Play all audios:
ABSTRACT There has been a surge of interest in covalent inhibitors for protein kinases in recent years. Despite success in oncology, the off-target reactivity of these molecules is still
hampering the use of covalent warhead-based strategies. Herein, we disclose the development of precision-guided warheads to mitigate the off-target challenge. These reversible warheads have
a complex and cyclic structure with optional chirality center and tailored steric and electronic properties. To validate our proof-of-concept, we modified acrylamide-based covalent
inhibitors of c-Jun N-terminal kinases (JNKs). We show that the cyclic warheads have high resilience against off-target thiols. Additionally, the binding affinity, residence time, and even
JNK isoform specificity can be fine-tuned by adjusting the substitution pattern or using divergent and orthogonal synthetic elaboration of the warhead. Taken together, the cyclic warheads
presented in this study will be a useful tool for medicinal chemists for the deliberate design of safer and functionally fine-tuned covalent inhibitors. SIMILAR CONTENT BEING VIEWED BY
OTHERS LINKING ATP AND ALLOSTERIC SITES TO ACHIEVE SUPERADDITIVE BINDING WITH BIVALENT EGFR KINASE INHIBITORS Article Open access 20 February 2024 REVERSIBLE LYSINE-TARGETED PROBES REVEAL
RESIDENCE TIME-BASED KINASE SELECTIVITY Article 19 May 2022 CATALYST-FREE LATE-STAGE FUNCTIONALIZATION TO ASSEMBLE Α-ACYLOXYENAMIDE ELECTROPHILES FOR SELECTIVELY PROFILING CONSERVED LYSINE
RESIDUES Article Open access 14 February 2024 INTRODUCTION While covalent targeting has long played a subordinate role in drug developments, there has been a recent surge of interest in the
application of the so-called targeted covalent inhibitors (TCIs) in potent and selective inhibition of kinases1. Of the 62 protein kinase inhibitors that are approved by the Food and Drug
Administration, over half a dozen of them are TCI, thus, this paradigm shift has become the basis of many current drug discovery programs2. TCIs are typically evolved from optimized
reversible ligands, which are then endowed with an electrophilic reactive group to form a covalent bond with the targeted enzyme via a specific, noncatalytic residue (mainly cysteine on
kinases)3,4,5. This unique combination of correct positioning and covalent binding leads to a drug with exceptional potency and selectivity. Moreover, this approach has also been effective
in counteracting resistance developed against some broadly used classical cancer drugs6,7,8,9. The cysteine-targeting acrylamides have become the gold standard for warheads in TCIs,
especially in the kinase field, thanks to their attenuated reactivity toward nucleophiles of the biological matrix and the relative ease of their synthetic elaboration10. Nevertheless, it
has become apparent during their usage that these open-chain electrophilic ligands could inhibit not only kinases, but also other proteins and elicit time-dependent off-target
reactivity11,12. These may lead to increased idiosyncratic drug reactions, enhanced risk of haptenization and therefore these acrylamide-tagged TCIs might increase the risk of side-effects.
Furthermore, an additional problem could potentially arise as cells contain free thiol nucleophiles (e.g., glutathione/GSH) in a concentration that is often many orders of magnitude higher
than any targeted sulfhydryl functionalities3,5. This competitive reactivity may evoke depletion of covalent inhibitors by GSH. These challenges have triggered the development of various
approaches to solve or mitigate the inherent shortcomings of previously developed acrylamide-based covalent warheads4. One of the most promising advances in this field was the introduction
of reversible inhibitors which possess more reactive but reversible cyanoacrylamide warheads13,14. In this modification, however, the enhanced reactivity toward GSH may still result in
reduced effective concentration of TCIs. Additionally, the covalent fragment screening discovery approach, which puts the emphasis on the electrophile-first concept instead of optimizing the
noncovalent moiety for a given target, has started to emerge and it is now facilitated by the development of chemoproteomic platforms15. Mitogen-activated protein kinases (MAPK) are
ubiquitous regulators of cellular physiology. Increased extracellular signal-regulated kinase (ERK1/2) activity is a hallmark of cancer due to its pivotal role in promoting cell
growth16,17,18. Although up-regulation of p38 or c-Jun N-terminal kinase (JNK) activity is also known to be associated with specific cancers, these kinases are better known to be associated
with acute or chronic inflammation due to their pivotal role in regulating cell death19,20,21,22. There are three distinct genes (_Jnk1_, _Jnk2_, and _Jnk3_) with about ten different
isoforms generated through alternative splicing: the predominant JNK1 and JNK2 isoforms are expressed ubiquitously while JNK3 is expressed primarily in the nervous system23. JNKs are
activated by phosphorylation at their activation loop by MAPK kinases (MAP2K) such as MKK4 and/or MKK7 and regulate their downstream substrates by phosphorylation. For example, the c-Jun and
ATF2 transcription factors are phosphorylated by JNK at Ser63/73 and Thr69/71, respectively. JNKs are activated when cells are exposed to stress conditions or cytokines, while they are
activated less to exposure to growth factors. Hyperactivation of JNK signaling is common in several disease states including cancer, inflammatory, and neurodegenerative diseases. Based on
genetic and pharmacological studies in animal models, JNK inhibitors have anti-inflammatory and neuroprotective effects23. Specific blocking of JNK activity could be particularly important
in treating diseases such as Alzheimer’s and Parkinson’s diseases, asthma, diabetes, and rheumatoid arthritis. Moreover, selective JNK inhibition would likely have a positive role on neuron
regeneration/repair after injury24. Thus, JNK is an attractive target for therapeutic intervention with small molecule kinase inhibitors. As a result, various inhibitors have been developed;
however, limitations are noted with these inhibitors including lack of specificity or cell toxicity25. JNKs play critical roles in development and homeostasis, and complete and sustained
JNK inhibition may not be desirable26. To date, there are no direct JNK inhibitors approved for use in human therapies. To block JNK activity selectively, an ATP binding scaffold – developed
from imatinib – had formerly been linked to an acrylamide warhead27. These compounds selectively target a specific cysteine through covalent bond formation; this residue is conserved in
JNK1-3 and is unique among MAPKs (cysteine 154 in JNK3, cysteine 116 in JNK1 or JNK2). The inhibitors – where the most selective JNK inhibitor was called JNK-IN-8 and will be referred to as
such henceforth – were shown to bind to JNK irreversibly. Thus, JNK-IN-8 proved to be a promising JNK-specific kinase inhibitor. Nevertheless, the foreseeable formation of irreversible C-S
cysteine adducts – formed by an acrylamide warhead – raises concerns about potential unwanted off-target effects and about the half-life in a biological environment (e.g., in the presence of
various S, N nucleophiles; 1–10 mM GSH)28. In this work we show through covalent warhead modification of JNK-IN-8 that our recently identified class of cyclic, tunable, and reversible
covalent warheads can overcome many shortcomings of previously utilized open-chain warheads29. First we compare JNK inhibitors with a cyclohexenone/pentenone warhead scaffold with other
formerly known open-chain Michael acceptor containing compounds and demonstrate that composite inhibitors with a cyclic warhead may have beneficial properties. We show that the inhibitors
form a reversible covalent adduct with the target cysteine on JNKs and that inhibitor residence time can be fine-tuned by adjusting the electronic properties and/or steric crowding around
the Michael acceptor. In addition to the specificity of the so-called directing group responsible mainly for classical noncovalent binding in the ATP-pocket, the cyclic warhead endows
composite drugs with increased kinase and JNK isoform specificity. Finally, we showcase the translational potential of the reversible covalent warheads and underscore their significance in
the development of specific reversible covalent inhibitors. RESULTS JNK INHIBITORS WITH DIFFERENT CHEMICAL WARHEADS A reversible warhead could provide a better alternative to an irreversible
electrophile (e.g., acrylamide), particularly if one considers that unwanted cross-reactions in cells, tissues, or in the body are likely to be more devastating if happened through
irreversible covalent bond formation. Despite the advances in reversible warhead development, however, there is still a need to broaden the list of easily available and effective warheads.
Thus, we envisioned that development and incorporation of warheads with tunable reactivity and enhanced 3D shape might result in more effective and selective inhibitors. In an
electrophile-first discovery approach targeting the common MAPK D-groove cysteine via a C-S covalent bond, we identified a class of cyclic, Michael acceptor-based, reversible covalent
warhead with a terpenoid-like structure29. Owing to their special structural feature, a state of frustration was expected to emerge, as enhanced steric crowding (substituents of C4 (γ))
around the reactive β-position can hamper Michael-adduct formation with thiol-groups. Additionally, the presence of nearby electron-withdrawing groups is expected to counteract the effect of
steric shielding, thus the binding properties (affinity, kinetics and specificity) could be custom-engineered. Gratifyingly, this frustrated state in these warheads allowed the development
of cysteine targeting drugs with (a) higher selectivity compared to open-chain (acyclic) covalent inhibitors with an acrylamide warhead and (b) prominent GSH tolerance. Further practical
advantage of these warheads is that they can be easily elaborated further using various orthogonal synthetic methods. Thus, the previously used late-stage, appendage approach to introduce
acrylamide warheads can be circumvented. Potentially, due to their more complex three-dimensional shape, the cyclic structure of our warheads allows the development of more selective and
improved cysteine targeting drugs. In contrast to free solvent exposed thiols, for example in glutathione, the sulfhydryl group of a cysteine from a protein domain is usually located in a
more specific chemical environment: surface topography and the chemical nature of the neighboring amino acid side chains will affect the intrinsic capacity of the α,β-unsaturated Michael
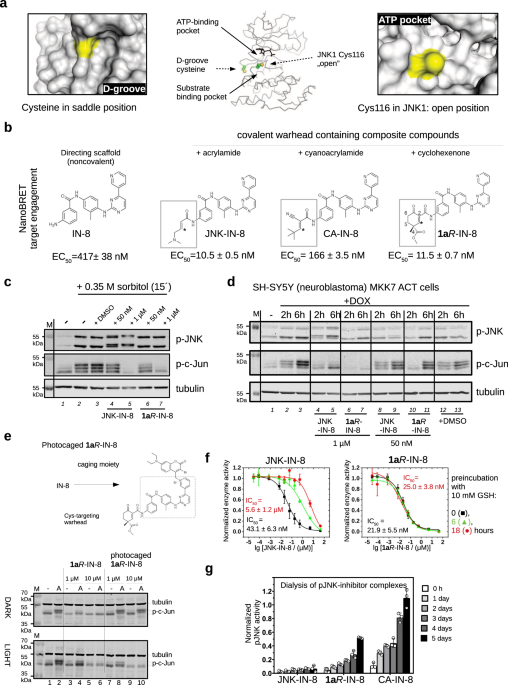
acceptor (C3) to form the covalent bond. The thiol of Cys116 in JNK1 is more accessible compared to the MAPK D-groove cysteine (Fig. 1a). Therefore, we envisioned that the advantageous
feature of our cyclic warhead designs may also be exploited in the inhibition of JNKs. Indeed, NanoBRET target engagement assay in live HEK293T cells showed that the irreversible
(acrylamide) JNK-IN-8 and the reversible (cyclohexenone) 1A_R_-IN-8 performed similarly (~ 10 nM), while reversible cyanoacrylamide-based CA-IN-8 was a weaker JNK binder ( ~ 200 nM); and, as
expected, the ATP binding moiety without any warhead was less effective (~ 500 nM) (Fig. 1b and Supplementary Fig. 1). Next, we assessed the inhibitory capacity of JNK-IN-8 and 1A_R_-IN-8
in cells on the JNK pathway. First, HEK293T cells were treated with sorbitol which is known to activate the JNK dependent signaling pathway30. Both inhibitors efficiently blocked JNK
mediated c-Jun phosphorylation when added in 1 µM concentration while JNK activation was unaffected. Since JNK-IN-8 binding to JNK is irreversible, the electrophoretic mobility of JNK on
SDS-PAGE was changed after inhibitor binding. In contrast, 1A_R_-IN-8 treatment did not affect the electrophoretic mobility of JNK, suggesting that the binding of this inhibitor to JNK is
indeed reversible (Fig. 1c). In further experiments we used an engineered human neuroblastoma cell line (SH-SY5Y MKK7 ACT) where specific JNK pathway activation was triggered by the addition
of an artificial inducer (doxycycline). This cellular system allows specific activation of JNKs while leaving other MAPK pathways unaffected31. JNK-mediated c-Jun(Ser73) phosphorylation was
examined after 2 or 6 h of doxycycline (DOX) treatment, where longer DOX induction triggered higher JNK activity flux. Experiments with inhibitors added in 50 nM or 1 µM concentrations
showed that both compounds can block c-Jun phosphorylation (Fig. 1d). Next, we devised a chemical biological strategy to demonstrate that the effects of the warhead (1A_R_) on c-Jun
phosphorylation in the context of the composite inhibitor (1A_R_-IN-8) indeed depends on JNK ATP-pocket binding, as this would be naturally anticipated. The crystal structure of the
acrylamide warhead containing inhibitor bound to JNK3 (PDB ID: 3V6S) showed that the pyridine ring of IN-8 is deeply buried in the ATP-pocket. To achieve light-controllable JNK activity, a
photoactivatable version of the 1A_R_-IN-8 compound was synthesized. The recently introduced 3-bromo-7-diethylaminocoumarin photoremovable protecting group was selected for caging the
pyridine nitrogen of IN-8 via quaternization to reduce binding into the narrow ATP-pocket due to steric blocking32. This photocage permits efficient photorelease of the pyridines upon blue
light (450 nm) irradiation generating in this case the original inhibitor in situ for JNK binding (Supplementary Fig. 2). HEK293 cells were treated with anisomycin that turned JNK and thus
c-Jun phosphorylation on, and the latter was found to be similar under dark or upon illumination as expected. This demonstrates that the light-activation protocol is neutral to cells.
