a, Supplementary Data 6, see the “Methods” section for details). Moreover, the expression of EPICATs was positively correlated with that of the assembled gene units (Supplementary
Fig. 13a–c). 73% of the transcripts associated with _met1_-activated EPICATs had more than one exons, of which 112 (~9%) shared splicing junctions with 75 reference gene units (Fig. 6a).
Surprisingly, about half (50/112) of these spliced transcripts possessed at least one active TSS in wild-type background, suggesting that their regular transcription, and consequently
downstream functions, can potentially be affected by the ectopic activation of EPICATs. We selected and experimentally confirmed the production of novel cryptic fusion transcripts at some of
these loci in _met1_ and/or _ddm1_ backgrounds, which include _SQN (AT2G15790)_, a gene critical for vegetative shoot maturation57, _COQ3 (AT2G30920)_, a gene encoding a
mitochondria-localized methyltransferase important for ubiquinone biosynthesis and embryo development58,59, and a gene of unknown function (_AT2G16050_) (Fig. 6b, c, Supplementary Fig. 14a,
b). To complement the CAGE-seq data, transcripts with significant alteration in promoter usage were analyzed using mRNA-seq data (see Methods section for details). Of the resulting
transcripts, 10 were found associated with _met1_-activated EPICATs at three gene loci (Supplementary Data 7). We also experimentally confirmed the production of a read-through fusion
transcript from the annotated TSS at the _AT5G28442_ gene locus, which harbored an EPICAT in _met1_ and _ddm1_ backgrounds (Supplementary Fig. 14a, b). Although it has been suggested that
repressive chromatin associated with TE insertions potentially imposes negative impacts on the transcription of nearby genes13,14, direct consequences of TE-encoded TSS activation on the
surrounding transcriptional environment remain obscure. Inspection of the loci producing cryptic fusion transcripts revealed that some of them concurrently exhibited reduced transcription
from their regular TSSs in the mutant backgrounds (Fig. 6b, Supplementary Fig. 14a). This suggests that, the activation of EPICATs may also quantitatively affect the transcription from
nearby regular TSSs. Therefore, wild-type active TSSs located in the vicinity (up to 3 kb) of EPICATs were examined to see how their expression is altered upon EPICAT activation. While some
showed increased expression, the majority were not significantly affected (Fig. 6d, e). Nevertheless, there were groups of TSSs whose expressions were significantly suppressed in concomitant
with the activation of nearby EPICATs (Fig. 6e, Supplementary Data 8). Of the gene loci associated with the TSSs suppressed in _met1_, five were selected for validation by qPCR. Except
_AT5G28442_, which could not be amplified, significant decreases in the expression at three out of the four loci in _met1_ and _ddm1_ were confirmed, which is consistent with the observation
from the CAGE-seq data (Fig. 6f, Supplementary Fig. 14c). These include _AT1G23935_, _SUS5_ (_AT5G37180_), and _PRB1_ (_AT2G14580_), a gene involved in response to abiotic stress in
_Arabidopsis_60. Taken together, these data demonstrate that the activation of cryptic TSSs has critical impacts on the transcriptome of _A. thaliana_, both qualitatively and quantitatively.
DISCUSSION To understand how transcription initiation in plants is epigenetically regulated, we have generated a comprehensive maps of TSSs in various epigenetic mutants of _A. thaliana_
using CAGE-seq. Compared to mammals, epigenetic mechanisms regulating transcription initiation in plants are much less clear, mainly due to a lack of suitable resources which allow the
investigation of the alteration of transcription initiation under different conditions25,26,56. This study, therefore, provides valuable reference data for research communities to enlighten
the impact of epigenetic regulation on transcription initiation landscapes in plants. Our study showed that, in epigenetic mutant backgrounds, thousands of cryptic TSSs are activated, in
which the mutant of maintenance DNA methylation _met1_ regulates the largest number of targets (Fig. 2a). A large number of cryptic TSSs reside in TE sequences, which are dominantly
contributed by members of the LTR/Gypsy, LTR/Copia, and DNA/En-Spm families (Fig. 5a). Interestingly, there is a clear difference in DNA methylation between TEs with and without EPICATs,
where the former accumulate higher DNA methylation (Fig. 5b, e). This suggests that the DNA methylation of TEs could be largely influenced by their potential to initiate transcription. On
the other hand, the analysis of LTR sequences indicated that the conservation of core promoter elements alone is not sufficient for transcription initiation (Fig. 5d, Supplementary Fig. 12d)
as their transcription levels are largely varied, even among LTRs with nearly identical sequences. The ability of TE-encoded TSSs to initiate transcription may, therefore, also be dependent
on their relative positions within TEs (e.g., whether they are located at the \({5}^{\prime}\)- or 3\({}^{\prime}\)-end of the TEs), and/or local chromatin environments, such as
higher-order chromatin conformation and long-range enhancer interactions61. In mammals, the loss of gene-body DNA methylation caused by _DNMT3b_ knockout triggers spurious RNAPII recruitment
and cryptic transcription initiation from intragenic regions25. The analysis of intragenic TSSs in the present study showed that a complete loss of gbM in the _met1_ mutant does not
profoundly activate intragenic transcription in the _Arabidopsis_ genome (Fig. 3a, 4, Supplementary Fig. 9a, b). Recruitment of DNMT3b to genic regions in mammals is dependent on histone
H3K36 methylation62. In yeast, H3K36 methylation (H3K36me) mediated by SET2 suppresses cryptic intragenic transcription initiation63. In plants, however, concurrent loss of both gbM and
H3K36me3 does not show significant difference in transcription between (BM) and unmethylated (UM) loci48. On the other hand, regulation of cryptic transcription from intronic heterochromatin
by the RdDM pathway64, and the suppression of intragenic antisense transcripts by histone H1 and DNA methylation65 have also recently been reported. These results suggest that plants may
employ additional layers of epigenetic regulation to prevent spurious transcription initiation, especially in intragenic regions. The activation of spurious transcription from cryptic TSSs
would inevitably alter transcription from nearby regular TSSs (Fig. 6, Supplementary Fig. 14). The data showed that such alteration may occur in several different scenarios. First, an
activated cryptic TSS located upstream may function as the major initiation site facilitating the formation of a read-through transcript, which can suppress transcription from a downstream
regular TSS, as observed at _AT2G16050_ and _SQN_ loci (Fig. 6b). This regulatory effect is likely facilitated by a less understood mechanism known as transcriptional interference66,67.