Addition of 1 or 10 μM free inhibitor (1A_R_-IN-8) greatly reduced or eliminated the effect of anisomycin under both dark and light-activation conditions. More importantly, cells treated
with the photocaged inhibitor displayed c-Jun phosphorylation blockade only upon blue light illumination (Fig. 1e). This experiment shows that the inhibitory capacity of 1A_R_-IN-8 directly
depends on JNK ATP-pocket engagement in cells, as expected from a JNK-specific ATP-competitive inhibitor. We also examined the inhibitory capacity of irreversible JNK-IN-8 and reversible
1A_R_-IN-8 after exposing them to high amounts of GSH. When these two compounds were incubated in the presence of 10 mM GSH for 6 or 18 h under physiological conditions before adding them
into the in vitro kinase assay (PhALC), the IC50 of JNK-IN-8 drastically dropped but 1A_R_-IN-8 stayed similarly active in blocking MAPK activity (Fig. 1f). These experiments demonstrate
that 1A_R_-IN-8 stays functionally intact even after 18 h of incubation in 10 mM GSH, while JNK-IN-8 becomes inactive and its efficacy dramatically drops within a few hours. This latter is
due to irreversible covalent adduct formation with GSH based on an analysis with electrospray mass spectrometry (Supplementary Fig. 3). Note that the GSH–1A_R_-IN-8 adduct also forms, which
we could only detect by shortening the LC step prior to electrospray mass spectrometry (see Supplementary Fig. 3). Moreover, the covalent adduct of 1A_R_-IN-8 with beta-mercaptoethanol (BME)
was also detected by 1H NMR, and a titration experiment indicated the Kchem to be ~1 mM for the free thiol (Supplementary Fig. 4). This value was corroborated by an additional electronic
circular dichroism measurement using a truncated anilide derivative (the Kchem value of 19’_S_ + BME reaction by EDC titration was also found to be ~ 1 mM)29. In brief, GSH does not lower
the effective concentration of the developed reversible covalent inhibitor since their adduct is transient and its affinity is low ( ~ mM) compared to the thiol adduct forming on the target
(JNK) with higher affinity ( ~ low nM) and with a greatly decreased koff. Next, the off-rate of phosphorylated JNK1–JNK-IN-8, –1A_R_-IN-8, and –CA-IN-8 complexes were examined in an in vitro
dialysis experiment. JNK-inhibitor complexes were preformed and then dialyzed up to 5 days. The reversible nature of the JNK1–1A_R_-IN-8 complex, in contrast to the irreversible
JNK1–JNK-IN-8 complex, was also confirmed by these experiments since some enzymatic activity of JNK1 ( ~ 50%) could be regained if the enzyme had bound to 1A_R_-IN-8 but not when to
JNK-IN-8. Moreover, faster recovery of JNK activity upon dialysis indicated that the off-rate of the reversible cyanoacrylamide based CA-IN-8 inhibitor is faster (Fig. 1g). Overall, these in
vitro experimental results suggest that 1A_R_-IN-8 might be a superior JNK inhibitor, due to its reversible cysteine targeting, resilience to GSH, and its similar in-cell potency compared
to JNK-IN-8, or to its more promising properties compared to the cyanoacrylamide based reversible covalent reference compound (CA-IN-8). CYCLIC MICHAEL ACCEPTOR BASED REVERSIBLE COVALENT
WARHEADS Cyclohexenone/pentenone scaffolds provide a platform for controlling intrinsic reactivity: the electronic or steric properties and ring strain could all be modified. We synthesized
a set of JNK inhibitors that all contained the same ATP-pocket binding moiety (IN-8) but had different warheads. The warheads were the following: acrylamide – acyclic irreversible
(JNK-IN-8)27, cyanoacrylamide (CA) – acyclic reversible, and cyclohexenone/pentenone – cyclic reversible (1A_R_, 1A_S_, 2, 3, and 4). These were linked to IN-8 in the same way so that the
electrophilic carbon (C3) atom of the α,β-unsaturated carbonyl system could selectively target a surface cysteine located next to the ATP-pocket (Cys116 in JNK1). We also generated two
additional 1A_R_-IN-8 derivatives, in which the ATP-pocket binding moiety was linked to the cyclic warhead scaffold via an additional methylene group (1A’_R_-IN-8), making the distance
between these two presumably less optimal, and this was further modified by changing the amide to an ester group at C2 (1A”_R_-IN-8; thus the latter has a more reactive Michael acceptor).
Finally, two more enantiomeric pairs – similar to 1A_R_/1A_S_, but distinct in one of the substituent groups at C4 – were synthesized: 5S/5 _R_ or 6_S,S_/6_R,R_ contained a sterically more
demanding isopropyl or a C4-C6 bridged skeleton instead of the methyl group, respectively (Table 1). These compounds thus display different tactics to fine-tune the covalent reactivity of
the warhead: either increasing sterical crowding (5) or constraining the flexibility of the cyclohexenone ring (6), which could both indirectly affect the axial-equatorial stereochemical
transition at C3 upon C-S covalent bond formation33. Note that the results of the different assays shown in Table 1 may not be directly compared since they report on the performance of the
inhibitors under different scenarios. The in vitro PhALC assay or the cell-based NanoBRET target engagement assay monitors only JNK1 specific effects under clean or more competitive
conditions, respectively, while the two p-c-Jun assay reports on overall JNK inhibition in the used cell line. The crystal structure of JNK1–1A_R_-IN-8, –1A_S_-IN-8, and –1A’_R_-IN-8
complexes were next determined to confirm the formation of the covalent adduct (Table 2). Comparison of these structures suggests that the cyclohexenone core may be used as a platform to
orient extra moieties, attached at C4 for example, towards different regions in the wide substrate binding crevice (Fig. 2a). To demonstrate that specific covalent bond formation contributes
to binding, JNK-inhibitor complex formation was examined by SPR using wild-type JNK1 or the C116S mutant. This showed that the warheads indeed work as covalent anchors if C116 is intact and
thus they decrease the koff as expected (Fig. 2.b). INTRINSIC REACTIVITY AND BINDING KINETICS To analyze the contribution of the warheads of IN-8-based JNK inhibitors more rigorously, the
binding kinetics of different composite inhibitors were further examined by SPR and the results were quantitatively analyzed (Supplementary Fig. 5). For the latter, a 2-step reversible
kinetic model was established (Fig. 3). The reversible binding scheme included the formation of the noncovalent kinase-inhibitor (K:I) complex as well as the covalent adduct (K:Icov). The
association and dissociation of K:I are described by the k1 and k2 rates, while the formation of K:Icov from K:I or the formation of K:I from K:Icov are described by k3 and k4, respectively.
To unambiguously determine all four parameters based on numerical fitting of the kinetic plots with the 2-step reversible kinetic model, k1 and k2 were first determined using the
cysteine-to-serine mutated protein (JNK1 Cys116Ser). Reversible inhibitor potency ranking can be based on the steady-state inhibition constant, Ki = (k2/k1) / (1+ (k3/k4)) in molarity (M)34.
This analysis can also be used to estimate the energy contribution of reversible covalent bond formation for each inhibitor (ΔΔG) (Supplementary Table 1). The kinetic constants related to
noncovalent binding (k1 and k2) varied only marginally as expected, since the common IN-8 moiety governs ATP-pocket binding for all inhibitors. In contrast, k3 and k4, describing the
contribution of the reversible covalent bond towards overall inhibitor behavior, greatly varied. In general, the reactivity of Michael acceptor – influenced by the presence of an
electron-withdrawing ester group at C4, an ester vs amide at C2, ring strain (5- versus 6-membered ring) – correlated with inhibitor potency. The value of ΔΔG, in addition to the above
parameters, was also affected by the configuration at C4 (1A_R_ vs 1A_S_) and the relative positioning of the warhead compared to the directing IN-8 moiety (1A_R_-IN-8 vs 1A’_R_-IN-8).
Importantly, properly tuned and well-positioned cyclohexenone/pentenone warheads all outperformed a similar and previously introduced cyanoacrylamide (CA) warhead design. The dissociation
rate of the compounds (k4) were lowered by about two orders of magnitude compared to k2, demonstrating that reversible covalent bond formation greatly increases occupancy as expected,
provided that the warhead is positioned by the IN-8 directing moiety next to the cysteine thiol properly; otherwise k4 drops close to k2 (compare values for 1A_R_-, 1A’_R_-, and 1A”_R_-IN-8)
(see Supplementary Table 1). Inhibitor residence could also be tuned by further sterical crowding effects at C4 (5_S_ vs 1A_R_) or by the rigidity of the cyclohexenone ring (6_S,S_ vs
1A_R_). In agreement with this, further sterical crowding by the isopropyl group at C4 gave a JNK inhibitor that bound and dissociated fast (due to enhanced frustration upon thiol adduct
formation), while the C4-C6 bridge slowed down both the association and the dissociation of the inhibitor (perfected for covalent residence) (see Supplementary Table 1). In summary, we
showed that the binding affinity as well as the residence time of the composite JNK inhibitors could be adjusted and fine-tuned by varying the substitution pattern of the warhead scaffold
and/or ring size. JNK INHIBITORS IN VITRO AND IN CELL-BASED TESTS Further studies in vitro and in cell-based assays were conducted to compare reversible and irreversible JNK inhibitors (see
Table 1). To test the inhibitory capacity of different inhibitors in vitro, a fast and quantitative assay for MAPK activity was developed which was based on the concept of phosphorylation
assisted luciferase fragment complementation (PhALC)35. JNK phosphorylates a protein construct containing a MAPK target motif (SENSOR) and phosphorylation of the target motif is recognized
by the recognition construct (RC) containing a phosphoserine/threonine binding domain. The assay requires ~1 μM purified SENSOR and RC constructs and nanomolar concentration (1–5 nM) of
phosphorylated JNK. The luminescence signal is generated by phosphorylation dependent luciferase complementation which is measured for 10–30 min giving a kinetic slope proportional to JNK
activity. We used this assay to measure the inhibitory capacity (IC50) of different covalent warhead containing JNK inhibitors in the presence of 10 mM GSH. In addition, some of the
inhibitors’ capacity to bind to the JNK1 ATP-pocket was also assessed by the NanoBRET target engagement assay in live HEK293T cells36. Finally, interference with c-Jun phosphorylation was
monitored by quantitative western blots in HEK293T cells (p-c-Jun IC50). We also tested some additional covalent inhibitors using the PhALC assay. These contained varieties of the IN-8
ATP-binding moiety (IN-7, IN-9, and IN-10)27 or a modified version of the IN-7 moiety in which one of the phenyl groups was replaced with a phenyl isostere, bicyclo[1.1.1]pentane, to improve
physicochemical and pharmacological properties (1A_R_-isoPHEN)37. These inhibitors performed similarly or slightly better compared to 1A_R_-IN-8 in the PhALC assay (Table 3 and
Supplementary Fig. 6). The IC50 value of irreversible JNK-IN-8 and reversible 1A_R_-IN-8 on c-Jun phosphorylation triggered by 6 h of DOX stimulation in the engineered neuroblastoma cell
line (SH-SY5Y MKK7 ACT; see Fig. 1d) was found to be similar (~ 0.7 μM in the western blot based p-c-Jun(Ser73) and ~ 100 nM in the HTRF p-c-Jun(Ser63) assay) (Supplementary Fig. 7 and 8).
We noticed that specific JNK activation in this neuroblastoma cell line leads to decreased cell survival after 72 h, likely initiating cell death (Supplementary Fig. 9)23,38. We then tested
whether this JNK-specific response in this cell line could be blocked by the addition of JNK-inhibitors. It was found that reversible warhead-containing compounds (1A_R/S_, 1A_R_-isoPHEN) as
well as the irreversible JNK-IN-8 inhibitor protected cells from cell death to a significantly greater extent compared to IN-8 (i.e. the ATP competitive moiety alone). This showed that the
reversible warhead could perform similarly to an irreversible one in this cellular setting and confirmed that covalent targeting indeed enhances the biological effect of a noncovalent
directing group (IN-8) (Fig. 4a). MAPKs are known to regulate gene expression by directly phosphorylating components of the AP-1 transcription factor (e.g., c-Jun, ATF or c-Fos)39. The
effect of JNK inhibitors on AP-1 mediated transcription was probed by using the AP-1 Reporter – HEK293 Recombinant Cell Line harboring an AP-1 promoter+luciferase reporter gene cassette
stably integrated into the genome. Promoter activity was stimulated by the addition of phorbol 12-myristate 13-acetate (PMA) and the transcription of the reporter gene was monitored by
measuring luminescence after 6 h (where inhibitors were co-administered with PMA). According to these experiments, JNK-IN-8 showed similarly weak inhibition as IN-8 (tested at 1, 3, and 10
μM concentrations), while 1A_R_-IN-8 and 1A_R-_isoPHEN clearly caused stronger inhibition. Moreover, the reversible cyanoacrylamide warhead in CA-IN-8 could mediate only modest inhibition
compared to the different cyclohexenone-based warheads from 5_S_-, 6_S,S_- and 1A_R_-IN-8 (Fig. 4b). Earlier in vitro experiments showed that cyclohexenone/pentenone based warheads greatly
decrease the koff. To demonstrate that the reversible covalent warheads increase the cellular residence time as expected, we carried out cell-based wash-out experiments using the
physiologically relevant AP-1 promoter-based reporter assay as well as the more direct NanoBRET target engagement assay. The inhibitors (IN-8a, JNK-IN-8, 1A_R_-IN-8, 6_S,S_-IN-8; 10 μM) were
washed out from AP-1 Reporter – HEK293 cells after 2 h of incubation and AP-1 promoter mediated luciferase reporter activity was measured after 6 h. These showed that all covalent
inhibitors, in contrast to IN-8a, were indeed resilient to the wash-out as expected (Fig. 4c). Moreover, longer target engagement compared to IN-8a (noncovalent, N-acetylated IN-8 core) or
even to CA-IN-8 (the reversible, cyanoacrylamide containing benchmark compound) was confirmed by similar wash-out experiments in HEK293T cells using the NanoBRET target engagement assay
(Fig. 4d). In these latter experiments cells were first transiently transfected with a plasmid allowing the expression of the luciferase-JNK1 chimera construct. After 24 h cells were
incubated with the different inhibitors for two hours and washed three times before the fluorescent ATP-competitive tracer was added at different times after the wash-out (0, 4 and 8 h). The
NanoBRET signal monitors the occupancy of the fluorescent dye on the target and reports on the level of free JNK1 (Supplementary Fig. 10). Results of these wash-out experiments suggest that
reversible covalent bond formation greatly increases the in-cell residence time of cyclohexenone warhead containing inhibitors and, in contrast to the cyanoacrylamide containing inhibitor,
the reversible warheads seem to be similarly as effective as the irreversible acrylamide warhead (at least up to the examined time, ~ 8 h). KINASE SPECIFICITY MAPK specificity of reversible
warhead containing inhibitors were assessed by using the PhALC assay, which showed that 1A_R_-IN-8 has close to 1000-fold specificity towards JNK1 compared to p38α or ERK2 (Supplementary
Fig. 11a). Next, we tested the specificity of the inhibitors in cells. We used a cell line that allows p38-specific activation (HEK293T MKK6EE) and monitored MK2 substrate kinase
phosphorylation with western blots using an anti-phosphoMK2 antibody40. These experiments showed that 1A_R_-IN-8, 1A’_R_-IN-8, and 1A”_R_-IN-8, similarly to JNK-IN-8, do not affect
p38-mediated MK2 phosphorylation (Supplementary Fig. 11b). Finally, we tested if 1A_R_-IN-8 may target a nearby surface cysteine (Cys163) located in the docking groove of JNK by using an in
vitro fluorescence polarization-based protein-peptide binding assay. 1A_R_-IN-8 did not affect docking peptide binding, thus suggesting that the reversible inhibitors selectively interfere
with JNK by specifically targeting its unique cysteine located next to the ATP binding pocket (Cys116 in JNK1) (Supplementary Fig. 11c). Compared to an acrylamide based (acyclic) warhead
moiety, the more complex (cyclic) structure around the α,β-unsaturated carbonyl in the cyclohexenone/pentenone scaffold could potentially lead to more selective cysteine targeting.
Therefore, we tested the performance of 1A_R_-IN-8 on a large portion of the human kinome to assess its specificity towards JNKs. The compound was tested in 1 µM concentration on the Wild
Type Kinase Panel of Reaction Biology (comprised of 340 human kinases)36,41. 1A_R_-IN-8 inhibited all three JNK isoforms (JNK1/2/3) as expected. In addition, this reversible compound also
inhibited LIMK1 and TNK1 more than 50% (remaining activity: 38% and 44%, respectively), but all other kinases were mostly unaffected (Fig. 5a and Supplementary Table 2). This data suggests
that 1A_R_-IN-8 is a uniquely specific JNK inhibitor. The modest inhibition of LIMK1 and TNK1 is presumably due to their surface cysteine located in their flexible P-loop that could be
targeted by the warhead moiety and to good compatibility with the ATP-competitive moiety (IN-8) by their nucleotide binding pocket. In agreement to this, the corresponding close homologs
(LIMK2 and ACK1/TNK2) that do not have a cysteine in their P-loop were less inhibited (remaining activity: 85% and 80%, respectively). Formerly, JNK-IN-8 was profiled in 1 µM concentration
using KINOMEScan methodology (DiscoverX, 442 distinct kinases)27. This platform is quite different compared to the Reaction Biology platform, since the former employs an active site-directed
competition binding assay to quantitatively measure interactions between test compounds and a kinase, while the latter quantitatively measures the compound’s capacity to inhibit model
substrate phosphorylation. There were 280 kinases common in the DiscoverX and Reaction Biology kinase panels and the inhibitory capacity of JNK-IN-8 and 1A_R_-IN-8 may be directly addressed
on this common human kinase set. There were 23 kinases in the JNK-IN-8 dataset that were strongly bound by the irreversible acrylamide-based inhibitor (including the three JNKs and 20
off-target kinases). Importantly, 1A_R_-IN-8 with its reversible cyclohexenone-based warhead inhibited none of the 20 off-target kinases (Supplementary Table 3). However, due to differences
between the two kinome scan platforms, a direct comparison of these data sets may be misleading. To this end, we have chosen 6 kinases from the common set (the JNK3 target, and 5 off-target
kinases: INSR, IRAK1, KIT, PDGFRB as off-targets from the JNK-IN-8 study and TNK1 as an off-target from the 1A_R_-IN-8 study) and an additional kinase, the homolog of TNK1 without a P-loop
cysteine (ACK1/TNK2) and had them tested in the Eurofins DiscoverX platform at 1 μM concentration with JNK-IN-8, 1A_R_-IN-8, and 6_S,S_-IN-8. All three inhibitors bound to the target JNK3
kinase well as expected (%Control was 0% for all three, where lower numbers indicate stronger interaction score). JNK-IN-8 had higher binding score for most off-targets, while 1A_R_-IN-8
performed better but off-target binding of 6_S,S_-IN-8 was even weaker (Fig. 5b). An explanation for the better performance of the latter two compounds against off-targets is provided below.
The Michael acceptor carbon atom is in a fundamentally different chemical environment in a cyclohexenone ring compared to an open-chain acrylamide-based warhead. Intuitively, the former has
generally more limited accessibility and steric crowding groups will limit the spatial availability of the reactive carbon center for covalent adduct formation. This intuition is indeed
supported by buried volume analysis where the accessibility of the reactive carbon was systematically analyzed in the most stable conformer42 (Fig. 5c, Supplementary Fig. 12, and
Supplementary Note 1). Moreover, this comparative analysis on acrylamide (a1), cyanoacrylamide (b1) and cyclohexenone-(C2)amide (c1) – and their sterically crowded versions (e.g.,
_N_,_N_-dimethyl-acrylamide (a2), _tert_-butyl-cyanoacrylamide (b2), or (c2) 4,4-dimethyl-, (c3) 4-methyl-4-carboxymethyl-, (c4) 4-isopropyl-4-carboxymethyl-, and (c5)
C4-C6-bridged-4-carboxymethyl-cyclohexenone-amide) – clearly suggested that the beta-carbon atom is notably hindered when the Michael acceptor moiety is in a substituted ring compared to
acrylamide-based structures. In summary, in JNK TCIs the cyclic warheads could be used as somewhat different and more specific chemical solutions for nucleophile targeting compared to
open-chain warheads. JNK ISOFORM SPECIFICITY Owing to the structure and inherent reactivity of the cyclic warheads, further structural modifications can be made to modulate selectivity of
the composite compound. To generate additional compounds, the ester group at C4 was modified by using azide-alkyne cycloaddition and Sonogashira coupling. Accordingly, the methyl ester group
was replaced with propargyl ester (1B_R_-IN-8, 1B_S_-IN-8), which was further orthogonally extended by either CuAAc click chemistry (1C_R_-IN-8, 1C_S_-IN-8) or by Sonogashira coupling
(1D_R_-IN-8). Based on the crystal structure of the JNK1–1A_R_-IN-8 complex, the pendant substituents at C4 were expected to project towards the shallow and wide substrate binding pocket of
JNKs. Importantly, the propargyl ester was well-tolerated in this position, since 1B_R_-IN-8 showed a similar IC50 value compared to that of 1A_R_-IN-8 in the PhALC assay or a similar EC50
in the cell-based p-c-Jun phosphorylation assay (Fig. 6a,b). Intriguingly, JNK isoforms (JNK1/2/3), with some distinct functional roles in vivo and in cancer progression43, show some
sequence differences by the base of the substrate binding cleft. This latter is encoded by exon 6 which differs somewhat between the JNK isoforms (Fig. 6c and Supplementary Fig. 13).