Secondly, the activation of a cryptic TSS located downstream may attenuate transcription initiated from an upstream regular TSS and trigger the production of spurious transcripts, as
observed at _AT2G14580_, _AT2G15042_, and _AT5G28442_ loci (Supplementary Fig. 14). Thirdly, when cryptic and regular TSSs are situated close to each other, but in divergent directions,
transcription from the regular TSS may also be suppressed (Fig. 6f). Such repressive impacts could be facilitated by competitive binding to regulatory sequences of transcription initiation
complexes associated with the two TSSs66, or by the mechanism suppressing transcription from divergent promoters68, or by the lack of a mechanism facilitating bi-directional transcription in
plants20 compared to mammals37. Whether the epigenetic regulation of cryptic TSSs brings any potential developmental and/or adaptive advantages or disadvantages to a plant species is of
great interest in plant research. As epigenetic information is relatively flexible and can be reprogrammed according to environmental stimuli, the mechanisms described here may provide
plants with a fast and efficient mean for tuning, or even inverting the polarity of regulatory inputs on, gene expression. In addition, potential activation and co-option of cryptic TSSs can
provide alternative promoters to the existing transcription units, as observed at _AT2G16050_ and _COQ3_ loci (Fig. 6b, c, Supplementary Fig. 14a, b), which may help plants customize gene
functions during development51,52. Such events can also create opportunities for plants to innovate their transcriptome in response to environmental changes. However, the mis-control of
cryptic TSSs encoded in TEs may trigger developmental abnormality in plants11,69. In addition, modulating 3\({}^{\prime}\) and/or \({5}^{\prime}\) UTRs of a transcript without changing its
coding potential can critically affect its function in response to pathogen attacks in _Arabidopsis_70. Epigenetic suppression of a cryptic TSS at the \({5}^{\prime}\) UTR of the LRR gene
_AT2G15042_ (Supplementary Fig. 14a) may, therefore, help maintain the proper response of _Arabidopsis_ to viral infection71. Importantly, activation of the cryptic TSS upstream of _SQN_
(_AT2G15790_), a gene important for vegetative shoot maturation in _Arabidopsis_57, leads to ectopic production of aberrant transcripts and a decreased accumulation of the normal one (Fig.
6b). Although the impacts of such transcriptional attenuation on plant development are to be confirmed, it has been shown in _A. thaliana_ that, light-induced regulation of alternative
promoters could generate proteins with differential localizations from the same genes, which help alleviate the impact of changing light conditions on the plant72. Our data, therefore,
demonstrate that the epigenetic regulation of cryptic TSSs would profoundly and critically affect proper responses of plant species to ever changing environmental conditions. Additionally,
as many protein coding genes in _A. thaliana_ possess multiple active upstream as well as intragenic TSSs, it would be interesting to investigate whether cryptic TSSs are still in the
process of being co-opted to become functional in the _Arabidopsis_ genome. METHODS PLANT MATERIALS _ddm1-1_, _met1-3_, _ibm1-4_, _ibm2-2_, and _edm2-9_ mutants have been described
previously16,73,74,75. _suvh456_ and _nrpe1-7_ seeds were kindly provided by Dr. Kakutani and Dr. Kanno, respectively. The T-DNA insertion line of _nrpd1a-3_ (SALK_128428) was obtained from
the Arabidopsis Biological Resource Center (https://abrc.osu.edu). All the mutants are in Columbia (Col) background. The second generation of homozygous _met1_, _ddm1_, _ibm1_, _ibm2_, and
_edm2_ were used for the RNA experiments described below. _nrpd1a_, _nrpe1_, and _suvh456_ were maintained as homozygous for at least three generations before the experiments. The seeds were
germinated and grown on 1/2 Murashige and Skoog (MS) plate under long-day conditions (16-h light; 8-h dark) at 22 ∘C. RNA EXTRACTION AND CAGE For CAGE analysis, 10-to-12-day-old whole
seedlings of wild-type Col and mutant plants were pooled for RNA extraction. Total RNA was extracted using RNAiso (TAKARA), and DNA was digested with TURBO DNase (Thermo Fisher Scientific),
followed by purification by RNeasy Plant Minikit (QIAGEN). Four technical replicates of WT Col and _met1_, and two technical replicates of other samples were prepared for CAGE. Single end
75bp CAGE libraries were prepared and sequenced in DNAFORM (Yokohama, Japan). RNA quality was assessed by Bioanalyzer (Agilent) to ensure that the RIN (RNA integrity number) was over 7.0,
and A260/280 and 260/230 ratios were over 1.7. CAGE SEQUENCING DATA ANALYSIS The CAGE sequencing (CAGE-seq) data were processed as follows: sequencing reads were trimmed using Trimmomatic
(v0.30)76 with the following parameters: HEADCROP:1, TRAILING:20, to remove nonspecific guanines38 and low quality bases at the read ends. These were then mapped to the
_A__r__a__b__i__d__o__p__s__i__s_ Col reference genome by HISAT2 (v2.0.0-beta)77, allowing up to ten alignments for a single read. Due to low mapping coverage, met1.4 replicate was excluded
from further analysis. met1.3 was also discarded due to its low correlations with two other replicates (met1.1 and met1.2). Then, uniquely mapped reads were used to identify TSSs at a single
base resolution (CTSSs) by CAGEr (v1.20.0)78 with the following parameters: sequencingQualityThreshold = 20, mappingQualityThreshold = 20. After being normalized to Tags Per Million (TPM),
CTSSs in each sample were grouped into tag clusters by the paraclu method, with threshold = 0.1, nrPassThreshold = 2, removeSingletons = TRUE, keepSingletonAbove = 0.3, minStability = 2,
maxLength = 100. Finally, tag clusters from individual samples were merged into a common set of consensus tag clusters by the aggregateTagCluster function, with threshold = 0.3, _q_Low =
NULL, _q_UP = NULL, maxDist = 100, excludeSignalBelowThreshold = TRUE. Each consensus tag cluster was then considered a single reliable TSS, represented by its dominant CTSS, to distinguish
from the TSSs annotated by TAIR10. Promoter width was defined by the distance between the 10th (_q_Low = 0.1) and 90th (_q_Up = 0.9) quantiles of the cummulative distribution of CAGE signal
along each tag cluster, as described in ref. 78. Raw tag counts were used to identify differentially expressed TSSs in the mutants compared to wild-type plants by DESeq2 (v1.22.2)79, with
significance cut-off threshold _p_adj ≤ 0.1. ANNOTATING TSSS IDENTIFIED BY CAGE-SEQ TAIR10 genome annotations of 19,891 TEs and 27,600 protein coding genes and non coding RNAs in _A_.