Notably, this ~12 amino acid long region may also differ due to the alternative splicing of exon 6, which has an a and b form23. In agreement to this, compounds derived from the propargyl
ester derivatives and extended by azide-alkyne cycloaddition or Sonogashira coupling showed notable differences in JNK isoform specificity (measured for JNK2a2, JNK1b1 and JNK3a1, referred
to below as JNK1, JNK2 and JNK3, respectively) (Supplementary Fig. 14). For example, inhibition of JNK1 was 10-fold higher compared to JNK2 by one of the two enantiomers of the propargyl
ester derivative (1B_R_-IN-8), while 1C_R_-IN-8, in which the more effective stereoisomer was further extended via azide-alkyne cycloaddition, had a more than tenfold selectivity for JNK3
over JNK2. Overall, these findings lend support to the notion that reversible and bulkier cyclohexenone-based warheads may be well-suited to sense subtle structural differences around the
targeted nucleophile, and thus they may also contribute to isoform specificity on their own right in TCIs. TRANSLATIONAL POTENTIAL OF JNK COMPOSITE INHIBITORS Synthetic elaboration of the
cyclic warhead in composite JNK inhibitors is straightforward and varying substituent groups at C4 could be used to increase potency. For example, compared to a simpler design (1A_R_-IN-8),
kinome-wide selectivity (see 1A_R_-IN-8 vs 6_S,S_-IN-8) or JNK isoform selectivity (see 1A_R_-IN-8 vs 1B_R_-IN-8), and even inhibitor efficacy on c-Jun substrate phosphorylation in cells
(see Table 1), could be improved by changing the warhead scaffold at this accessible position. Moreover, the C4 of the rigid cyclohexenone ring appears to be an excellent exit vector for
further synthetic elaboration of the warhead part of the inhibitors via click chemistry. This prompted us to test if these JNK binding compounds may also be used as JNK-specific moieties in
protein targeting chimeras (PROTACs) and to affect protein function by an independent mechanism, namely by lowering the cellular level of JNK. The 1A_R_-IN-8 moiety was linked to a E3
ubiquitin ligase binding moiety specific for cereblon (CRBN) using a PEG-linker and a click chemistry-based (CuAAC) approach (PRT_1). This heterobifunctional molecule was effective in
lowering the cellular level of JNK1 in HeLa cells in a proteasome-dependent manner (by ~65%), confirmed using the MG132 proteasome blocker compound, while p38 levels remained unaffected as
expected (Supplementary Fig. 15). This shows that the C4 position of the cyclohexenone warhead is an easily variable exit point for different linkers and additional protein binding moieties
even when these are introduced in the late stage of the synthesis. According to our knowledge, the metabolic stability of double-activated cyclohexenone based compounds have not yet been
tested. To this end, we examined the hepatic stability of 1A_R_-IN-8 parallel to JNK-IN-8 using rat primary hepatocyte model. In addition, we also carried out the same analysis on a more
simple warhead compound (PK_test), which was the C2 ester analogue of the warhead moiety present in 1A_R_-IN-8 (referred to as compound 3 in ref. 29). Based on measured in vitro
pharmacokinetics values (Clint in rat primary hepatocyte cultures), the in vivo hepatic clearance (ClH) in rat was predicted to be 10.1, 28.4 and 29.6 ml/min/kg for JNK-IN-8, 1A_R_-IN-8, and
PK_test, respectively (Supplementary Table 4)44. According to this analysis, JNK-IN-8 was considered to have low, while 1A_R_-IN-8 has intermediate hepatic extraction. Faster extraction of
the latter is likely due to the warhead moiety, since PK_test was metabolized similarly to the composite 1A_R_-IN-8 inhibitor. Next, we examined the stability of a more extended set of
compounds using blood plasma from rats (see Supplementary Table 4). The half-life values of JNK-IN-8, PK_test and five composite JNK inhibitors, containing differently functionalized
cyclohexenone warheads at C4, were determined from their depletion profiles by using mass spectrometry. Some of our warhead designs contained an ester at C4, which made the warhead more
potent, but the known high esterase activity of blood plasma could decrease potency. The half-life values of ester containing compounds such as PK_test, 1A_R_-IN-8, or 6_S,S_-IN-8 were found
to be indeed very short (a few minutes), while JNK-IN-8 or 2-IN-8, containing no ester groups, were found to be stable in plasma. Importantly, the half-life could be greatly increased by
extending the carboxymethyl group (1C_R_-IN-8) or replacing the C4 methyl group with a bulkier isopropyl group (5_S_-IN-8). Increased stability of these latter two compounds is likely due to
their decreased compatibility with the reactive centers of blood esterases because of increased steric congestion at C4. In summary, this PK analysis demonstrated that the PK properties of
the cyclic warheads could be greatly improved via simple synthetic elaboration of the core scaffold. DISCUSSION We carried out a proof-of-concept study on the use of designer Michael
acceptor covalent warhead with cyclic structure and extended 3D shape to inhibit JNKs, where an accessible cysteine located next to the ATP-pocket/substrate binding site was specifically
targeted. These studies showed that the cyclohexenone/pentenone scaffold can be engineered to fine-tune binding affinity, residence time, and specificity by modifying steric and electronic
properties of the warhead. For specific targeting, conjugation of the warheads to suitable noncovalent directing groups that fit into adjacent binding pockets is required in addition to
introducing substituent groups at key ring positions. Such composite drugs with cyclic warheads can have greatly altered/enhanced properties compared to other cysteine targeting solutions.
The warhead provides a synthetically feasible alternative to other irreversible or reversible covalent warheads (e.g., acrylamide or cyanoacrylamide) as it can be similarly linked to various
directing groups: a _tert_-butyl group or IN-8 to direct it to the MAPK D-groove or next to the JNK ATP-pocket, respectively29. A comparative analysis showed that the cyclic warhead is
comparable to, or may even supersede, its tested alternatives (irreversible acrylamide or reversible cyanoacrylamide) in the case of Cys116 in JNK1. Moreover, for the D-groove cysteine of
ERK2 (Cys161), the cyclic warhead also performed better compared to its alternatives29. The stereochemical requirements for covalent bond formation as well as for noncovalent contact
compatibility are more rigorous and more specific around a Michael acceptor in a ring system compared to previously utilized acyclic warheads. Conversely, a more rigid, cyclic, and thus
structurally more constrained reversible warhead may contribute more to binding affinity and specificity on its own right, far beyond that of a flexible, open-chain reversible covalent
moiety (e.g. cyanoacrylamide). The advantage is more apparent when the cyclic warhead can target a cysteine in a surface pocket that matches up to its size and/or stereochemical
configuration well. This positive selection mechanism is possibly the reason why cyclohexenone-based warheads were so efficient in targeting the MAPK D-groove cysteine29. Conversely, the
more complex ring structure can also hinder reactivity towards off-target cysteines due to higher probability for incompatibility with local surface topography (negative selection). In
addition to less off-target effects, reversible cysteine engagement enables fine-tuning of inhibitor binding kinetics and thus residence time, which is a key feature in therapeutic
applications requiring sustained target engagement45, similarly to applications where more rapid target disengagement is more favorable46. According to the 2-step binding/reaction mechanism,
the stable covalent complex forms via a more labile noncovalent complex (see Fig. 3), and for irreversible inhibitors k4 (the kinetic dissociation constant of the covalent complex) is 0,
while for reversible covalent inhibitors k4 > 0. When the noncovalent complex first forms then this complex may dissociate fully (k2) or progress to make the covalent complex (k3). For
reversible covalent JNK inhibitors, in vitro potency (Ki) is set mainly by the characteristics (k3, k4) of the cyclohexenone/pentenone-based warhead designs (and the impact of k1 or k2 is
relatively less since this is mainly governed by the ATP-pocket binding moiety shared by all inhibitors used for this comparison). In addition to the irreversible JNK-IN-8 reference
compound, we also tested an open-chain reversible cyanoacrylamide based compound (CA-IN-8). In vitro characterization showed that this compound is also much more effective compared to its
ATP-competitive directing moiety alone (IN-8 or IN-8a) as expected. However, this compound behaved poorly in most cell-based tests, particularly in the cell-based p-c-Jun phosphorylation
assay. In summary, despite that CA-IN-8 could potentially form a strong, slowly dissociating covalent complex in a biochemical experiment (as in the dialysis experiments, see Fig. 1g), it is
not as efficient as other composite JNK inhibitors with electronically similarly tuned cyclic Michael acceptors in the cell. Naturally, this could be due to inefficient cellular uptake or
to weakened binding to the signaling relevant pJNK-substrate complex because of its flexible aliphatic terminal appendage next to its reactive carbon but may also be due to higher reactivity
of cyanoacrylamide compared to less reactive acrylamide. This higher reactivity may allow more (protein) off-target thiol interactions to take place causing a decrease in the effective
cellular concentration compared to more rigid and specific cyclic Michael acceptors. Another important finding of this comparative study is that a more rigid, cyclic reversible covalent
warhead scaffold, if electronically properly tuned, may become similarly as effective as an irreversible covalent inhibitor. Additionally, we also demonstrated that the intrinsic reactivity
(Kchem) of the reversible warhead scaffold in JNK composite inhibitors against free thiols is low and their reaction with GSH is dynamic and this off-target thiol adduct is only transient.
Naturally, this is in stark contrast to an irreversible acrylamide-thiol adduct. Briefly, the transient nature of the reversible GSH adduct of the warhead scaffold is the mechanistic basis
underlying their resilience to GSH in vitro as well as in cells. We posit that the cysteine-targeting cyclic warhead complements and adds further specificity to the ATP-pocket binding moiety
on its own right. Moreover, direct elaboration of properly oriented substituent groups through various orthogonal chemistries provides further opportunities for the design of more complex
JNK inhibitors: these composite molecules distinguish even between some of the JNK isoforms, which remained elusive so far with conventional warheads. Moreover, reversible warhead containing
JNK-specific compounds may also be used, in addition to their capacity to block JNK activity, to lower the cellular levels of JNKs as the key specificity component in JNK-specific PROTACs.
This provides an additional modality to affect signaling protein function and the reversibility of the warhead could potentially be an advantageous property in such applications because JNK
PROTACs can potentially be salvaged after proteosomal degradation of the adduct48. The linker or the concrete structure of the ubiquitin ligase binding moiety are generally important for
producing effective PROTACs, and the potency of a particular JNK PROTAC design may be optimized by varying these47,48. The pharmacokinetics of the compounds (e.g., hepatic and non-hepatic
metabolism) need to be tested systematically, but our first characterization suggests that cyclohexenone-based warheads may have sufficient metabolic stability in vivo, moreover, their
pharmacokinetic properties could be optimized in further work. Knowing that JNKs play the role of a double-edged sword in controlling many physiological processes, where too much or too less
JNK activity or altered kinetics of the JNK response may all lead to pathological conditions depending on cell type, the capacity of being able to control the strength and/or the kinetics
of cellular JNK inhibition via synthetic elaboration of the same reversible covalent scaffold will be a valuable asset in producing effective drugs against JNK-controlled diseases/processes
(e.g. inflammation, neurodegeneration, cancer or resistance against chemotherapy). In summary, the cyclic warhead scaffold provides a structural platform to fine-tune JNK inhibitor
reactivity and selectivity by using different substitution patterns in rationally selected ring positions. We anticipate that our cyclic, structurally more complex warhead, which according
to our knowledge previously had not been demonstrated as a practical solution to target cysteines on proteins, could be exploited far beyond the first two cases presented for the MAPK
D-groove or for the JNK ATP-pocket29. METHODS PREPARATION OF PROTEIN SAMPLES Activated human ERK2, p38α, JNK1 and JNK2 for the biochemical assays were produced by co-expressing the MAPKs
with constitutively active GST-tagged MAP2Ks in _E. coli_ with bicistronic plasmids (GST-MKK1_3D/ERK2, GST-MKK6_EE/p38α, GST-MKK7_EE/JNK1/2)49. Double phosphorylation of the activation loop
of MAPKs was confirmed in western blots with phosphoMAPK-specific antibody and/or mass spectrometry. MAPKs were expressed with an N-terminal His6-tag that was cleaved off by the TEV protease
after purification with Ni-NTA affinity resin, and then samples were further purified using ion-exchange chromatography (MonoQ, GE Healthcare). Dephosphorylated MAPKs were produced with
GST-tagged λ-phage phosphatase and purified similarly. Human JNK3 was expressed alone using a simpler bacterial expression vector, and its activated form was produced by incubating the
protein with constitutively active version of MKK7 in vitro before kinase activity measurements49. GST-MKK7EE-His6 was expressed in bacteria and double-affinity purified. Biotinylated JNK1
for the SPR experiments was co-expressed with the BirA ligase in _E. coli_ and purified as described above but this construct retained an N-terminal AviTag after TEV cleavage. The JNK1 C116S
mutant was generated by two-step PCR using DNA oligonucleotides to introduce the amino acid replacement (C116S_F: GCTCATGGATGCAAATCTTAGCCAAGTGATTCAGATGGA and C116S_R:
TCCATCTGAATCACTTGGCTAAGATTTGCATCCATGAGC). Peptides were chemically synthesized using solid phase peptide synthesis (on Rink Amid resin, PS3 peptide synthesizer, Protein Technologies) with
Fmoc/_t_Bu strategy and purified by RP-HPLC using a Jupiter 300 Å C18 column (Phenomenex). Quality of the peptides were checked by HPLC-MS (Shimadzu LCMS-2020). Dialysis experiments to
confirm reversibility/irreversibily of different JNK-inhibitor complexes were done the following way. 50 nM phosphorylated JNK1 (p-JNK1) was incubated with 10 μM inhibitor in 50 mM HEPES pH
= 7.4, 100 mM NaCl, 5 mM MgCl2, 0.02% IGEPAL, 5 % glycerin, 5 mM TCEP, 0.5 m/mL BSA (1 mL) for 1.5 h at room temperature, then samples were loaded into dialysis tubes and dialyzed in 50 mM
HEPES pH = 7.4, 100 mM NaCl, 5 mM MgCl2, 2 mM DTT. 50 μL aliquots were taken from the samples before the start of the dialysis experiment (0) and after the indicated times (1, 2, 3,4, 5
days) and 5 μL were tested for enzymatic activity using the PhALC assay. The dialysis buffer (500 mL) was changed 2 h after the start of the experiment and then daily till the finish of the
experiment. The control sample was treated the same way but no inhibitor was added in the incubation step before the dialysis experiment. X-RAY CRYSTALLOGRAPHY For crystallographic analysis
C-terminally truncated human JNK1_ΔC20 was expressed in _E. coli_ and purified with His6-tag affinity, ion-exchange, and a gel-filtration step as described earlier49. JNK1 was concentrated
to 15 mg/ml, supplemented with 2 mM TCEP, and mixed in a 1:1.25 molar ratio with the inhibitors. Samples were crystallized in a hanging drop setup with 1 M NaCl in the reservoir.
JNK1–1A’_R_-IN-8 crystals grew in 2–4 days in 8% PEG 3350, 0.1 M HEPES pH = 7.5 and the best crystals with JNK1–1A_R_-IN-8 or JNK1–1A_S_-IN-8 were grown in 12% PEG 3000, 2–4% MPD, 0.1 M
HEPES pH=7.5. Crystals were flash-cooled in 25% glycerol. Data were collected at EMBL PETRA III beamline P14, Hamburg (see Table 2). Crystal structures were solved by molecular replacement
using JNK as the search model from the JNK3–JNK-IN-7 crystallographic complex (PDB ID: 3V6S). The crystallographic models have three JNK1-inhibitor complexes in the asymmetric unit.
Inhibitors were built using JLigand 1.0.4050. Merohedral twinning was detected in the JNK1-inhibitor crystals (0.46, 0.45, 0.42 for JNK1–1A_R_-IN-8, –1A_S_-IN-8, and –1A’_R_-IN-8 calculated
by PHENIX Xtriage, respectively) giving higher looking hexagonal symmetry, while the true space group proved to be P31. Structures were refined by using the -h,-k, l twin operator and NCS
restraints with PHENIX51. The pose of the compounds regarding the functional groups at C4 was clear even from the beginning of the refinement for 1A_R_-IN-8, since the relative orientation
of the methyl versus carboxymethyl groups could be decided based on unbiased maps even at the early stages of the refinement process. Due to somewhat poorer quality of the electron density
map for 1A_S_-IN-8 or for 1A’_R_-IN-8 the binding poses had to be decided by generating the different alternatives and analyzing the peaks in the Fo-Fc density maps. The two different
stereoisomers emerging due to forming the C-S bond at C3 were drawn up in Jligand and whichever gave a better fit into the density was retained and the refinement was then finalized with the
better fitting stereoisomer at C2 (since the covalent bond generates a new center at C2 as well). Density features were suitable to decide on the more likely stereoisomer at the A or B
complexes. However, the asymmetric unit contains three different JNK-inhibitor adducts (A, B, C) and C was worse and had significantly higher B factors. Unfortunately, the density for this
chain was not good enough to decide on the concrete stereochemistry at C2 and C3 without any bias. The stereoisomer from chain A and B were used to fit the weaker adduct density for C,
therefore the final configurations in C need to be handled carefully for all JNK-inhibitor complexes. PROTEIN-PEPTIDE BINDING ASSAY Fluorescence polarization (FP) based protein-peptide
binding experiments were used to assess binding into the MAPK docking groove49. A known JNK docking groove binding peptide (evJIP1) was N-terminally labeled by carboxyfluorescein and 50 nM
labeled reporter peptide was mixed with the MAPK in a concentration to achieve ~50–80% complex formation. The unlabeled competitor (peptide or small molecule was added in increasing amounts
and the FP signal was measured in a Cytation 3 (BioTek Instruments) fluorescence plate reader in 384-well plates in 20 µL volume. The Ki for the competitor was determined by fitting the data
to a competition binding equation. Titration experiments were carried out in triplicates, and the average FP signal was used for fitting the data in Origin 201849. IN VITRO KINASE ASSAY AND
IN VITRO IC50 DETERMINATION The Phosphorylation-Assisted Luciferase Complementation assay (PhALC) was developed to measure JNK activity in vitro for fast and cost-effective
identification/characterization of any compound blocking MAPK activity35. The principle of this assay is the following: the general MAPK phosphorylation target motif (S/TP) is positioned
C-terminal from a MAPK binding D-motif and this SENSOR construct is fused with the small fragment of the luciferase enzyme (NanoBiT small subunit, deep sea shrimp, _Oplophorus
glacilirostris_, Promega). In the Recognition Construct (RC), the WW domain (from Pin1) binding specifically to the phosphorylated MAPK target motif is fused with the large fragment of the
luciferase enzyme (NanoBiT large subunit)52. Upon SENSOR phosphorylation the RC will bind the SENSOR and triggers the assembly of the luciferase enzyme which will produce photons as it turns
over its substrate (coelenterazine). Both constructs are produced with N-terminal maltose binding protein (MBP) and C-terminal histidine tag for high-yield bacterial expression and for
simple affinity-resin purification. The two purified constructs (SENSOR and RC) were mixed with activated MAPKs, the reaction was started by injecting ATP into the reaction mix containing
the luciferase enzyme substrate coelenterazine, and the luminescence signal was monitored in time. The PhALC assay was used as a semi-high throughput, microplate compatible biochemical assay
to obtain IC50 values of any compounds blocking MAPK activity. In more details: The PhALC constructs (SENSOR and RC) were expressed in _E. coli_ using a modified pET expression vector
(pET-MBP) and standard bacterial expression conditions in LB where the expression of proteins was induced by the addition of 0.2 mM IPTG at 25 °C for 4 h. Proteins were purified on Ni-NTA
resin which was followed by another affinity purification step using maltose resin. RC and SENSOR were typically used in 1 µM concentrations with 1-10 nM double-phosphorylated MAPKs produced
and purified as described earlier53. The coelenterazine concentration was 200 µM and the reaction was started by adding 0.1 mM ATP. The kinase assay buffer contained 20 mM Tris pH = 8, 150
mM NaCl, 0.1% IGEPAL, 5 mM MgCl2. The luminescence signal was monitored up to 30 min and the slope at the linear range (typically up to 5 min) was calculated based on linear regression. The
p38 and ERK SENSOR contained the MEF2A D-motif (SRKPDLRVVIPP) and the JNK sensor had the more JNK-specific pepPDE4B motif (GDGISRPTTLPLTTLP)49,53. SENSORs contained the same MAPK
phosphorylation target sequence compatible with WW domain binding: VPRTPVS. This sequence motif was positioned C-terminal from the D-motif in SENSOR, separated by a flexible linker
(HMGSGSSGGSSGSGSVD). For IC50 determination the competitor was added in increasing concentrations and the normalized slope of the luminescence signal was fitted to the dose-response equation
in Origin 2018. For the GSH resilience experiments the inhibitors were added in different concentrations into PhALC kinase assay buffer (without the ATP), the samples were incubated for 0,
6 or 18 h in the presence of 10 mM GSH before PhALC assay reagents were added (activated JNK1, SENSOR, and RC). The reaction was started by adding ATP. (The concentration of the inhibitors
during incubation with 10 mM GSH was ~ 33% more compared to their final concentration used in the PhALC reaction mix). JNK enzymatic activity was measured as described above with the PhALC
assay. (0 h incubation practically means ~10 min, since this was the lag time of the measurement, i.e. before enzyme activity could be determined.) KINETIC BINDING EXPERIMENTS For surface
plasmon resonance (SPR) measurements, biotinylated proteins were captured on a Biacore CAP sensor chip using a Biacore S200 instrument (GE-Healthcare). All measurements were done at room
temperature using single-cycle setup with the standard Biacore method for CAP chip including double referencing. The KD was determined based on response unit (RU) values corresponding to
different concentrations of the analyte. Sensorgrams were fit to a 1:1 binding model using the BiaEvaluation software (GE Healthcare). The kinetic binding parameters (koff) were determined
from sensorgrams obtained at an analyte concentration corresponding to the equilibrium binding constant (KD). The SPR running buffer was the following: 1 mM HEPES, 150 mM NaCl, 0.05 % TWEEN,
1 mM GSH, 1% DMSO. JNK1 was expressed biotinylated on an N-terminal AviTag in bacteria, purified, and immobilized on a Biacore CAP sensor chip. For more accurate kinetic analysis of
covalent JNK inhibitors, the determination of k1, k2 (noncovalent kon and koff, respectively) and k3, k4 (covalent kon and koff, respectively) were done the following way. Inhibitors were
injected over the JNK1(Cys116Ser) mutant surface at three different concentrations (100 nM, 300 nM, and 1000 nM) and this kinetic data was globally fit with to 1:1 binding model using the
BiaEvaluation software (GE Healthcare), which gave the k1 and k2 value for each inhibitor. Next, the inhibitors were injected at three different concentrations (depending on their KD
determined earlier by independent equilibrium measurements) over the CAP sensor chip loaded with wild-type JNK1 and k3 and k4 values were determined by fitting the kinetic binding curves
using the COPASI biochemical modeling software based on a 2-step reversible binding scheme, where k1 and k2 values were fixed54. To assess the precision of this quantitative analysis, we
carried out the following parallel measurements and determined/calculated some of the parameters listed in Supplementary Table 1. 1) k1 and k2 were determined (100, 300, 1000 nM) for
JNK-IN-8 on the JNK C116S surface (the values for this were 1492000 M−1s−1 and 0.1676 s−1 compared to the results of the first measurement 1482000 M−1s−1 and 0.15360 s−1, respectively, 2) k3
for irreversible JNK-IN-8 was determined based on a second experiment (1, 3, 15 nM) on the JNK WT surface and it was calculated to be 0.03181 s−1 (where the original value was 0.02952 s−1)
(moreover, using the newly determined k1, k2 a new k3 based on the new experiment was calculated: 0.03454 s−1), 3) k3 and k4 for the reversible covalent CA-IN-8 inhibitor were determined on
the JNK WT surface based on two new experiments (a: 15, 40, 70 nM; b: 30, 60 nM) giving the following new values: a, k3 = 0.01137 s−1 and k4 = 0.00033 s−1; b, k3 = 0.01230 s−1 and k4 =
0.00035 s−1 (and the original values were 0.01140 s−1 and 0.00026 s−1, respectively), 4) the k3 and k4 for 1A_R_-IN-8 were calculated based on a new experiment (2, 5, 10 nM): k3 = 0.02453
s−1 and k4 = 0.00035 s−1 (and the values for the first experiment were 0.02617 s−1 and 0.00034 s−1, respectively), and finally 5) all four values for 6_S,S_-IN-8 were determined based on new
experiments (100, 300, 1000 nM on the JNK C116S and 3, 6 nM on the JNK WT surface): k1 = 129000 M−1s−1, k2 = 0.05387 s−1, k3 = 0.00630 s−1, k4 = 0.00004 s−1 (and the original values were k1
= 164000 M−1s−1, k2 = 0.03460, k3 = 0.01064, k4 = 0.00006). We note that k1 and k2 values, which were determined based on the SPR experiments on the JNK C116S chip surface, are technically
difficult to measure precisely since both kinetic parameters for the noncovalent mechanism are very fast (and thus give rapidly ascending or descending sensorgrams). Despite this, based on
these measurements, the precision of the k1-4 values listed in Supplementary Table 1 is good ( ~ 50 %) in the light of the technical challenges of the measurements and the complexity of the
calculation. CELL CULTURE METHODS 50,000 HEK293T (ATCC, CRL-3216) cells were seeded into 24-well plates and were grown till confluence (typically 24 h) in 500 µl DMEM supplemented with 10 %
FBS and were serum-starved (0% FBS) for 16 h before sorbitol (0.35 M) treatment for 15 min. The plate was put on ice and cells were resuspended, pelleted by centrifugation, and washed with