_t__h__a__l__i__a__n__a_ were obtained from ref. 14. Araport11 version of genome annotations were also downloaded from The Arabidopsis Information Resource (TAIR)
(https://www.arabidopsis.org/). Promoters were defined as the regions of 1 kb upstream of the TAIR-annotated TSSs. A TSS identified by CAGE-seq was annotated based on genomic location of its
dominant CTSS, in the following order: promoter, \({5}^{\prime}\) UTR, 3\({}^{\prime}\) UTR, intron, exon, antisense, TE, intergenic. TSSs identified by PEAT method were obtained from ref.
31. Then, the nearest distance between the dominant CTSS of each CAGE-seq tag cluster and the mode locations of PEAT TSSs in the same direction was calculated. PEAT TSSs, which exactly
matched with CAGE-seq TSSs (distance = 0 nt), were used as the proxy to estimate interquantile widths for each shape category defined in ref. 31, including NP, broad with peak (BP), and weak
peak (WP). MRNA SEQUENCING DATA ANALYSIS Paired-end mRNA sequencing (mRNA-seq) data were prepared following the method described in ref. 14 and processed as follows: reads were trimmed by
Trimmomatic to remove sequencing bias and adapter sequences, then mapped to the _A__r__a__b__i__d__o__p__s__i__s_ Col reference genome by HISAT2, allowing up to ten alignments for a read
pair. The featureCounts function in the package Rsubread (v1.14.2)80 was used to identify the number of read pairs uniquely mapped to genes and TEs. The outputs of mRNA-seq mapping were also
used for transcript assembly as follows: first, transcripts of each individual sample were assembled by Cufflinks (v2.2.1)81. Low-expressed transcripts (smaller than the 10th percentile of
expression of all the assembled transcripts) were then removed. The remaining transcripts from all samples were merged to create a unified set of transcripts. They were then compared to
reference transcripts in TAIR10 by the cuffcompare function to identify splicing patterns. Differential promoter usage was assessed by the cuffdiff function. To identify assembled
transcripts associated with EPICATs, overlap tests were conducted between the transcripts and genomic regions centering around the EPICATs’ dominant CTSSs (extended 180 bp into both sides,
regarding that a TSS identified by CAGE-seq could be associated with a nearby transcript (Supplementary Fig. 2b)). The results were given in Supplementary Data 6. CHIP SEQUENCING DATA
ANALYSIS ChIP sequencing (ChIP-seq) data of histone modifications, including H3K27me1/3, H3K9me2, H3K36me3, and H3K4me3, in wild-type plants were retrieved from a previous study82.
Paired-end Chip-seq data of RNAPII in wild-type plants and mutants were prepared as follows: Two-week-old whole seedlings of wild-type Col and _met1_ and _ddm1_ were fixed in a fixation
buffer (10 mM Tris-HCl (pH 7.5), 50 mM NaCl, 0.1 M sucrose, 1% formaldehyde) for 20-min, followed by quenching by 125 mM Glycine. Nuclei isolation was performed as previously described83.