ice cold PBS. The cell pellet was lysed in 80 µl 1X SDS-PAGE loading buffer, 10 µl was loaded onto 10% Tris-glycine SDS-PAGE gels and gels were blotted to nitrocellulose membrane. Inhibitors
(1 µl dissolved in 50 % DMSO) were added 2 h before sorbitol treatment. SH-SY5Y MKK7 ACT cells (created from SH-SY5Y, ATCC CRL-2266; see later), allowing specific activation of JNK upon
addition of an inducer, were handled similarly but JNK activation was induced by the addition of 3 µg/ml doxycycline. Inhibitors (1 µl dissolved in 50% DMSO) were added together with
doxycycline and samples were harvested for western blot analysis as described above. Western blot results were analyzed using Odyssey CLx imaging system (Li-Cor) and fluorescently labeled
secondary antibodies: IRDye 680RD (Goat anti-mouse IgG; Li-cor #926-68070; 1:0000) or IRDye 800CW (Goat anti-rabbit IgG, Li-Cor #926-32211; 1:5000). The primary antibodies were the
following: anti-phospho JNK (Cell Signaling #9251; 1:1000), anti-phospho-c-Jun(Ser73) (Cell Signaling #9164; 1:1000), anti-phospho p38 (Cell Signaling #9215, 1:3000), anti-phospho MK2 (Cell
Signaling #3007, 1:1000), anti-α-tubulin (Sigma #T6199; 1:10000), or anti-FLAG (Sigma #F1804; 1:10000). Anti-α-tubulin antibody was used as the load control and the anti-FLAG antibody was
used to monitor the expression of the transgene responsible for specific MAPK activation. The HEK293T MKK6EE cell line (allowing p38-specific activation by doxycycline inducible expression
of the constitutively active upstream activator kinase, MK6EE) was described earlier40. The SH-SY5Y (neuroblastoma; ATCC CRL-2266) MKK7 ACT cell line was generated using lentiviral
transduction with an MKK7(ACT)-MLK3 construct containing full-length human MKK7 fused to the kinase domain of human MLK3 (aa. 117-379). The generated Tet-ON inducible stable cell line was
evaluated by time dependent transgene induction and downstream target phosphorylation upon doxycycline treatment in DMEM F12 (Gibco) media supplemented with 1% FBS, as described
earlier31,40. For the determination of JNK inhibitor mediated c-Jun phosphorylation, cells were stimulated with 2 or 3 μg/mL doxycycline (DOX), co-administered with different amounts of
inhibitors, and samples were subjected to quantitative western blot (WB) analysis after 6 h. To obtain the EC50 value, the normalized phospho-c-Jun WB signal was fitted with the DoseResponse
function of Origin 2018. For the p-c-Jun HTRF measurements ~40,000 SH-SY5Y MKK7 ACT cells were seeded into a 96-well plate in DMEM F12 + 1% FBS and the HTRF Phospho-c-Jun(Ser63) Detection
Kit (#64JUNPEG, PerkinElmer) was used to measure the level of phosphorylated c-Jun after 6 h of DOX treatment. To obtain the IC50 value, the HTRF ratio (emission at 665 / emission at 620 nm)
was fitted with the DoseResponse function of Origin 2018. The HTRF signal was measured using a BioTek Cytation 3 multi-mode reader equipped with an HTRF measuring cube (Europium cyptate
donor / Red acceptor readout setup; Agilent Technologies). The PrestoBlue Cell Viability Reagent (ThermoFisher, P50200) was used to assess cell viability. 3,000 SH-SY5Y MKK7 ACT cells were
plated in 96-well plates in DMEM with 10% FBS. Next day the medium was changed for new media (DMEM with 0.1% of FBS) and cells were incubated with inhibitors for 72 h. The medium was then
removed and PrestoBlue reagent was added to live cells, incubated for 45 min, and then the fluorescence signal (ex: 560 nm, em: 600 nm) was measured in a plate reader. Linearity of the
detection was checked in a parallel experiment where control cells were serially diluted, and the signal was measured according to the manufacturer’s protocol. 20,000 Reporter AP-1 – HEK293
cells (BPS Bioscience, #60405) were seeded into a 96-well plate in 100 µl DMEM (Gibco) supplemented with 10% FBS. After cells become adherent (confluency: 80%) the inhibitors were added in
Opti-MEM (Gibco) supplemented with 1% FBS and following 2 h of incubation cells were stimulated with 6 ng/ml PMA solution for 6 h. The luminescence signal was read out using a BioTek
Cytation 3 microplate reader after adding freshly prepared Steadylite Plus solution (PerkinElmer) according to the manufacturer’s protocol. For the wash-out experiments cells were similarly
handled but washed three times with Opti-MEM + 1% FBS (100 μl) before adding 100 μl fresh media with 6 ng/ml PMA solution without any inhibitor for 6 h (Wash-out). Control cells were treated
the same way but the fresh media after the wash-out contained the original inhibitors and PMA. For the experiments with the photocaged 1A_R_-IN-8 compound, Reporter AP-1 – HEK293 cells were
seeded into a microplate as described above. Cells were pre-treated with the inhibitor in the dark (DARK plate) two hours before stimulation with anisomycin. The LIGHT plate was treated
differently: 30 min after inhibitor addition, the plate was subjected to four cycles of illumination (15 s) and recovery (30 s) with blue light (λ = 450 nm) using a custom-made led
illumination device allowing water cooling of the plate (output power: 210 mW)55, and then cells were incubated in the dark for 1.5 h. Cells from the DARK and LIGHT plates were then
stimulated with 10 μg/mL anisomycin for 30 min to turn JNK activity on, lysed with SDS loading buffer, and prepared for western blot analysis. Uncaging of Photocaged 1A_R_-IN-8 by
illumination was confirmed in vitro (see Supplementary Fig. 2). NANOBRET ASSAY The NanoBRET target engagement assay was carried out by Reaction Biology Corp (Malvern, USA; 4000 cells/well,
384-well format) or performed by us (40,000 cells/well, 96-well format) according to the protocol of the NanoBRET® TE Intracellular Kinase Assays (#N2500, Promega) using the NanoLuc-MAPK8
fusion vector with the K5 tracer (see Supplementary Fig. 1). Briefly, HEK293 cells were transfected with JNK1 and transfection carrier DNA (1:9). The transfected cells were treated with
compounds (8–10-dose with 3-fold dilution starting at 1, 10 or 100 μM). JNK1 target engagement was measured by NanoBRET assay. The K-5 tracer concentration was 0.5 μM. Compound treatment
time was 2 h. To obtain the EC50 value, the normalized BRET response (normalized BRET ratio to untreated cells) was fitted with the DoseResponse function of Origin 2018. In the wash-out
experiments the NanoLuc-MAPK8 fusion vector (NV1701, Promega) was transfected into HEK293T cells using Lipofectamine 3000 (Invitrogen), otherwise the nanoBRET measurement was carried out
according to the protocol of the supplier (NanoBRET Target Engagement Intracellular Kinase Assay, adherent format) using the K-10 tracer (0.4 μM). 40,000 transfected cells were seeded into a
96-well plate and after 24 h cells were preincubated with inhibitors for 2 h, washed 3 times with 100 μl Opti-MEM + 1% FBS before adding fresh media to cells (see Supplementary Fig. 10).
The BRET ratio (emission 650 nM/emission 450 nM) was measured using a BioTeK Cytation 3 multi-mode reader equipped with a nanoBRET cube (#8040592, Agilent Technologies). KINASE SPECIFICITY
PROFILING The human kinase specificity profile of 1A_R_-IN-8 was tested by Reaction Biology Europe GmbH (Freiburg, Germany) using the Wild Type Kinase Panel comprised of 340 active human
kinases. Briefly, profiling was done using the 33PanQinaseTM assay which measures the inhibition of phosphorylation of kinase substrate peptides in a radioactive assay format40. The data is
given as per cent remaining activity compared to the control experiment without the inhibitor. The inhibitor was used in 1 μM concentration and ATP was used at a concentration matching its
apparent KM for each kinase. Test compound, kinase, ATP, and a natural substrate peptide for each kinase were incubated in FlashPlate microtiter plates (PerkinElmer) coated with a
scintillant. Reactions were stopped after one hour. Assays were carried out in duplicates. The off-target kinase panel was tested by Eurofins DiscoverX® Products LLC (Fremont, US) using the
ScanELECT service on the indicated kinases at 1 μM compound concentration. The assay was carried out in duplicates according to the KINOMEscan procedure as described earlier56. MASS
SPECTROMETRY The molecular weights of the conjugates of GSH were identified using a Triple TOF 5600+ hybrid Quadrupole-TOF LC/MS/MS system (Sciex, USA) equipped with a DuoSpray IonSource
coupled with a Shimadzu Prominence LC20 UFLC (Shimadzu, Japan) system consisting of quaternary pump, an autosampler and a thermostated column compartment. Data acquisition and processing
were performed using Analyst TF software version 1.7.1 (Sciex). Chromatographic separation was achieved on a Phenomenex Luna Omega PS C18 (50 mm × 2,1 mm, 3 µm, 100 Å) HPLC column. Sample
was eluted in gradient elution mode using solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in ACN). The initial condition was 10% B followed by a linear gradient to 55 %
B by 12 min, to 95 % B by 3 min, 15 to 17.5 min 95% B was retained; and from 17.5 to 18 min back to initial condition with 10% eluent B and retained from 18 to 20 min. Flow rate was set to
0.4 ml/min. The column temperature was 40 °C and the injection volume was 5 µl. UV-VIS spectrometer was used at 254 nm wavelength. Nitrogen was used as the nebulizer gas (GS1), heater gas
(GS2), and curtain gas with the optimum values set at 30, 30 and 35 (arbitrary units), respectively. Data were acquired in positive electrospray mode in the mass range of m/z = 300 to 2500,
with 1 s accumulation time. The source temperature was 350 °C and the spray voltage was set to 5500 V. Declustering potential value was set to 80 V. Peak View SoftwareTM V.2.2 (Sciex) was
used for deconvoluting the raw electrospray data to obtain the neutral molecular masses. The procedure described above allowed the identification of the irreversible GSH–JNK-IN-8 adduct
only. For the identification of the reversible GSH–1A_R_-IN-8 adducts the LC step had to be shortened and the details of these differences are described below. For this adduct there was only
a short separation carried out and samples were only retained on a short security guard cartridge (Phenomenex C18, 4 × 3 mm, Phenomenex, USA). The goal of this solution was focusing and
desalting the protein samples prior to ionization. A 4 min gradient (both in solvent composition and flow rate) was used with initial flow of 0.5 mL/min and 10% eluent B. This was held for
0.5 min for washing out the salts from the sample. A 1.5 min linear increase was applied to reach the final flow of 1 mL/min and maximum eluent composition of B at 65%. These parameters were
held for 0.5 min and a quick 0.1 min linear gradient was used to reach the initial flow rate and eluent composition. This was followed by a 1.4 min equilibrating part. Water containing 0.1%
formic acid (eluent A) and acetonitrile containing 0.1% formic acid) were used for chromatography. The injected volumes were 5 µL. The wavelength in UV detection was 280 nm. The MS step was
carried out similarly as described above. NMR MEASUREMENTS ON COVALENT ADDUCT FORMATION NMR experiments were conducted on a Varian NMR System spectrometers operating at 600 MHz. Off-target
adduct formation was analyzed through the reaction between 1A_R_-IN-8 and beta-mercaptoethanol (BME) by 1H NMR. All reagents were dissolved and the experiments were carried out in PBS (D2O,
pH ~ 7.2) with 75% DMSO. Details of the measurements, the assignment of the spectra and Kchem determination are described in Supplementary Note 2. PHARMACOKINETIC ANALYSIS In vitro
pharmacokinetic behaviour of the JNK inhibitors was determined in rat (Wistar) plasma and primary hepatocytes. Blood plasma and hepatocytes isolated from male Wistar rats were obtained from
Toxi-Coop Toxicological Research Centre Ltd. (Budapest, Hungary). Primary hepatocytes were isolated by using collagenase perfusion method of Bayliss and Skett57. Briefly, the liver tissues
were perfused through the portal vein with Ca2+-free medium (Earle’s balanced salt solution) containing EGTA (0.5 mM) and then with the same medium without EGTA, finally with the perfusate
containing collagenase (Type IV, 0.25 mg/ml) and Ca2+ at physiological concentration (2 mM). The yield and percent of cell viability according to the trypan blue exclusion test were
determined58. Time courses of the unchanged JNK inhibitors (JNK-IN-8, 1A_R_-IN-8 and PK_test) in primary hepatocytes were obtained. Each compound was incubated with cell suspension (2 × 106
cells/ml) at 37 °C in a humid atmosphere containing 5 % CO2. The parent compounds dissolved in dimethylsulphoxide (DMSO) were added directly to the cell culture medium (William’s E medium:
Ham’s F12 medium= 1:1) at the final concentration of 1 or 10 μM. The final concentration of DMSO was 0.1%. At various time points (at 0, 10, 20, 30, 60, 90, 120, 180, 240 min), the
incubation mixtures were sampled (aliquots: 0.1 ml) and terminated by the addition of ice-cold acetonitrile (0.3 ml). JNK inhibitors were also incubated in cell-free medium and sampled at 0
and 240 min. The samples were centrifuged and analyzed by LC-MS/MS for quantitation of the parent compound (Supplementary Note 3). Intrinsic clearance (_Cl_int), the in vitro pharmacokinetic
parameter was calculated from the decrease in the concentration of the parent compound, whereas the in vivo parameters (hepatic clearance _Cl_H, hepatic extraction ratio _E_,
bioavailability _F_) were predicted from _Cl_int according to the previously published method58. The cut-off value of hepatic extraction ratio between low and intermediate was considered to
be 0.3, whereas 0.7 between intermediate and high extraction compounds. Time courses of the JNK inhibitors (JNK-IN-8, 1A_R_-IN-8, 2-IN-8, 5_S_-IN-8, 1C_R_-IN-8, _6S,S_-IN-8 and PK_test) in
rat plasma were also obtained. The parent compounds dissolved in DMSO were added directly to the blood plasma at the final concentration of 1 or 10 μM. At various time points (at 0, 10, 20,
30, 60, 120 min), the incubation mixtures were sampled (aliquots: 0.05 ml) and terminated by the addition of 0.15 ml ice-cold acetonitrile. The samples were centrifuged and analyzed by
LC-MS/MS for quantitation of the parent compound, and elimination half-life values (_t__1/2_) were calculated from the decrease in the concentration of the parent compound. CHARACTERIZATION
AND SYNTHESIS OF SMALL MOLECULES All reactions were carried out using oven-dried glassware and anhydrous solvents unless noted otherwise. Flash silica chromatography was performed on silica
gel (ZEOprep 60 25-40 µm, ZEOCHEM) with the indicated eluents. Reverse phase chromatography was performed using a gradient method on a Gemini® 5 µm C18 110 Å column (H2O:MeCN = 95:5 (0.1%
HCOOH) to 100% MeCN (0.1% HCOOH)). Thin-layer chromatography was performed on silica plates (Kieselgel 60 F254 Merck). Compounds on TLC were visualized by UV (254 nm) or KMnO4 or
p-anisaldehyde staining. Chemical shifts are referenced to the residual solvent signals (CHCl3: δ = 7.26 ppm for 1H, δ = 77.0 ppm for 13C; DMSO: δ = 2.50 ppm for 1H, δ = 39.5 ppm for 13C;
CH3OH: δ = 4.87 ppm and 3.31 ppm for 1H, δ = 49.0 ppm for 13C). Data are reported as follows: chemical shifts (ppm), multiplicity (s = singlet, d=doublet, t=triplet, q=quartet,
sept.=septet, br=broad, m=multiplet, dd=doublet of doublets, ddd=doublet of doublet of doublets, dddd=doublet of doublet of doublet of doublets, dt=doublet of triplets, ddt=doublet of
doublet of triplets, td=triplet of doublets), coupling constants (Hz), and integration. All spectra were recorded with the standard spectrometer pulse sequences and settings using a Varian
300, 500 and 600 MHz spectrometer. The enantiomeric excess of the products was determined on a chiral stationary phase HPLC (Daicel Chiralpak ID 250 × 4.6 mm, 5 µm column) using
Hexane:iPrOH=9:1 as eluent. All starting materials were purchased from Aldrich, TCI, or Fluorochem and used without further purification unless stated otherwise. Anhydrous THF and toluene
were distilled from sodium/benzophenone, while anhydrous DMF and DMSO were kept under Ar on 4 Å molecular sieves. JNK-IN-8 was purchased from Selleck Chemicals. The synthetic procedures for
compounds and intermediates thereof used in this study (e.g., IN-8, IN-8a, CA-IN-8, 1A_R_-IN-8, 1A_S_-IN-8, Photocaged 1A_R_-IN-8, 1A’_R_-IN-8, 1A”_R_-IN-8, 2-IN-8, 3-IN-8, 4-IN-8,
5_S_-IN-8, 5_R_-IN-8, 6_S,S_-IN-8, 6 _R,R_-IN-8, 1A_R_-IN-7, 1A_R_-IN-9, 1A_R_-IN-10, 1A_R_-isoPHEN, 1B_R_-IN-8, 1B_S_-IN-8, 1C_R_-IN-8, 1C_S_-IN-8, 1D_R_-IN-8) are described in
Supplementary Note 4. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY Crystal
structures of JNK1–1A_R_-IN-8, –1A_S_-IN-8, and –1A’_R_-IN-8 are deposited in the Protein Data Bank with accession codes 8PTA, 8PT9, and 8PT8. The following X-ray structures are available
from the PDB: 3V6S, 2ERK and 3S95. Source data are provided with this paper. REFERENCES * Boike, L., Henning, N. J. & Nomura, D. K. Advances in covalent drug discovery. _Nat. Rev. Drug
Discov._ 21, 881–898 (2022). Article CAS PubMed PubMed Central Google Scholar * Roskoski, R. Orally effective FDA-approved protein kinase targeted covalent inhibitors (TCIs).
_Pharmacol. Res._ 165, 105422 (2021). Article CAS PubMed Google Scholar * Krishnan, S. et al. Design of reversible, cysteine-targeted michael acceptors guided by kinetic and
computational analysis. _J. Am. Chem. Soc._ 136, 12624–12630 (2014). Article CAS PubMed PubMed Central Google Scholar * Gehringer, M. & Laufer, S. A. Emerging and Re-Emerging
Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. _J. Med. Chem._ 62, 5673–5724 (2019). Article CAS PubMed Google Scholar * Resnick, E.
et al. Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. _J. Am. Chem. Soc._ 141, 8951–8968 (2019). Article CAS PubMed PubMed Central Google Scholar * Liu, Q. et al.
Developing irreversible inhibitors of the protein kinase cysteinome. _Chem. Biol._ 20, 146–159 (2013). Article ADS PubMed PubMed Central Google Scholar * Miller, R. M. & Taunton, J.
Targeting protein kinases with selective and semipromiscuous covalent inhibitors. _Methods Enzymol._ 548, 93–116 (2014). Article CAS PubMed PubMed Central Google Scholar * Zhao, Z.,
Liu, Q., Bliven, S., Xie, L. & Bourne, P. E. Determining Cysteines Available for Covalent Inhibition Across the Human Kinome. _J. Med. Chem._ 60, 2879–2889 (2017). Article CAS PubMed
PubMed Central Google Scholar * Chaikuad, A., Koch, P., Laufer, S. A. & Knapp, S. The Cysteinome of Protein Kinases as a Target in Drug Development. _Angew. Chem. - Int. Ed._ 57,
4372–4385 (2018). Article CAS Google Scholar * Jackson, P. A., Widen, J. C., Harki, D. A. & Brummond, K. M. Covalent Modifiers: A Chemical Perspective on the Reactivity of
α,β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions. _J. Med. Chem._ 60, 839–885 (2017). Article CAS PubMed Google Scholar * Klaeger, S. et al. The target
landscape of clinical kinase drugs. _Science_ 358, eaan4368 (2017). Article PubMed PubMed Central Google Scholar * Lanning, B. R. et al. A road map to evaluate the proteome-wide
selectivity of covalent kinase inhibitors. _Nat. Chem. Biol._ 10, 760–767 (2014). Article CAS PubMed PubMed Central Google Scholar * Serafimova, I. M. et al. Reversible targeting of
noncatalytic cysteines with chemically tuned electrophiles. _Nat. Chem. Biol._ 8, 471–476 (2012). Article CAS PubMed PubMed Central Google Scholar * Bradshaw, J. M. et al. Prolonged and
tunable residence time using reversible covalent kinase inhibitors. _Nat. Chem. Biol._ 11, 525–531 (2015). Article CAS PubMed PubMed Central Google Scholar * Spradlin, J. N., Zhang, E.
& Nomura, D. K. Reimagining Druggability Using Chemoproteomic Platforms. _Acc. Chem. Res._ 54, 1801–1813 (2021). Article CAS PubMed Google Scholar * Ullah, R., Yin, Q., Snell, A. H.
& Wan, L. RAF-MEK-ERK pathway in cancer evolution and treatment. _Semin. Cancer Biol._ 85, 123–154 (2022). Article CAS PubMed Google Scholar * Fu, L., Chen, S., He, G., Chen, Y.
& Liu, B. Targeting Extracellular Signal-Regulated Protein Kinase 1/2 (ERK1/2) in Cancer: An Update on Pharmacological Small-Molecule Inhibitors. _J. Med. Chem._ 65, 13561–13573 (2022).
Article CAS PubMed Google Scholar * Lavoie, H., Gagnon, J. & Therrien, M. ERK signalling: a master regulator of cell behaviour, life and fate. _Nat. Rev. Mol. Cell Biol._ 21, 607–632
(2020). Article CAS PubMed Google Scholar * Davis, R. J. Signal Transduction by the JNK Group of MAP Kinases. _Cell_ 103, 239–252 (2000). Article CAS PubMed Google Scholar *
Johnson, G. L. & Nakamura, K. The c-Jun Kinase/Stress-activated Pathway: Regulation, Function and Role in Human Disease. _Biochim. Biophys. Acta_ 1773, 1341 (2007). Article CAS PubMed
PubMed Central Google Scholar * Canovas, B. & Nebreda, A. R. Diversity and versatility of p38 kinase signalling in health and disease. _Nat. Rev. Mol. Cell Biol._ 22, 346–366 (2021).
Article CAS PubMed PubMed Central Google Scholar * Wagner, E. F. & Nebreda, Á. R. Signal integration by JNK and p38 MAPK pathways in cancer development. _Nat. Rev. Cancer_ 9,
537–549 (2009). Article CAS PubMed Google Scholar * Zeke, A., Misheva, M., Reményi, A. & Bogoyevitch, M. A. JNK signaling: Regulation and functions based on complex protein-protein
partnerships. _Microbiol. Mol. Biol. Rev._ 80, 793–835 (2016). Article CAS PubMed PubMed Central Google Scholar * Schellino, R., Boido, M. & Vercelli, A. JNK Signaling Pathway
Involvement in Spinal Cord Neuron Development and Death. _Cells_ 8, 1576 (2019). Article CAS PubMed PubMed Central Google Scholar * Duong, M. T. H., Lee, J. H. & Ahn, H. C. C-Jun
N-terminal kinase inhibitors: Structural insight into kinase-inhibitor complexes. _Comput. Struct. Biotechnol. J._ 18, 1440–1457 (2020). Article CAS PubMed PubMed Central Google Scholar
* Bogoyevitch, M. A. & Arthur, P. G. Inhibitors of c-Jun N-terminal kinases: JuNK no more? _Biochim. Biophys. Acta_ 1784, 76–93 (2008). Article CAS PubMed Google Scholar * Zhang,
T. et al. Discovery of potent and selective covalent inhibitors of JNK. _Chem. Biol._ 19, 140–154 (2012). Article CAS PubMed PubMed Central Google Scholar * Meister, A. Glutathione
metabolism and its selective modification. _J. Biol. Chem._ 263, 17205–17208 (1988). Article CAS PubMed Google Scholar * Póti, A. L. et al. Targeting a key protein-protein interaction
surface on mitogen-activated protein kinases by a precision-guided warhead scaffold. _Nat. Commun_. https://doi.org/10.1038/s41467-024-52574-1 (2024). * Bogoyevitch, M. A., Ketterman, A. J.