PolII ChIP was performed for two replicates for each genotype (about 1 g tissue/IP) by SimpleChIP Plus Kit (Cell Signaling Technology) according to the manufacturer’s instructions. Anti-RNA
polymerase II CTD repeat YSPTSPS (phospho S2) (Abcam ab5095) and Anti-RNA polymerase II CTD repeat YSPTSPS (phospho S5) (Abcam ab5408) antibodies were used for IPs (4 μg/IP). Precipitated
DNA samples were sequenced by Hiseq 4000 in the 150 bp paired-end mode in OIST SQC. Due to the large overlap between two reads, only one read (read 1) in each pair was used for downstream
analysis. Reads were trimmed to remove sequencing bias and adapter sequences using Trimmomatic, then mapped to the _A__r__a__b__i__d__o__p__s__i__s_ Col reference genome by Bowtie
(v1.0.0)84. Reads mapped to an identical position were collapsed into a single read, and only the best alignment was kept for a read mapped to multiple locations. Mapping results were given
in Supplementary Data 9. ChIP-seq data of PolIV (NRPD1) and the list of NRPD1 binding loci were obtained from ref. 85. Genomic locations of NRPD1 binding loci were then converted from TAIR8
to TAIR9 coordinates using the _update_coordinates.pl_ script provided by TAIR. ChIP-seq data of RNAPII in _pol4_ and corresponding wild-type plants were obtained from ref. 50. These data
were processed as described above. Preprocessed RNAPII Ser5P ChIP-seq data (in bigwig format) in _pol5_ were downloaded from ref. 64 and directly used for visualization. BISULFITE SEQUENCING
DATA ANALYSIS Whole-genome bisulfite sequencing (WGBS) MethylC-Seq data of wild-type plants and epigenetic mutants were retrieved from ref. 9. High quality reads (_q_ ≥ 28), trimmed to
remove adapter effects and sequencing bias, were mapped to the _Arabidopsis_ Col reference genome using Bismark (v0.12.1)86 allowing up to two mismatches. Bases covered by fewer than 3 reads
were excluded, and only uniquely mapped reads were used for further analysis. Methylation levels were calculated using MethylKit (v0.5.7)87. The list of BM, intermediate methylated (IM),
and unmethylated (UM) genes were obtained from ref. 44. To exclude the potential impacts of non-CG methylation on the activation of intragenic EPICATs, only _met1_-activated intragenic
EPICATs with low (less than 10%) CHG methylation in the 101 bp regions centering around their dominant CTSSs were examined (Supplementary Data 4). SMALL RNA SEQUENCING DATA ANALYSIS
Sequencing data of 24 nt small interference RNAs (siRNAs) in wild-type and _nrpd1_ mutant plants were obtained from ref. 85 and trimmed by TrimGalore (v0.4.5)88 with Cutadapt (v1.8.3)89,
using the following parameters: stringency:4, quality:20, length:15, max_length:30. PolIV-dependent small RNAs (P4RNAs) longer than 27 nt in _dcl2/3/4_ and corresponding wild-type plants
were obtained from ref. 50 and trimmed by Trimmomatic. These data were then mapped to the _A__r__a__b__i__d__o__p__s__i__s_ Col reference genome by Bowtie (v1.0.0), allowing up to two
mismatches. Only uniquely mapped reads were used for further analysis. SEQUENCE MOTIF ANALYSIS De novo motif analysis and search of motif instances were conducted using MEME suite (v4.11.2)
with default parameters90. GYPSY LTR ANALYSIS Gypsy family sequences were retrieved from the TAIR database and aligned to obtain the full-length sequence for each family. LTR regions were
then determined by comparing \({5}^{\prime}\) and 3\({}^{\prime}\) ends of TE sequences and also checked by LTR_FINDER (v1.0.2)91. Several copies from each family were used to obtain
consensus sequences of LTRs (Supplementary Data 10). Consensus sequences of Gypsy LTRs were used to search for LTR sequences in the _Arabidopsis_ genome (TAIR10) using BLAST (v2.0)92. BLAST
hits shorter than 100 bp were discarded. LTR sequences were then aligned using ClustalW (v2.1)93, and edited using Jalview (v2.11.0)94. DATA VISUALIZATION Figures were created using
deepTools (v3.3.0)95, Integrated Genome Browser (IGB) (v9.1.2)96 with the Araport11 version of genome annotations, Excel, and the R package ggplot2 (v2.3.1)97. DNA methylation files were
firstly converted from bedGraph into bigWig format by the bedGraphToBigWig function (http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/), then used to generate heatmap and metaplot
figures using deepTools. mRNA-seq data were normalized to reads per million (RPM), and a single replicate was used to create IGB track. ChIP-seq signals were normalized to log2(ChIP/input),
and a single replicate of RNAPII (both Ser5P and Ser2P) were used for visualization in IGB. Small RNA sequencing data and RNAPII ChIP-seq data with no input samples were normalized to counts
per million (CPM). 5′-RACE AND QUANTITATIVE PCR \({5}^{\prime}\)-RACE was performed by SMARTer RACE kit (TAKARA) according to the manufacturer’s instructions. Quantitative PCR (qPCR) was
performed following the method described in ref. 75. All primers used in this study are listed in Supplementary Data 11. REPORTING SUMMARY Further information on research design is available
in the Nature Research Reporting Summary linked to this article. DATA AVAILABILITY Sequencing data have been deposited to the DDBJ Sequence Read Archive under the accession codes DRA009134
and DRA009847. Processed CAGE-seq data are also accessible via the following web link: https://plantepigenetics.oist.jp/. The source data underlying Figs. 1b, 2c, 3b, 4d–e, 5b, and 6c, d, f
and Supplementary Figs. 6b–c, 7d, and 14b–c are provided as a Source Data file. CODE AVAILABILITY In-house R codes and bash scripts customized for analyzing data are available from the
authors upon request. REFERENCES * Fedoroff, N. V. Transposable elements, epigenetics, and genome evolution. _Science_ 338, 758–767 (2012). ADS CAS PubMed Google Scholar * Lisch, D. How
important are transposons for plant evolution? _Nat. Rev. Genet._ 14, 49 (2013). CAS PubMed Google Scholar * Chuong, E. B., Elde, N. C. & Feschotte, C. Regulatory activities of
transposable elements: from conflicts to benefits. _Nat. Rev. Genet._ 18, 71 (2017). CAS PubMed Google Scholar * Slotkin, R. K. & Martienssen, R. Transposable elements and the
epigenetic regulation of the genome. _Nat. Rev. Genet._ 8, 272–285 (2007). CAS PubMed Google Scholar * Law, J. A. & Jacobsen, S. E. Establishing, maintaining and modifying DNA
methylation patterns in plants and animals. _Nat. Rev. Genet._ 11, 204–220 (2010). CAS PubMed PubMed Central Google Scholar * Zhang, H., Lang, Z. & Zhu, J.-K. Dynamics and function
of DNA methylation in plants. _Nat. Rev. Mol. Cell Biol._ 19, 489–506 (2018). CAS PubMed Google Scholar * Du, J. et al. Dual binding of chromomethylase domains to H3K9ME2-containing
nucleosomes directs DNA methylation in plants. _Cell_ 151, 167–180 (2012). CAS PubMed PubMed Central Google Scholar * Zemach, A. et al. The _Arabidopsis_ nucleosome remodeler DDM1 allows
dna methyltransferases to access H1-containing heterochromatin. _Cell_ 153, 193–205 (2013). CAS PubMed PubMed Central Google Scholar * Stroud, H., Greenberg, M. V., Feng, S.,
Bernatavichute, Y. V. & Jacobsen, S. E. Comprehensive analysis of silencing mutants reveals complex regulation of the _Arabidopsis methylome_. _Cell_ 152, 352–364 (2013). CAS PubMed
PubMed Central Google Scholar * Liu, J., He, Y., Amasino, R. & Chen, X. siRNAs targeting an intronic transposon in the regulation of natural flowering behavior in _Arabidopsis_. _Genes
Dev._ 18, 2873–2878 (2004). CAS PubMed PubMed Central Google Scholar * Kinoshita, Y. et al. Control of FWA gene silencing in _Arabidopsis thaliana_ by sine-related direct repeats.
_Plant J._ 49, 38–45 (2007). CAS PubMed Google Scholar * Henderson, I. R. & Jacobsen, S. E. Tandem repeats upstream of the _Arabidopsis_ endogene SDC recruit non-cg DNA methylation
and initiate sirna spreading. _Genes Dev._ 22, 1597–1606 (2008). CAS PubMed PubMed Central Google Scholar * Hollister, J. D. & Gaut, B. S. Epigenetic silencing of transposable
elements: a trade-off between reduced transposition and deleterious effects on neighboring gene expression. _Genome Res._ 19, 1419–1428 (2009). CAS PubMed PubMed Central Google Scholar *
Le, T. N., Miyazaki, Y., Takuno, S. & Saze, H. Epigenetic regulation of intragenic transposable elements impacts gene transcription in _Arabidopsis thaliana_. _Nucleic Acids Res._ 43,
3911–3921 (2015). CAS PubMed PubMed Central Google Scholar * Saze, H., Shiraishi, A., Miura, A. & Kakutani, T. Control of genic DNA methylation by a JMJC domain-containing protein in
_Arabidopsis thaliana_. _Science_ 319, 462–465 (2008). ADS CAS PubMed Google Scholar * Saze, H. et al. Mechanism for full-length RNA processing of _Arabidopsis_ genes containing
intragenic heterochromatin. _Nat. Commun._ 4, 2301 (2013). ADS PubMed Google Scholar * Lei, M. et al. _Arabidopsis_ EDM2 promotes _IBM1_ distal polyadenylation and regulates genome DNA
methylation patterns. _Proc. Natl Acad. Sci. USA_ 111, 527–532 (2014). ADS CAS PubMed Google Scholar * Ni, T. et al. A paired-end sequencing strategy to map the complex landscape of
transcription initiation. _Nat. Methods_ 7, 521–527 (2010). CAS PubMed PubMed Central Google Scholar * Takahashi, H., Lassmann, T., Murata, M. & Carninci, P. 5’ end-centered
expression profiling using cap-analysis gene expression and next-generation sequencing. _Nat. Protoc._ 7, 542–561 (2012). CAS PubMed PubMed Central Google Scholar * Hetzel, J., Duttke,
S. H., Benner, C. & Chory, J. Nascent RNA sequencing reveals distinct features in plant transcription. _Proc. Natl Acad. Sci. USA_ 113, 12316–12321 (2016). CAS PubMed PubMed Central
Google Scholar * Tokizawa, M. et al. Identification of _Arabidopsis_ genic and non-genic promoters by paired-end sequencing of TSS tags. _Plant J._ 90, 587–605 (2017). CAS PubMed Google
Scholar * Lu, Z. & Lin, Z. Pervasive and dynamic transcription initiation in _Saccharomyces cerevisiae_. _Genome Res_. https://doi.org/10.1101/gr.245456.118 (2019). * Fort, A. et al.
Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. _Nat. Genet._ 46, 558–566 (2014). CAS PubMed Google
Scholar * Hashimoto, K. et al. Cage profiling of ncrnas in hepatocellular carcinoma reveals widespread activation of retroviral LTR promoters in virus-induced tumors. _Genome Res._ 25,
1812–1824 (2015). CAS PubMed PubMed Central Google Scholar * Neri, F. et al. Intragenic DNA methylation prevents spurious transcription initiation. _Nature_ 543, 72–77 (2017). ADS CAS
PubMed Google Scholar * Brocks, D. et al. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. _Nat. Genet._ 49, 1052 (2017). CAS PubMed
PubMed Central Google Scholar * Yamamoto, Y. Y. et al. Differentiation of core promoter architecture between plants and mammals revealed by LDSS analysis. _Nucleic Acids Res._ 35,
6219–6226 (2007). CAS PubMed PubMed Central Google Scholar * Mejía-Guerra, M. K. et al. Core promoter plasticity between maize tissues and genotypes contrasts with predominance of sharp
transcription initiation sites. _Plant Cell._ 27, 3309–3320 (2015). PubMed PubMed Central Google Scholar * Yamamoto, Y. Y. et al. Identification of plant promoter constituents by analysis
of local distribution of short sequences. _BMC Genomics_ 8, 67 (2007). PubMed PubMed Central Google Scholar * Yamamoto, Y. Y. et al. Heterogeneity of _Arabidopsis_ core promoters
revealed by high-density TSS analysis. _Plant J._ 60, 350–362 (2009). CAS PubMed Google Scholar * Morton, T. et al. Paired-end analysis of transcription start sites in _Arabidopsis_
reveals plant-specific promoter signatures. _Plant Cell._ 26, 2746–2760 (2014). CAS PubMed PubMed Central Google Scholar * Fejes-Toth, K. et al. Post-transcriptional processing generates
a diversity of 5’-modified long and short rnas: affymetrix/cold spring harbor laboratory encode transcriptome project. _Nature_ 457, 1028–1032 (2009). ADS CAS PubMed Central Google
Scholar * Mercer, T. R. et al. Regulated post-transcriptional RNA cleavage diversifies the eukaryotic transcriptome. _Genome Res._ 20, 1639–1650 (2010). CAS PubMed PubMed Central Google
Scholar * Nielsen, M. et al. Transcription-driven chromatin repression of intragenic transcription start sites. _PLoS Genet._ 15, e1007969 (2019). CAS PubMed PubMed Central Google
Scholar * Soppe, W. J. et al. The late flowering phenotype of FWA mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. _Mol. Cell_ 6, 791–802 (2000). CAS PubMed
Google Scholar * Lippman, Z. & Martienssen, R. The role of RNA interference in heterochromatic silencing. _Nature_ 431, 364–370 (2004). ADS CAS PubMed Google Scholar * Seila, A.
C. et al. Divergent transcription from active promoters. _Science_ 322, 1849–1851 (2008). ADS CAS PubMed PubMed Central Google Scholar * Carninci, P. et al. Genome-wide analysis of
mammalian promoter architecture and evolution. _Nat. Genet._ 38, 626–635 (2006). CAS PubMed Google Scholar * Shu, H., Wildhaber, T., Siretskiy, A., Gruissem, W. & Hennig, L. Distinct
modes of DNA accessibility in plant chromatin. _Nat. Commun._ 3, 1281 (2012). ADS PubMed Google Scholar * Zhang, T., Marand, A. P. & Jiang, J. PlantDHS: a database for DNase I
hypersensitive sites in plants. _Nucleic Acids Res._ 44, D1148–D1153 (2015). PubMed PubMed Central Google Scholar * Eick, D. & Geyer, M. The rna polymerase II carboxy-terminal domain
(CTD) code. _Chem. Rev._ 113, 8456–8490 (2013). CAS PubMed Google Scholar * Xiao, J. et al. _Cis_ and _trans_ determinants of epigenetic silencing by polycomb repressive complex 2 in
_Arabidopsis_. _Nat. Genet._ 49, 1546–1552 (2017). CAS PubMed Google Scholar * Deleris, A. et al. Loss of the DNA methyltransferase MET1 induces H3K9 hypermethylation at PcG target genes
and redistribution of H3K27 trimethylation to transposons in _Arabidopsis thaliana_. _PLoS Genet._ 8, e1003062 (2012). CAS PubMed PubMed Central Google Scholar * Takuno, S. & Gaut,
B. S. Body-methylated genes in _Arabidopsis thaliana_ are functionally important and evolve slowly. _Mol. Biol. Evol._ 29, 219–227 (2011). PubMed Google Scholar * Bewick, A. J. &
Schmitz, R. J. Gene body DNA methylation in plants. _Curr. Opin. Plant Biol._ 36, 103–110 (2017). CAS PubMed PubMed Central Google Scholar * Zilberman, D., Gehring, M., Tran, R. K.,
Ballinger, T. & Henikoff, S. Genome-wide analysis of _Arabidopsis thaliana_ DNA methylation uncovers an interdependence between methylation and transcription. _Nat. Genet._ 39, 61–69
(2007). CAS PubMed Google Scholar * Horvath, R., Laenen, B., Takuno, S. & Slotte, T. Single-cell expression noise and gene-body methylation in _Arabidopsis thaliana_. _Heredity_ 123,
81–91 (2019). * Bewick, A. J. et al. On the origin and evolutionary consequences of gene body DNA methylation. _Proc. Natl Acad. Sci. USA_ 113, 9111–9116 (2016). CAS PubMed PubMed Central
Google Scholar * Rigal, M., Kevei, Z., Pélissier, T. & Mathieu, O. DNA methylation in an intron of the IBM1 histone demethylase gene stabilizes chromatin modification patterns. _EMBO
J._ 31, 2981–2993 (2012). CAS PubMed PubMed Central Google Scholar * Zhai, J. et al. A one precursor one siRNA model for Pol IV-dependent siRNA biogenesis. _Cell_ 163, 445–455 (2015).
CAS PubMed PubMed Central Google Scholar * Faulkner, G. J. et al. The regulated retrotransposon transcriptome of mammalian cells. _Nat. Genet._ 41, 563–571 (2009). CAS PubMed Google
Scholar * Batut, P., Dobin, A., Plessy, C., Carninci, P. & Gingeras, T. R. High-fidelity promoter profiling reveals widespread alternative promoter usage and transposon-driven
developmental gene expression. _Genome Res._ 23, 169–180 (2013). CAS PubMed PubMed Central Google Scholar * Settles, A. M., Baron, A., Barkan, A. & Martienssen, R. A. Duplication and
suppression of chloroplast protein translocation genes in maize. _Genetics_ 157, 349–360 (2001). CAS PubMed PubMed Central Google Scholar * Butelli, E. et al. Retrotransposons control
fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. _Plant Cell_ 24, 1242–1255 (2012). CAS PubMed PubMed Central Google Scholar * Tsukahara, S. et al. Bursts of
retrotransposition reproduced in _Arabidopsis_. _Nature_ 461, 423–426 (2009). ADS CAS PubMed Google Scholar * Hirsch, C. D. & Springer, N. M. Transposable element influences on gene
expression in plants. _Biochim Biophys. Acta Gene Regul. Mech._ 1860, 157–165 (2017). CAS PubMed Google Scholar * Prunet, N. et al. SQUINT promotes stem cell homeostasis and floral
meristem termination in _Arabidopsis_ through APETALA2 and CLAVATA signalling. _J. Exp. Bot._ 66, 6905–6916 (2015). CAS PubMed Google Scholar * Avelange-Macherel, M.-H. & Joyard, J.
Cloning and functional expression of ATCOQ3, the _Arabidopsis_ homologue of the yeast COQ3 gene, encoding a methyltransferase from plant mitochondria involved in ubiquinone biosynthesis.
_Plant J._ 14, 203–213 (1998). CAS PubMed Google Scholar * Meinke, D. W. Genome-wide identification of EMBRYO-DEFECTIVE (EMB) genes required for growth and development in _Arabidopsis_.
_New Phytol_. 14, 306–325 (2019). Google Scholar * Santamaria, M., Thomson, C. J., Read, N. D. & Loake, G. J. The promoter of a basic PR1-like gene, ATPRB1, from _Arabidopsis_
establishes an organ-specific expression pattern and responsiveness to ethylene and methyl jasmonate. _Plant Mol. Biol._ 47, 641–652 (2001). CAS PubMed Google Scholar * Todd, C. D.,
Deniz, Ö., Taylor, D. & Branco, M. R. Functional evaluation of transposable elements as enhancers in mouse embryonic and trophoblast stem cells. _eLife_ 8, e44344 (2019). CAS PubMed
PubMed Central Google Scholar * Teissandier, A. & Bourc’his, D. Gene body DNA methylation conspires with H3K36ME3 to preclude aberrant transcription. _EMBO J._ 36, 1471–1473 (2017).
CAS PubMed PubMed Central Google Scholar * Carrozza, M. J. et al. Histone H3 methylation by set2 directs deacetylation of coding regions by RPD3S to suppress spurious intragenic
transcription. _Cell_ 123, 581–592 (2005). CAS PubMed Google Scholar * Zhou, J. et al. Intronic heterochromatin prevents cryptic transcription initiation in _Arabidopsis_. _Plant J_. 101,
1185–1197 (2019). PubMed Google Scholar * Choi, J., Lyons, D. B., Kim, M. Y., Moore, J. D. & Zilberman, D. DNA methylation and histone h1 jointly repress transposable elements and
aberrant intragenic transcripts. _Mol. Cell._ 77, 310–323 (2020). CAS PubMed Google Scholar * Shearwin, K. E., Callen, B. P. & Egan, J. B. Transcriptional interference–a crash course.
_Trends Genet._ 21, 339–345 (2005). CAS PubMed PubMed Central Google Scholar * Palmer, A. C., Egan, J. B. & Shearwin, K. E. Transcriptional interference by rna polymerase pausing
and dislodgement of transcription factors. _Transcription_ 2, 9–14 (2011). PubMed Google Scholar * Wu, A. C. et al. Repression of divergent noncoding transcription by a sequence-specific
transcription factor. _Mol. Cell._ 72, 942–954 (2018). CAS PubMed PubMed Central Google Scholar * Hedtke, B. & Grimm, B. Silencing of a plant gene by transcriptional interference.
_Nucleic Acids Res._ 37, 3739–3746 (2009). CAS PubMed PubMed Central Google Scholar * Wang, Y.-H. & Warren Jr, J. T. Mutations in retrotransposon atcopia4 compromises resistance to
hyaloperonospora parasitica in _Arabidopsis thaliana_. _Genet Mol. Biol._ 33, 135–140 (2010). CAS PubMed PubMed Central Google Scholar * Diezma-Navas, L. et al. Crosstalk between
epigenetic silencing and infection by tobacco rattle virus in _Arabidopsis_. _Mol. Plant Pathol_. 20, 1439–1452 (2019). * Ushijima, T. et al. Light controls protein localization through
phytochrome-mediated alternative promoter selection. _Cell_ 171, 1316–1325 (2017). CAS PubMed Google Scholar * Vongs, A., Kakutani, T., Martienssen, R. A. & Richards, E. J.
_Arabidopsis thaliana_ DNA methylation mutants. _Science_ 260, 1926–1928 (1993). ADS CAS PubMed Google Scholar * Saze, H., Scheid, O. M. & Paszkowski, J. Maintenance of CPG
methylation is essential for epigenetic inheritance during plant gametogenesis. _Nat. Genet._ 34, 65–69 (2003). CAS PubMed Google Scholar * Osabe, K., Harukawa, Y., Miura, S. & Saze,
H. Epigenetic regulation of intronic transgenes in _Arabidopsis_. _Sci. Rep._ 7, 45166 (2017). ADS CAS PubMed PubMed Central Google Scholar * Bolger, A. M., Lohse, M. & Usadel, B.
Trimmomatic: a flexible trimmer for illumina sequence data. _Bioinformatics_ 30, 2114–2120 (2014). CAS PubMed PubMed Central Google Scholar * Kim, D., Langmead, B. & Salzberg, S. L.
Hisat: a fast spliced aligner with low memory requirements. _Nat. Methods_ 12, 357–360 (2015). CAS PubMed PubMed Central Google Scholar * Haberle, V., Forrest, A. R., Hayashizaki, Y.,
Carninci, P. & Lenhard, B. Cager: precise TSS data retrieval and high-resolution promoterome mining for integrative analyses. _Nucleic Acids Res._ 43, e51 (2015). PubMed PubMed Central
Google Scholar * Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. _Genome Biol._ 15, 550 (2014). PubMed PubMed
Central Google Scholar * Liao, Y., Smyth, G. K. & Shi, W. The R package rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads.
_Nucleic Acids Res._ 47, e47 (2019). CAS PubMed PubMed Central Google Scholar * Trapnell, C. et al. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and
isoform switching during cell differentiation. _Nat. Biotechnol._ 28, 511–515 (2010). CAS PubMed PubMed Central Google Scholar * Luo, C. et al. Integrative analysis of chromatin states
in _Arabidopsis_ identified potential regulatory mechanisms for natural antisense transcript production. _Plant J._ 73, 77–90 (2013). CAS PubMed Google Scholar * Saleh, A.,
Alvarez-Venegas, R. & Avramova, Z. An efficient chromatin immunoprecipitation (CHIP) protocol for studying histone modifications in _Arabidopsis_ plants. _Nat. Protoc._ 3, 1018 (2008).
CAS PubMed Google Scholar * Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. _Genome
Biol._ 10, R25 (2009). PubMed PubMed Central Google Scholar * Law, J. A. et al. Polymerase IV occupancy at RNA-directed DNA methylation sites requires SHH1. _Nature_ 498, 385–389 (2013).
ADS CAS PubMed PubMed Central Google Scholar * Krueger, F. & Andrews, S. R. Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. _Bioinformatics_ 27,
1571–1572 (2011). CAS PubMed PubMed Central Google Scholar * Akalin, A. et al. methylkit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. _Genome
Biol._ 13, R87 (2012). PubMed PubMed Central Google Scholar * Krueger, F. Trim galore (Babraham Bioinformatics, 2015). * Martin, M. Cutadapt removes adapter sequences from high-throughput
sequencing reads. _EMBnet. J._ 17, 10–12 (2011). Google Scholar * Bailey, T. L. et al. Meme suite: tools for motif discovery and searching. _Nucleic Acids Res._ 37, W202–W208 (2009). CAS
PubMed PubMed Central Google Scholar * Xu, Z. & Wang, H. Ltr_finder: an efficient tool for the prediction of full-length LTR retrotransposons. _Nucleic Acids Res._ 35, W265–W268
(2007). PubMed PubMed Central Google Scholar * Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. _J. Mol. Biol._ 215, 403–410
(1990). CAS PubMed Google Scholar * Thompson, J. D., Higgins, D. G. & Gibson, T. J. Clustal w: improving the sensitivity of progressive multiple sequence alignment through sequence
weighting, position-specific gap penalties and weight matrix choice. _Nucleic Acids Res._ 22, 4673–4680 (1994). CAS PubMed PubMed Central Google Scholar * Clamp, M., Cuff, J., Searle, S.
M. & Barton, G. J. The jalview java alignment editor. _Bioinformatics_ 20, 426–427 (2004). CAS PubMed Google Scholar * Ramírez, F., Dündar, F., Diehl, S., Grüning, B. A. & Manke,
T. deeptools: a flexible platform for exploring deep-sequencing data. _Nucleic Acids Res._ 42, W187–W191 (2014). PubMed PubMed Central Google Scholar * Freese, N. H., Norris, D. C. &
Loraine, A. E. Integrated genome browser: visual analytics platform for genomics. _Bioinformatics_ 32, 2089–2095 (2016). CAS PubMed PubMed Central Google Scholar * Wickham, H. _ggplot2:
Elegant Graphics for Data Analysis_ (Springer-Verlag, New York, 2016). MATH Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by JSPS KAKENHI Grant Number
19K06619 to H.S., and by Okinawa Institute of Science and Technology Graduate University. We thank the Arabidopsis Biological Resource Center and the Salk Institute Genomic Analysis
Laboratory for providing _Arabidopsis_ T-DNA insertion mutants, OIST SQC for RNA-seq, ChIP-seq, and BS-seq sequencing services, Dr. Tetsuji Kakutani and Dr. Tatsuo Kanno for providing mutant
seeds, Dr. Shohei Takuno for kindly sharing the list of BM genes in _A. thaliana_, OIST Infrastructure Section for technical supports in building web interface to access data, and OIST
English editing service for proofreading of the manuscript. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Plant Epigenetics Unit, Okinawa Institute of Science and Technology (OIST), 1919-1
Tancha, Onna-son, Kunigami-gun, Okinawa, 904-0495, Japan Ngoc Tu Le, Yoshiko Harukawa, Saori Miura & Hidetoshi Saze * Wageningen University & Research, Droevendaalsesteeg 4, 6708 PB
Wageningen, Netherlands Damian Boer * Faculty of Life Sciences, Kyoto Sangyo University, Kyoto, 603-8555, Japan Akira Kawabe Authors * Ngoc Tu Le View author publications You can also search
for this author inPubMed Google Scholar * Yoshiko Harukawa View author publications You can also search for this author inPubMed Google Scholar * Saori Miura View author publications You
can also search for this author inPubMed Google Scholar * Damian Boer View author publications You can also search for this author inPubMed Google Scholar * Akira Kawabe View author
publications You can also search for this author inPubMed Google Scholar * Hidetoshi Saze View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS
Experiments were designed by N.T.L. and H.S., and performed by Y.H., S.M., and H.S. Data analysis was performed by N.T.L., with the support of D.B. for gene expression analysis using
mRNA-seq data. LTR sequences were analyzed by A.K. The manuscript was prepared by N.T.L. and H.S. CORRESPONDING AUTHOR Correspondence to Hidetoshi Saze. ETHICS DECLARATIONS COMPETING
INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PEER REVIEW INFORMATION _Nature Communications_ thanks the anonymous reviewers for their contribution to the peer
review of this work. Peer review reports are available. PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional
affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 SUPPLEMENTARY DATA
3 SUPPLEMENTARY DATA 4 SUPPLEMENTARY DATA 5 SUPPLEMENTARY DATA 6 SUPPLEMENTARY DATA 7 SUPPLEMENTARY DATA 8 SUPPLEMENTARY DATA 9 SUPPLEMENTARY DATA 10 SUPPLEMENTARY DATA 11 REPORTING SUMMARY
SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation,
distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and
indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to
the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will
need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE
CITE THIS ARTICLE Le, N.T., Harukawa, Y., Miura, S. _et al._ Epigenetic regulation of spurious transcription initiation in _Arabidopsis_. _Nat Commun_ 11, 3224 (2020).
https://doi.org/10.1038/s41467-020-16951-w Download citation * Received: 09 December 2019 * Accepted: 01 June 2020 * Published: 26 June 2020 * DOI: https://doi.org/10.1038/s41467-020-16951-w
SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy
to clipboard Provided by the Springer Nature SharedIt content-sharing initiative