& Sugden, P. H. Cellular stresses differentially activate c-Jun N-terminal protein kinases and extracellular signal-regulated protein kinases in cultured ventricular myocytes. _J. Biol.
Chem._ 270, 29710–29717 (1995). Article CAS PubMed Google Scholar * Kirsch, K. et al. Co-regulation of the transcription controlling ATF2 phosphoswitch by JNK and p38. _Nat. Commun._ 11,
5769 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Tang, X. J. et al. Photorelease of Pyridines Using a Metal-Free Photoremovable Protecting Group. _Angew. Chem. Int.
Ed. Engl._ 59, 18386–18389 (2020). Article CAS PubMed Google Scholar * Deslongchamps, P. Reactions on SP2 type unsaturated systems. in _Stereoelectronic Effects in Organic Chemistry_
221–241 (Pergamon Press Ltd, 1983). * Mons, E., Roet, S., Kim, R. Q. & Mulder, M. P. C. A Comprehensive Guide for Assessing Covalent Inhibition in Enzymatic Assays Illustrated with
Kinetic Simulations. _Curr. Protoc._ 2, e419 (2022). Article CAS PubMed Google Scholar * Póti, Á. L. et al. Phosphorylation-Assisted Luciferase Complementation Assay Designed to Monitor
Kinase Activity and Kinase-Domain-Mediated Protein-Protein Binding. _Int. J. Mol. Sci._ 24, 14854 (2023). Article PubMed PubMed Central Google Scholar * Anastassiadis, T., Deacon, S. W.,
Devarajan, K., Ma, H. & Peterson, J. R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. _Nat. Biotechnol._ 29, 1039–1045 (2011).
Article CAS PubMed PubMed Central Google Scholar * Stepan, A. F. et al. Application of the bicyclo[1.1.1]pentane motif as a nonclassical phenyl ring bioisostere in the design of a
potent and orally active γ-secretase inhibitor. _J. Med. Chem._ 55, 3414–3424 (2012). Article CAS PubMed Google Scholar * Le-Niculescu, H. et al. Withdrawal of survival factors results
in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. _Mol. Cell. Biol._ 19, 751–763 (1999). Article CAS PubMed PubMed Central Google Scholar
* Whitmarsh, A. J. & Davis, R. J. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. _J. Mol. Med. (Berl.)._ 74, 589–607 (1996).
Article CAS PubMed Google Scholar * Sok, P. et al. MAP Kinase-Mediated Activation of RSK1 and MK2 Substrate Kinases. _Structure_ 28, 1101–1113.e5 (2020). Article CAS PubMed PubMed
Central Google Scholar * Wang, Y. & Ma, H. Protein kinase profiling assays: a technology review. _Drug Discov. Today Technol._ 18, 1–8 (2015). Article PubMed Google Scholar *
Falivene, L. et al. Towards the online computer-aided design of catalytic pockets. _Nat. Chem._ 11, 872–879 (2019). Article CAS PubMed Google Scholar * Bogoyevitch, M. A. The
isoform-specific functions of the c-Jun N-terminal Kinases (JNKs): differences revealed by gene targeting. _Bioessays_ 28, 923–934 (2006). Article CAS PubMed Google Scholar * Tóth, K. et
al. Utility of in vitro clearance in primary hepatocyte model for prediction of in vivo hepatic clearance of psychopharmacons. _Microchem. J._ 136, 193–199 (2018). Article Google Scholar
* Copeland, R. A. The dynamics of drug-target interactions: drug-target residence time and its impact on efficacy and safety. _Expert Opin. Drug Discov._ 5, 305–310 (2010). Article CAS
PubMed Google Scholar * Knockenhauer, K. E. & Copeland, R. A. The importance of binding kinetics and drug-target residence time in pharmacology. _Br. J. Pharmacol_.
https://doi.org/10.1111/BPH.16104 (2023) * Gabizon, R. et al. Efficient Targeted Degradation via Reversible and Irreversible Covalent PROTACs. _J. Am. Chem. Soc._ 142, 11734–11742 (2020).
Article CAS PubMed PubMed Central Google Scholar * Bemis, T. A., La Clair, J. J. & Burkart, M. D. Unraveling the Role of Linker Design in Proteolysis Targeting Chimeras. _J. Med.
Chem._ 64, 8042–8052 (2021). Article CAS PubMed PubMed Central Google Scholar * Garai, A. et al. Specificity of linear motifs that bind to a common mitogen-activated protein kinase
docking groove. _Sci. Signal._ 5, ra74 (2012). Article PubMed PubMed Central Google Scholar * Lebedev, A. A. et al. JLigand: a graphical tool for the CCP4 template-restraint library.
_Acta Crystallogr. D. Biol. Crystallogr._ 68, 431–440 (2012). Article ADS CAS PubMed PubMed Central Google Scholar * Adams, P. D. et al. The Phenix software for automated determination
of macromolecular structures. _Methods_ 55, 94–106 (2011). Article CAS PubMed PubMed Central Google Scholar * Dixon, A. S. et al. NanoLuc Complementation Reporter Optimized for
Accurate Measurement of Protein Interactions in Cells. _ACS Chem. Biol._ 11, 400–408 (2016). Article ADS CAS PubMed Google Scholar * Zeke, A. et al. Systematic discovery of linear
binding motifs targeting an ancient protein interaction surface on MAP kinases. _Mol. Syst. Biol._ 11, 837 (2015). Article PubMed PubMed Central Google Scholar * Hoops, S. et al. COPASI
- A COmplex PAthway SImulator. _Bioinformatics_ 22, 3067–3074 (2006). Article CAS PubMed Google Scholar * Egyed, A. et al. Turning Red without Feeling Embarrassed-Xanthenium-Based
Photocages for Red-Light-Activated Phototherapeutics. _J. Am. Chem. Soc._ 145, 4026–4034 (2023). Article CAS PubMed PubMed Central Google Scholar * Fabian, M. A. et al. A small
molecule-kinase interaction map for clinical kinase inhibitors. _Nat. Biotechnol._ 23, 329–336 (2005). Article CAS PubMed Google Scholar * Bayliss, K. M. & Skett, P. Isolation and
culture of human hepatocytes. in _Human Cell Culture Protocols_ (ed. Jones, G. E.) 369–390 (Humana Press, 1996). * Berry, M. N. et al. Initial determination of cell quality. in _Isolated
Hepatocytes_ 45–47 (Elsevier, 1991). Download references ACKNOWLEDGEMENTS This work was supported by the National Research Development and Innovation Office (NKFIH) grants (KKP 126963
awarded to A.R., NKFIH-PD-135121 awarded to M.B., NKFIH-PD-146252 awarded to D.C., and NKFIH-K-142486 awarded to I.P.), VEKOP-2.3.3-15-2016-00011, and by the Hungarian Academy of Sciences
(KEP-10/2019), and by the National Research, Development and Innovation Fund (2020-1.1.2-PIACI-KFI-2020-00021 awarded to K.M.). We acknowledge support at the EMBL beamlines at PETRA III
storage ring, Hamburg. The authors are thankful to Krisztina Németh for HRMS measurements. A.R. is thankful to Marie Bogoyevitch for useful discussions on JNKs in the early phases of the
project. AUTHOR INFORMATION Author notes * These authors contributed equally: Dániel Bálint, Ádám Levente Póti. AUTHORS AND AFFILIATIONS * Organocatalysis Research Group, Institute of
Organic Chemistry, Research Centre for Natural Sciences, 1117, Budapest, Hungary Dániel Bálint, Lili Torda, Roberta Palkó & Tibor Soós * Hevesy György PhD School of Chemistry, Eötvös
Loránd University, 1117, Budapest, Hungary Dániel Bálint * Biomolecular Interaction Research Group, Institute of Organic Chemistry, Research Centre for Natural Sciences, 1117, Budapest,
Hungary Ádám Levente Póti, Anita Alexa, Péter Sok, Krisztián Albert, Dóra Földesi-Nagy, Eszter Szarka & Attila Reményi * Doctoral School of Biology, Eötvös Loránd University, 1117,
Budapest, Hungary Ádám Levente Póti * Theoretical Chemistry Research Group, Institute of Organic Chemistry, Research Centre for Natural Sciences, 1117, Budapest, Hungary Dániel Csókás &
Imre Pápai * NMR Research Laboratory, Centre for Structural Science, Research Centre for Natural Sciences, 1117, Budapest, Hungary Gábor Turczel * MS Metabolomic Research Laboratory, Centre
for Structural Science, Research Centre for Natural Sciences, 1117, Budapest, Hungary Tímea Imre & Pál Szabó * Metabolic Drug-interactions Research Group, Institute of Molecular Life
Sciences, Research Centre for Natural Sciences, 1117, Budapest, Hungary Ferenc Fekete & Katalin Monostory * European Molecular Biology Laboratory, EMBL, Hamburg, Germany Isabel Bento *
Chemical Biology Research Group, Institute of Organic Chemistry, Research Centre for Natural Sciences, 1117, Budapest, Hungary Márton Bojtár Authors * Dániel Bálint View author publications
You can also search for this author inPubMed Google Scholar * Ádám Levente Póti View author publications You can also search for this author inPubMed Google Scholar * Anita Alexa View author
publications You can also search for this author inPubMed Google Scholar * Péter Sok View author publications You can also search for this author inPubMed Google Scholar * Krisztián Albert
View author publications You can also search for this author inPubMed Google Scholar * Lili Torda View author publications You can also search for this author inPubMed Google Scholar * Dóra
Földesi-Nagy View author publications You can also search for this author inPubMed Google Scholar * Dániel Csókás View author publications You can also search for this author inPubMed Google
Scholar * Gábor Turczel View author publications You can also search for this author inPubMed Google Scholar * Tímea Imre View author publications You can also search for this author
inPubMed Google Scholar * Eszter Szarka View author publications You can also search for this author inPubMed Google Scholar * Ferenc Fekete View author publications You can also search for
this author inPubMed Google Scholar * Isabel Bento View author publications You can also search for this author inPubMed Google Scholar * Márton Bojtár View author publications You can also
search for this author inPubMed Google Scholar * Roberta Palkó View author publications You can also search for this author inPubMed Google Scholar * Pál Szabó View author publications You
can also search for this author inPubMed Google Scholar * Katalin Monostory View author publications You can also search for this author inPubMed Google Scholar * Imre Pápai View author
publications You can also search for this author inPubMed Google Scholar * Tibor Soós View author publications You can also search for this author inPubMed Google Scholar * Attila Reményi
View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Á.L.P., D.B., T.S. and A.R. conceived of the project idea, made intellectual contributions,
designed experiments, analyzed and interpreted the data and wrote the manuscript. D.B., L.T. and R.P. synthesized compounds, Á.L.P. characterized the compounds by biochemical measurements,
A.A. and E.SZ carried out cell-based assays and K.A. did SPR measurements. P.S. determined the crystal structures and I.B. collected X-ray data. D.C. and I.P. carried out the theoretical
buried volume calculations. M.B. synthesized the photocaged 1A_R_-IN-8 compound and D.F-N. characterized it in cells. F.F. and K.M. carried out in vitro pharmacokinetic studies in rat
hepatocytes and plasma. G.T. carried out the NMR measurements required for the analysis of the off-target BME adduct. T.I. and P.SZ. carried out MS measurements. CORRESPONDING AUTHORS
Correspondence to Tibor Soós or Attila Reményi. ETHICS DECLARATIONS COMPETING INTERESTS A.R., T.S., Á.L.P., D.B., A.A., P.S., L.T., K.A., T.I., E.SZ. and R.P. are inventors in a patent
application pending approval (PCT/HU2023/050079) on the use of cyclic designer scaffolds for the covalent targeting of proteins via Michael addition. The remaining authors declare no
competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is
available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission
under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons
licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Bálint, D., Póti, Á.L., Alexa, A. _et al._ Reversible covalent c-Jun
N-terminal kinase inhibitors targeting a specific cysteine by precision-guided Michael-acceptor warheads. _Nat Commun_ 15, 8606 (2024). https://doi.org/10.1038/s41467-024-52573-2 Download
citation * Received: 22 August 2023 * Accepted: 13 September 2024 * Published: 04 October 2024 * DOI: https://doi.org/10.1038/s41467-024-52573-2 SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative