Play all audios:
ABSTRACT Autoantibodies have been shown to be implied in COVID-19 but the emerging autoantibody repertoire remains largely unexplored. We investigated the new-onset autoantibody repertoire
in 525 healthcare workers and hospitalized COVID-19 patients at five time points over a 16-month period in 2020 and 2021 using proteome-wide and targeted protein and peptide arrays. Our
results show that prevalent new-onset autoantibodies against a wide range of antigens emerged following SARS-CoV-2 infection in relation to pre-infectious baseline samples and remained
elevated for at least 12 months. We found an increased prevalence of new-onset autoantibodies after severe COVID-19 and demonstrated associations between distinct new-onset autoantibodies
and neuropsychiatric symptoms post-COVID-19. Using epitope mapping, we determined the main epitopes of selected new-onset autoantibodies, validated them in independent cohorts of neuro-COVID
and pre-pandemic healthy controls, and identified sequence similarities suggestive of molecular mimicry between main epitopes and the conserved fusion peptide of the SARS-CoV-2 Spike
glycoprotein. Our work describes the complexity and dynamics of the autoantibody repertoire emerging with COVID-19 and supports the need for continued analysis of the new-onset autoantibody
repertoire to elucidate the mechanisms of the post-COVID-19 condition. SIMILAR CONTENT BEING VIEWED BY OTHERS DIVERSE FUNCTIONAL AUTOANTIBODIES IN PATIENTS WITH COVID-19 Article 19 May 2021
HIGH-RESOLUTION EPITOPE MAPPING AND CHARACTERIZATION OF SARS-COV-2 ANTIBODIES IN LARGE COHORTS OF SUBJECTS WITH COVID-19 Article Open access 22 November 2021 HIGH-RESOLUTION ANALYSIS OF
LONG-TERM SERUM ANTIBODIES IN HUMANS FOLLOWING CONVALESCENCE OF SARS-COV-2 INFECTION Article Open access 31 May 2022 INTRODUCTION In SARS-CoV-2 infection1 and other pulmonary viral
infections2, preexisting anti-type I interferon autoantibodies have been detected in 5–20% of severe disease cases and may affect therapeutic strategies3,4. Several studies have detected the
presence of established autoantibodies in COVID-19 patients5,6,7,8,9,10,11, although their clinical significance remains unclear. In addition, autoantibodies against a wide range of
extracellular antigens have been detected in COVID-19, and a subset of these have been shown to antagonize cytokine signaling, be associated with increased viral loads and decreased T-cell
and B-cell populations, and to increase disease severity in mouse models of COVID-1912. The total number of these autoantibodies in COVID-19 patients has been associated with disease
severity12. However, autoantibody repertoires are notoriously individual-specific in both health and disease13,14, rendering associations to clinical symptoms and outcomes difficult without
a longitudinal study design. To this end, some studies of hospitalized patients with COVID-19 have investigated the development of autoantibodies against a selection of previously
described15 or extracellular16 antigens. While these studies demonstrated the existence of new-onset autoantibodies in patients with severe COVID-19, they were constrained by the use of
baseline samples collected after hospitalization and short follow-up times. This presented limitations in the evaluation of the persistence of new-onset autoantibodies and their association
with the course of COVID-19. In the months following COVID-19, an estimated 6% of individuals experience lasting symptoms such as cognitive dysfunction, fatigue, and shortness of
breath17,18. These symptoms are collectively known as long COVID, post-acute sequelae of COVID-19, or post-COVID-19 condition and may occur after mild as well as severe acute disease17.
There are many theories on the etiology of the post-COVID-19 condition, including viral persistence, persistent inflammation, and autoimmunity, including the emergence of new-onset
autoantibodies3,19,20,21. In particular, neurological symptoms after SARS-CoV-2 infection, termed neuro-COVID, are suspected to stem from a dysregulated immune response with autoantibody
involvement5,22,23,24,25, similar to other post-infectious neurological disorders25,26. However, a notable cross-sectional study of immune disruption in the post-COVID-19 condition could not
identify any associations with autoantibodies27, indicating the need for a longitudinal study of this immune compartment. During the COVID-19 pandemic, we developed a highly specific and
sensitive multiplex bead array for SARS-CoV-2 serology28 which we have used to profile the serological response in several research projects, where the COMMUNITY (COVID-19 Immunity) study is
a longstanding collaboration29,30,31. This ongoing longitudinal study enrolled 2149 healthcare workers (HCW) and 118 admitted COVID-19 patients at Danderyd Hospital, Sweden, between April
and May 2020, with follow-up visits every four months. In the present study, we extend the analysis within a subgroup of the COMMUNITY study cohort by profiling the dynamics of autoantibody
repertoires across SARS-CoV-2 infection using proteome-wide and targeted in-house developed planar and bead arrays. The results reveal prevalent new-onset autoantibodies against a wide range
of antigens which remain elevated for at least 12 months, are associated with neuropsychiatric symptoms post-COVID-19, and invoke molecular mimicry with the SARS-CoV-2 Spike protein fusion
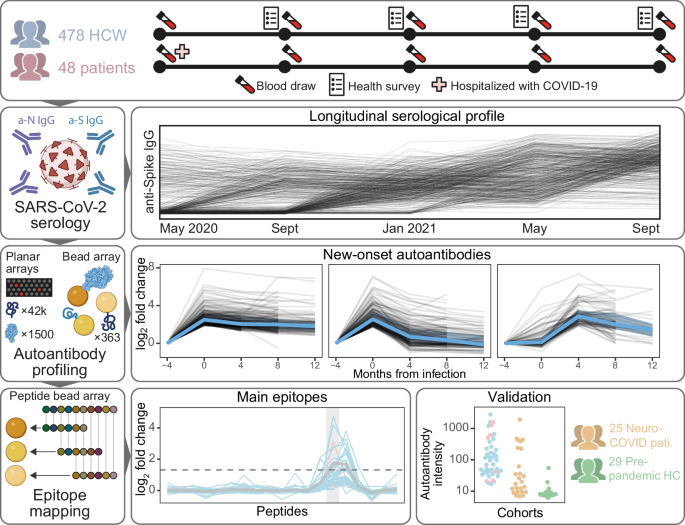
peptide. RESULTS In this study, we have profiled the autoantibody repertoire of 478 HCW and 47 hospitalized COVID-19 patients and validated our results in 25 neuro-COVID patients and 29
pre-pandemic healthy controls (HCs) (Supplementary Table 1). An overview of the study is shown in Fig. 1. In summary, samples in the discovery cohorts were collected across 3–5 visits (mean
4.8) over 16 months, for a total of 2532 samples analyzed in the present study. In HCW, 20% (_n_ = 96) were seropositive (had anti-SARS-CoV-2 immunoglobulin G (IgG)) at study inclusion in
May 2020. Among the remaining 382 baseline seronegative HCW, 109 were seroconverted (first display of anti-SARS-CoV-2 IgG) in Sept 2020, 233 in Jan 2021, and 40 in May 2021. All HCW
seroconverted before receiving their first SARS-CoV-2 vaccine dose. Among the patients, 85% (_n_ = 40) were seropositive at the first sampling after admission (May 2020), and the remaining
15% (_n_ = 7) had seroconverted at the first sampling after discharge (Sept 2020). PROTEOME-WIDE AUTOANTIBODY PROFILING REVEALS DIVERSE AUTOANTIBODIES IN COVID-19 To explore the
proteome-wide autoantibody landscape emerging with COVID-19, we screened blood sample from 32 healthcare workers (HCW) with self-reported symptoms post-COVID-19, and 16 hospitalized COVID-19
patients, on our in-house developed planar array platforms. Plasma samples from the 32 HCW were divided into eight groups of four individuals each, defined by specific symptoms
post-COVID-19 (Supplementary Table 2). Similarly, plasma samples from the 16 patients were divided into four groups of four individuals each, based on sex and comorbidities (Supplementary
Table 2). Within each group, plasma samples were combined and analyzed on the arrays. HCW and patient groups were analyzed on the proteome-wide arrays containing 42,000 protein fragments,
and HCW groups were, in addition, analyzed on the Secretome arrays containing 1522 full-length proteins. In total, IgG binding was detected towards 215 protein fragments and 22 full-length
proteins, with 14 to 36 reactive autoantibodies in each group. Autoantibody profiles were highly specific to each set of combined samples, with 6% (15 of 237) of autoantibodies being
reactive in two groups and 3% (8 of 237) being reactive in three to six groups (Supplementary Fig. 1). The reactive antigens were selected for further investigation in the full HCW and
patient cohorts. In addition, antigens were selected by combining evidence from multiple groups and arrays with prior knowledge from literature and in-house studies. The final panel included
307 protein fragments and 56 full-length proteins. Together, these 363 antigens represented proteins from 315 genes (Supplementary Data 1). PREVALENT AND PERSISTENT NEW-ONSET AUTOANTIBODIES
EMERGE WITH COVID-19 For the initial investigation of new-onset autoantibodies, data from the full HCW and patient cohorts were filtered to include individuals with three consecutive
samplings before, at, and after seroconversion (_n_ = 369: 362 HCW and seven hospitalized patients). Individual autoantibody trajectories were defined by calculating fold change (FC) of
autoantibody levels at the two later time points relative to the seronegative baseline. Clustering of all obtained trajectories revealed three distinct categories of new-onset autoantibodies
shown in Fig. 2a: stable (_n_ = 225), transient (_n_ = 177), and delayed (_n_ = 103) new-onset autoantibodies. Four additional clusters of relatively unchanging trajectories were detected
and not classified as new-onset (Supplementary Fig. 2). The new-onset autoantibody landscape of individuals and antigens with at least one detected new-onset autoantibody (_n_ individuals =
204, _n_ antigens = 187) is displayed in Supplementary Fig. 3. These autoantigens represent extracellular (_n_ = 57) and intracellular (_n_ = 119) proteins corresponding to 176 genes as
classified in the Human Protein Atlas32. We further examined the persistence of new-onset autoantibodies in the 160 trajectories where 12-month follow-up data was available (stable and
transient trajectories in 63 individuals). Persistence, defined as FC ≥2 at 12 months compared to baseline, was observed for the majority of autoantibodies with stable trajectories (95%
(78/82)), while only 23% (18/78) of transient new-onset autoantibodies remained elevated. In total, 60% of new-onset autoantibodies remained elevated 12 months after onset. Further
investigation of the trajectories revealed that new-onset autoantibodies were found in 204 of the 369 individuals and that they targeted a total of 187 antigens. Most individuals displayed
single new-onset autoantibodies (_n_ = 107), but three individuals we found to have 30 to 33 (Fig. 2b). In line with previous reports12,14,33, most autoantibodies (99 of 187 detected) were
rare and occurred in single individuals (Fig. 2c). The 22 most prevalent new-onset autoantibodies, detected in >1% of the cohort (>4 individuals), were subjected to further analysis
(Table 1 and Supplementary Data 2). The corresponding antigens represented both intracellular (_n_ = 16, 73%) and extracellular (_n_ = 6, 27%) proteins. Autoantibodies which have previously
been reported in autoimmune diseases or COVID-19 patients were present among the most prevalent new-onset autoantibodies, including anti-TPO (thyroid peroxidase)11, anti-AQP4 (aquaporin-4)9
and anti-IFNA1 IgG. However, the emergence of these autoantibodies with COVID-19 has not been reported previously. Several of the most prevalent new-onset autoantibodies have, to our
knowledge, not been described previously, including the three with the highest prevalence, i.e., anti-CCDC63 (coiled-coil domain-containing protein 63), anti-TRIM63 (E3 ubiquitin-protein
ligase TRIM63), and anti-SNURF (SNRPN upstream reading frame protein) IgG. In total, 150 of the 369 baseline seronegative individuals (41%) developed at least one of the 22 most prevalent
new-onset autoantibodies. Considering the demonstrated impact of antibodies targeting interferons (IFNs) in COVID-191, we specifically investigated the prevalence of new-onset autoantibodies
across IFN subtypes. In total, we detected new-onset anti-IFN IgG in 10 of 362 baseline seronegative HCW and 1 of the 7 COVID-19 patients that were seronegative at admission.
Anti-interferon alpha (IFNA) IgG was predominant (Fig. 2d), and 1 individual had new-onset autoantibodies targeting more than one IFN subtype (IFNA, interferon epsilon (IFNE), and interferon
omega (IFNW)). PREVALENT NEW-ONSET AUTOANTIBODIES IN INDIVIDUALS WITHOUT PRE-INFECTIOUS SAMPLES Next, we aimed to explore associations of the 22 most prevalent new-onset autoantibodies to
COVID-19 severity and symptoms post-COVID-19. To include individuals who were seropositive at study inclusion (94 HCW and 39 hospitalized patients), we first developed a model for
classification of new-onset based on autoantibody levels at seroconversion and four- and eight-month follow-up. The model used the aggregated categories acute new onset (stable or transient)
and delayed new-onset. This multinomial linear regression (MNL) model was trained on the autoantibody trajectories of the previously assessed baseline seronegative individuals that had an
eight-month follow-up sample after seroconversion (_n_ individuals = 282; _n_ trajectories for training = 6204). Using an 80%/20% training/testing split, we found that the model performed
well on the test set and was highly specific although moderately sensitive (specificity = 0.997, sensitivity = 0.667, AUC = 0.83). Applied to the baseline seropositive individuals, the model
classified 98 autoantibody trajectories as acute new-onset and 56 as delayed new-onset in 79 individuals (59%) (Supplementary Fig. 4). The high specificity and moderate sensitivity of the
model indicates that it may underestimate new-onset autoantibody prevalence in individuals without baseline seronegative samples. Still, the 22 new-onset autoantibodies were significantly
more prevalent in individuals without baseline seronegative samples than in individuals with baseline seronegative samples (average prevalence 5.3 and 2.9%, respectively; odds ratio (OR)
(95% confidence interval (CI)) = 1.7 (1.3–2.1), _p_ = 4 × 10−6, _n_ trajectories = 11,044). This could indicate increased autoimmune signatures in hospitalized COVID-19 patients. Summarizing
the new-onset autoantibody landscape in both seronegative and seropositive baseline individuals, 43% of HCW (_n_ = 196/456) and 72% of hospitalized COVID-19 patients (_n_ = 33/46) displayed
at least one of the most prevalent new-onset autoantibodies (Fig. 3a). As shown in Fig. 3b, 10 of the 22 were significantly more prevalent in the patients than the HCW, regardless of the
number of symptoms post-COVID-19 (logistic regression model adjusted for age and sex, Benjamini–Hochberg correction, _q_ ≤ 0.05). NEW-ONSET AUTOANTIBODIES ARE ASSOCIATED WITH
NEUROPSYCHIATRIC SYMPTOMS POST-COVID-19 Next, we investigated whether the most prevalent new-onset autoantibodies were associated with neuropsychiatric symptoms post-COVID-19 in HCW. Using a
proportional odds logistic regression model, we identified three new-onset autoantibodies associated with increased odds of higher severity of neuropsychiatric symptoms lasting for at least
2 months post-COVID-19, shown in Fig. 3c; anti-CALU (calumenin), anti-MYO16 (unconventional myosin-XVI), and anti-SNURF IgG (OR (95% CI) = 3.3 (1.4–7.4), 5.0 (1.1–24), 2.4 (1.1–5.1); _p_ =
0.004, 0.04, 0.02, respectively). In addition, we asked whether the most prevalent new-onset autoantibodies were associated with other post-COVID-19 symptoms. Using logistic regression, we
identified eight autoantibodies associated with 11 other reported moderate or severe symptoms post-COVID-19, with the association between anti-PCYT1B (choline-phosphate cytidylyltransferase
B) IgG and impaired hearing being the strongest (OR (95% CI) = 41 (8.3–220), _q_ = 0.002; Fig. 3d). Interestingly, there was also a moderate association between autoantibodies towards the
muscle protein CCDC63 and muscle and joint pain (OR (95% CI) = 2.3 (1.1–4.9), _p_ = 0.03), although this association was not significant after Benjamini–Hochberg correction. ANTI-SNURF IGG
INCREASES AFTER INFECTION AND AFTER VACCINATION While all 456 HCW that were assessed for new-onset autoantibodies had seroconverted before vaccination, 362 (79%) received their first
SARS-CoV-2 vaccine dose during the study period. The majority received the Pfizer/BioNTech vaccine (68%, _n_ = 248), 30% received the AstraZeneca vaccine (_n_ = 107), and 2% received the
Moderna vaccine (_n_ = 7). With this in mind, we asked whether any of the 22 most prevalent new-onset autoantibodies not only increase after infection, but also after vaccination.
Considering changes at the level of log2 FC ≥2, we identified 12 autoantibodies in 45 individuals (Supplementary Fig. 5). Notably, anti-SNURF IgG was the most commonly increasing
autoantibody after vaccination (67%; detected in 30 of 45 HCW displaying an increase). As seen in Fig. 4, individuals with as well as without previous new-onset anti-SNURF IgG displayed
increases in anti-SNURF IgG at vaccination (Fig. 4b) at comparable levels that of new-onset (Fig. 4a). However, the odds were greater for HCW with previous new-onset anti-SNURF IgG (5 of 28
vs 25 of 332, respectively; OR (95% CI) = 3.4 (1.0 – 9.5), _p_ = 0.03, _n_ = 360). As four of these five individuals had received the AstraZeneca SARS-CoV-2 vaccine, we investigated whether
the odds of autoantibody increase at vaccination was influenced by any interaction effect of previous new-onset autoantibodies and vaccine type and did not find sufficient evidence to
support this notion (OR (95% CI) = 10 (0.94–250), _p_ = 0.08, _n_ = 360). MAIN EPITOPES OF NEW-ONSET AUTOANTIBODIES Furthermore, we asked what epitopes were targeted by new-onset
autoantibodies. To address this question, we epitope mapped eight of the 22 most prevalent new-onset autoantibodies: the highly prevalent anti-CALU, CCDC63, SNURF, and TRIM63 IgG; the
previously described anti-IFNA6, ANO2 (anoctamine 2), and TPO IgG; and anti-NPC1 (NPC intracellular cholesterol transporter 1) IgG for which the antigen has sequence overlap with TPO. The
epitope mapping was performed on samples from the 142 individuals that had one or more of the eight selected new-onset autoantibodies, using an array of custom-designed 14- and 15-mer
peptides with an overlap of 10 to 13 amino acid residues (Supplementary Data 3). Five autoantibodies displayed main epitopes that were common to individuals with the corresponding new-onset
autoantibody. The epitopes correspond to the peptides CCDC63|175-189, NPC1|566–580, SNURF|50−64, TPO|918–932, TRIM63|234–247, TRIM63|236–249, and ANO2|135-149. Aside from the main epitopes,
other epitopes occurred individually (Fig. 5a). We next investigated the correspondence of autoantibodies targeting the main epitopes and autoantibodies detected against the full antigen. We
observed significantly elevated log2 FC of autoantibodies against the main epitopes in individuals with vs without the respective new-onset autoantibody, except in anti-ANO2|135-149 IgG
which therefore was excluded from further analysis (Supplementary Fig. 6). The correlation of autoantibodies detected using different antigen representations varied from a general
correlation, e.g., for the SNURF antigens, to a weak correlation in the CCDC63 antigens (Fig. 5c). VALIDATION OF AUTOANTIBODIES AGAINST THE MAIN EPITOPES IN BLOOD AND CEREBROSPINAL FLUID To
validate our findings, we analyzed two independent cohorts of pre-pandemic HCs, (_n_ = 29) and neuro-COVID patients (_n_ = 25) for the presence of autoantibodies against the detected main
epitopes. Cohort demographics are presented in Supplementary Table 1. Compared with levels in pre-pandemic HCs, the levels of autoantibodies against the main epitopes were significantly
elevated in the individuals with the corresponding new-onset autoantibody (CCDC63|175–189: _q_ = 1 × 10−8, _f_ = 0.9; NPC1|566-580: _q_ = 6 × 10−7, _f_ = 1; SNURF|50-64: _q_ = 6.4 × 10−12,
_f_ = 0.97; TPO|918-932: _q_ = 4.3 × 10−9, _f_ = 0.99; TRIM63|234–247: _q_ = 7.2 × 10−12, _f_ = 0.98; TRIM63|236–249: _q_ = 1.9 × 10−12, _f_ = 1). In addition, anti-NPC1|566-580 (_q_ = 2.6 ×
10−2, _f_ = 0.68), anti-SNURF|50–64 (_q_ = 6.7 × 10−4, _f_ = 0.77), anti-TRIM63|234–247 (_q_ = 1.6 × 10−5, _f_ = 0.84), and anti-TRIM63|236–249 (_q_ = 5.4 × 10−6, _f_ = 0.86) IgG levels
were significantly increased in neuro-COVID patients compared to pre-pandemic HCs. Notably, the only autoantibody for which the pre-pandemic HCs had levels above background was the
previously described autoantibody anti-TPO IgG (Fig. 5b). As we were specifically interested in the possible neurological pathology of autoantibodies, we asked whether epitope-directed
autoantibodies could also be found in the cerebrospinal fluid (CSF) of individuals with COVID-19. In the validation cohorts, paired CSF and blood samples were available for 23 neuro-COVID
patients and 21 pre-pandemic HCs. As seen in Fig. 5d, epitope-directed autoantibody signals correlated in CSF and blood of both validation cohorts (neuro-COVID: _ρ_ = 0.82, _p_ < 2.2 ×
10−16; HCs: _ρ_ = 0.56, _p_ = 9.5 × 10−13). THE MUSCLE PROTEINS TRIM63 AND CCDC63 ALIGN WITH THE SARS-COV-2 FUSION PEPTIDE One possible explanation for the emergence of new-onset
autoantibodies is molecular mimicry between viral and human proteins. Therefore, we examined any amino acid sequence similarity of the main epitopes and the SARS-CoV-2 Spike glycoprotein34
(UniProt accession P0DTC2) using BLAST35. We found sequence similarities between the Spike glycoprotein sequence 816-SFIEDLLFNK-825 and TRIM63|234–247 (_E_ = 0.022), TRIM63|236-249 (_E_ =
0.022), and CCDC63|175–189 (_E_ = 0.012) (Fig. 5e). This sequence is proximal to the S2’ cleavage site of Spike protein fusion peptide. Furthermore, we found an alignment of NPC1|566-580 and
the Spike S1 C-terminal domain sequence 656-VNNSY-660 (_E_ = 0.60), which is exposed at the surface of S1. In addition, NPC1|566–580 and TPO|918–932 share five residues (Fig. 5f).
DISCUSSION In the present work, we have characterized the autoantibody response emerging with COVID-19 using proteome-wide autoantibody screening in longitudinal and independent cohorts. As
the scope of our study was a proteome-wide analysis of autoantibody repertoires, we searched for autoantibodies towards intracellular as well as extracellular and secreted antigens. Although
the pathogenic mechanisms of antibodies towards intracellular antigens remain unclear, they are frequently observed in screening studies and can be of established clinical importance36.
While previous studies have indicated the existence of new-onset autoantibodies in COVID-1915,16 and in other pulmonary infections37, we have systematically charted the temporal dynamics of
the emerging self-directed humoral response in COVID-19 and showed that 60% of new-onset autoantibodies remained elevated for at least 12 months after infection. In addition, we have shown
that new-onset autoantibody prevalence corresponds to acute disease severity. Taken together, these results indicate that a dysregulated humoral immune response is a marked feature of acute
and post-acute COVID-19. Furthermore, our study shows that there is large diversity and interindividual heterogeneity of new-onset autoantibodies in COVID-19, corroborating previous findings
in cross-sectional autoantibody studies in health14 and disease33,38, including COVID-1912. The detected new-onset autoantibodies target a wide range of antigens across the proteome,
illustrating the breadth of the autoantibody response emerging after COVID-19. The dynamics of detected new-onset autoantibodies followed three distinct patterns: stable, transient, and
delayed onset. The two acute onset types reflect different autoantibody persistence, while delayed onset may reflect other parameters. As 31% (49 of 159) of delayed new-onset autoantibodies
emerged in individuals who were vaccinated between seroconversion and the subsequent visit, it is not possible to deconvolve the influence of the events on the onset of these autoantibodies.
While the processes behind the remaining 69% are not clear, we speculate that a short time between infection and blood sampling could prevent immediate detection of new-onset autoantibodies
despite detection of seroconversion, possibly due to the high sensitivity of the serological assay28. Alternatively, delayed autoantibody onset could reflect autoantibody emergence in late
affinity maturation. Further work is needed to shed light on the mechanisms underlying the emergence of new-onset autoantibodies. As anti-IFN IgG is implied in severe COVID-19, we
specifically characterized the new-onset anti-IFN IgG response in our study. Considering all IFN subtypes, we found new-onset anti-IFN IgG in 3% of the baseline seronegative cohort, mainly
consisting of anti-type I IFN IgG. These results are corroborated by previous studies indicating that anti-IFN IgG in COVID-19 are mainly directed against type I IFNs and that these
autoantibodies typically existed prior to COVID-191, although results suggesting new-onset anti-IFN antibodies have also been reported39. Since neurological symptoms after COVID-19 are
commonly occurring and often debilitating, we made directed efforts in understanding this group of symptoms post-COVID-19. We found three new-onset autoantibodies associated with increased
severity of neuropsychiatric symptoms post-COVID-19: anti-CALU, MYO16, and SNURF IgG. CALU is a membrane-bound or secreted calcium-binding protein mainly expressed in the heart and skeletal
muscle. MYO16 is a cytoplasmic unconventional myosin with enhanced brain expression, and may be involved in the extension of neuronal membrane processes40. SNURF is a small (71 aa) nuclear
protein of unknown function, primarily expressed in brain and muscle tissues and cardiomyocytes. In addition, we found associations of 11 self-assessed non-neurological symptoms
post-COVID-19 to 8 of the most prevalent new-onset autoantibodies. However, these associations were tentative, with only the association of anti-PCYT1B IgG and impaired hearing remaining
significant after FDR correction. Our epitope mapping identified the main epitopes of five autoantigens. These epitopes showed a marked increase in reactivity after infection. Among these,
the main epitopes of TRIM63 and CCDC63 displayed a sequence alignment to the fusion peptide of the SARS-CoV-2 spike glycoprotein. While not sufficient in isolation, the high post-infectious
reactivity to these autoantigens combined with their sequence alignment to a viral protein are together consistent with a molecular mechanism invoking molecular mimicry. The fusion peptide
is crucial for viral entry into the host cell and is highly conserved across the family of coronaviruses. Furthermore, it is accessible to antibody binding in the post fusion state, after
engaging the ACE2 receptor41. Concordantly, two studies have independently found that human antibodies that broadly neutralize coronaviruses are targeting the fusion peptide41,42. This
raises the possibility that the observed mimicry also might be found in infections with other coronaviruses. To our knowledge, however, these antigens have not previously been investigated
in this context. Notably, similar evidence of molecular mimicry between sorting nexin-8 (SNX8) and the SARS-CoV-2 Nucleocapsid protein has recently been reported in multisystem inflammatory
syndrome in children following COVID-1943. We did, however, not find any evidence suggestive of molecular mimicry with Nucleocapsid in our study. With the independent cohort, we validated
the presence of anti-TRIM63|234–247, and anti-TRIM63|236–249 IgG in patients with neuro-COVID but not in pre-pandemic HCs. In the discovery cohort, anti-TRIM63 IgG prevalence was increased
in hospitalized COVID-19 patients. In addition, we observed a moderate association between anti-CCDC63 IgG and self-reported muscle and joint pain. Taken together, there is evidence
suggestive of molecular mimicry between the immunologically important Spike protein fusion peptide and the muscle proteins TRIM63 and CCDC63 alongside associations with muscular symptoms
post-COVID-19 and COVID-19 severity. Despite not showing any sequence similarity with SARS-CoV-2, anti-SNURF IgG emerged in 9% of HCW (_n_ = 40) and 20% of hospitalized COVID-19 patients
(_n_ = 9). Furthermore, antibodies against the main epitope of SNURF (aa 50–64) were validated in the blood and CSF of patients with neuro-COVID (compared to pre-pandemic HCs). Our detected
correlation of autoantibody levels in the blood and CSF corroborates findings of blood-brain barrier disruption in patients with cognitive impairment post-COVID-1944. It is worth noting that
SNURF, like the other two most prevalent autoantibody targets TRIM63 and CCDC63, is expressed in heart and skeletal muscle. In addition, SNURF is expressed in several other tissues,
including the brain, corroborating the association of anti-SNURF IgG and neuropsychiatric symptoms post-COVID-19. Like CCDC63 and many classical autoantigens, SNURF is located in the
nucleus, indicating that epitope spreading may be the source of anti-SNURF IgG45. Furthermore, anti-SNURF IgG was by far the most common autoantibody increasing at vaccination. While the
data did not sufficiently support an interaction effect of previous new-onset anti-SNURF IgG and vaccine type on anti-SNURF IgG increase at vaccination, this could be due to small sample
sizes. However, a recent study reported that autoantibodies remained stable after vaccination with SARS-CoV-2 mRNA vaccines and were not elevated in patients with vaccine-associated
myocarditis16. Similarly, other studies reported that established autoantibodies do not increase after vaccination46,47. In line with this, our study does not provide any evidence linking
the herein-detected autoantibodies with any adverse effects following vaccination. Previous longitudinal studies have reported preexisting but not new-onset autoantibodies to the clinically
important autoantigen TPO in COVID-19 patients15. In contrast, we found new-onset anti-TPO IgG in 4% (_n_ = 22) of the longitudinal cohort, with a higher prevalence in hospitalized COVID-19
patients than in HCW with mild to moderate disease. In addition, we showed that new-onset anti-TPO IgG co-occurred with new-onset anti-NPC1 IgG in 10 of 22 cases, and anti-NPC1 IgG appeared
without anti-TPO IgG in only four individuals. Subsequent epitope mapping and sequence alignment revealed that the main epitopes of TPO and NPC1, which display elevated autoantibody levels
after infection, have a sequence similarity containing five identical residues. In addition, the N-terminal residues of the main epitope of NPC1 align with the amino acid sequence
656-VNNSY-660 at the C-terminal domain of the Spike glycoprotein S1 subunit. Together, this raises the possibility of molecular mimicry of NPC1 and Spike S1, with epitope spreading through
molecular linkage yielding anti-TPO IgG45. Although anti-TPO IgG benignly occurs in around 10% of the healthy population48, our discovery of new-onset antithyroid antibodies emerging with
mild to severe COVID-19 is concerning given that large epidemiological studies have shown increased incidence of autoimmune and autoinflammatory disorders, including autoimmune thyroid
diseases, following COVID-1949,50. However, we did not detect any anti-TPO IgG increases at vaccination. To our knowledge, anti-NPC1 IgG has not previously been identified following
SARS-CoV-2 infection. In contrast, the NPC1 protein has previously been suggested51,52 and recently reported53 as an alternative and inhibitable infective entry point of SARS-CoV-2.
Together, these findings warrant further research on NPC1, anti-NPC1 IgG, and antithyroid antibodies in COVID-19. Comparing autoantibody prevalence between studies is an interesting but
challenging task due to differences in antigen representation and response criteria. However, we note that only one of the five epitope-mapped autoantibodies, anti-TPO IgG, have been
previously reported in proteome-scale autoantibody studies of COVID-19, with a reported prevalence ranging from 012,16,27,43,54 to 2%15. These differences may arise from different
collections of antigen representations varying by, e.g., length, proteome coverage, and post-translational modifications, thus representing different sets of antigenic space. Similarly, the
design of the analytical approach, e.g., longitudinal or cross-sectional study design, control sample set, and response criteria, can contribute to differences between studies. Validation of
detected autoantibodies is an important matter requiring careful consideration. Here, we performed new-onset autoantibody discovery using protein fragments and thoroughly validated them
using 14- and 15-mer peptides, as this approach enables detection of the specific epitopes of interest. Additional validation on full-length proteins or clinical assays has the potential to
aid clinical translation. This can, however, be confounded by the larger epitope space of larger antigens unless an experimental step to enrich the antibodies of interest is used, i.e.,
antibody enrichment on the peptide representing the epitope of interest. As antibody enrichment experiments require more sample volume than that which is available for the present cohorts,
this additional validation is left for future studies. Certainly, our study has limitations. Although powerful, the Proteome-wide arrays do not detect autoantibodies towards epitopes that
are not covered by the protein fragments on the arrays, or conformational epitopes. Furthermore, as symptoms post-COVID-19 were of moderate prevalence and obtained by self-assessment, and
their pre-pandemic prevalence in the cohort is not known, clinical associations are limited and require validation in cohorts where symptoms post-COVID-19 are more prevalent and have been
assessed by a clinician. Moreover, as our controls were healthy and without other infections, the detected new-onset autoantibodies might also develop after other infections than SARS-CoV-2
infection. In addition, the functional properties of the presented autoantibodies remain tentative, and cross-reactivity should be further evaluated in future work using competitive assays.
In summary, our study shows that new-onset autoantibodies are prevalent and persistent following mild to severe COVID-19 and correspond to disease severity. Furthermore, some are found to be
associated with neuropsychiatric symptoms post-COVID-19 and could be detected in both plasma and CSF of patients with neuro-COVID. In addition, we demonstrate that anti-TRIM63 and
anti-CCDC63 IgG develop in 10% of the study cohort and reveal their main epitopes using epitope mapping. The main epitopes display sequence similarities indicative of molecular mimicry with
the highly conserved fusion peptide of the SARS-CoV-2 Spike glycoprotein, which is essential for viral entry and the target of broadly neutralizing antibodies. Conversely, anti-SNURF IgG is
highly prevalent without evidence of molecular mimicry, which may indicate epitope spreading to nuclear antigens. Our work reveals the complexity of the autoantibody repertoire that emerges
with COVID-19 and shows its potential impact on the course of acute viral infection and post-viral syndromes. This provides a strong rationale for further exploration of new-onset
autoantibody repertoires in other infectious diseases, as well as for continued investigation of the herein presented new-onset autoantibodies. METHODS STUDY COHORTS The COMMUNITY study is
an ongoing longitudinal study which enrolled 2149 HCW and 118 COVID-19 patients admitted to Danderyd Hospital, Stockholm, Sweden, between April and May/June 2020 (HCW/patients)29,30,31. The
cohort is followed with blood sampling every four months and we assess anti-SARS-CoV-2 IgG using a multiplex bead array of SARS-CoV-2 proteins28. In addition, HCW reported symptoms
post-COVID-19 through electronic self-assessment forms at selected visits, including visits 3 to 5. The symptoms for self-assessment were anxiety, brain fatigue, impaired concentration,
cough, depressed mood, diarrhea, dyspnea, dizziness, fatigue, fever, hair loss, headache, impaired hearing, impaired memory, ageusia, muscle/joint pain, nausea, numbness, anosmia,
palpitations, skin disorders, sleep disturbance, and stomach ache. Symptom severity was graded as mild, moderate, or severe. Neuropsychiatric symptoms were defined as reporting one or
several of anxiety, brain fatigue, impaired concentration, depressed mood, and impaired memory. The disease severity of the hospitalized patients ranged from mild to severe. Disease severity
and comorbidities are presented in Supplementary Table 1. At the start of the study in April 2020, all patients currently hospitalized with confirmed COVID-19 and all employees were
eligible for inclusion, with no exclusion criteria. In the present study, we retrospectively considered visits 1 to 5, i.e., May 2020 to September 2021. We selected a subgroup of 478 HCW and
47 hospitalized COVID-19 patients based on previous serological results and reported symptoms post-COVID29,30,31. HCW were selected in two steps. First, we selected HCW who were
seronegative for anti-SARS-CoV-2 IgG at the first visit in Apr-May 2020, seroconverted prior to vaccination, and had participated in the visits immediately before and after seroconversion
(_n_ = 381). Second, we selected HCW who were seropositive at the first visit, had participated in all of the four subsequent visits, and had reported several symptoms post-COVID-19 (_n_ =
97). Patients were selected based on participation in all of the first four follow-up visits (_n_ = 47). In total, 525 individuals were selected, with an average of 4.8 samples per
individual, yielding 2532 samples for autoantibody analysis. Demographics are presented in Supplementary Table 1. Ethical approval for the COMMUNITY study was obtained from the Swedish
Ethical Review Authority (Nos. 2020-01653 and 2021-04113). All healthcare workers left written informed consent for study participation. For the patients, oral informed consent was obtained
instead of written informed consent due to the risk of contagion. In the case of incapacity, informed consent was obtained from patients’ next of kin. Oral informed consent was recorded in
each patient’s medical record as well as in a separate file held by the responsible researcher. The use of oral consent was approved by the Swedish Ethical Review Authority. The pre-pandemic
healthy control group was university employees and students who had not received psychiatric care during their lifetime and were recruited in the timeframe April 2014–April 2017 as healthy
controls for the Uppsala Psychiatric Patient Samples (UPP) cohort. Ethical approval for the collection of CSF was acquired by the Regional Ethical Review Board in Uppsala, Sweden (Nos.
2012/081 and 2014/148). Oral and written consent was obtained from all controls. All available controls with matched CSF and serum were selected for this study. Participants underwent a
clinical health examination, including blood pressure and body mass index (BMI), and answered questionnaires on socio-demographics, medical history, heredity, and current medication, as well
as an interview to evaluate any psychiatric symptoms. While no controls had ongoing symptoms that required specialist psychiatric care, the frequency of prior or ongoing mild and
subclinical states of psychiatric conditions was higher than expected. All samples were acquired pre-pandemic. No other exclusion criteria were applied55. Demographics are presented in
Supplementary Table 1. The neuro-COVID population has been described previously56,57. Patients were prospectively included between April 2020 and June 2021. Inclusion was based on a positive
PCR for SARS-CoV-2 in upper and/or lower airway samples, and at least one new-onset neurological symptom and presence of anti-SARS-CoV-2 IgG in serum, or typical COVID-19 symptoms in
combination with pulmonary ground-glass opacities and consolidations on computed tomography scan of the thorax. Patients with previous central nervous system insults were excluded. One
patient had a previous cerebrovascular insult. Clinical neurological evaluation was performed by an experienced neurologist. All lumbar punctures were performed as part of the clinical
routine for neurological investigations. Neurological manifestations at the time of lumbar puncture included cranial nerve affection, central or peripheral paralysis, extrapyramidal, sensory
symptoms, altered mental status including confusion, encephalopathy, and reduced level of consciousness. All patients were hospitalized. The disease severity at the lumbar puncture was
moderate to severe. Disease severity, comorbidities, and other demographics are presented in Supplementary Table 1. Written informed consent was obtained from each patient, or next-of-kin if
a patient was unable to give consent. The collection and analysis of neuro-COVID and healthy control samples was approved by the Swedish Ethical Review Authority (No. 2017-043 with
amendments 2019-00169, 2020-01623, 2020-02719, 2020-05730, 2021-01469, and 2020-01883; and No. 2022-00526-01). The Declaration of Helsinki and its subsequent revisions were followed.
SARS-COV-2 SEROLOGY Serological classifications for sample selection were obtained from previous studies of the cohorts29,30,31. For increased resolution in the upper ranges of the data,
samples were re-analyzed at a higher dilution (1:5000 vs 1:50) while otherwise following the same procedure28. PLANAR ARRAYS Initial exploration of autoantibody repertoires was performed
using two sets of in-house developed protein arrays. The Proteome-wide planar array contains 42,000 protein fragments from the Human Protein Atlas (proteinatlas.org) that represent 18,000
proteins and cover ~40% of the amino acid residues of the human proteome33,58. The Secretome array contains 1522 full-length secreted or extracellular proteins from 1482 genes representing
58% of the human secretome, i.e., the proteins secreted by human cells32. The experimental procedure has been described previously14. In brief, plasma samples were pooled within the
described groups and diluted 1:100 in before applying them to planar arrays. After incubation and washes, fluorescently labeled secondary detection antibodies were applied to the array for
detection of autoantibody binding and detection of array microspots. Readout was performed in a microarray scanner, where the red channel readout corresponds to autoantibodies and the green
channel readout to array microspots. Grid alignment and data acquisition from the scanned images was performed using GenePix Pro 5.1. Selected protein fragments are available on reasonable
request. BEAD ARRAYS Bead arrays were used for the investigation of new-onset autoantibodies in the COMMUNITY cohort and for epitope mapping in the COMMUNITY and validation cohorts. Bead
array construction and assays were performed as previously described14,59. For an exploration of new-onset autoantibodies, the coupled antigens had been selected from planar arrays as
described below. For epitope mapping, the coupled antigens were 93 custom-synthesized biotinylated peptides of 14 to 15 amino acid residues (GenScript Biotech). Assay readout was performed
using Luminex FLEXMAP 3D® instruments with xPONENT® software (Luminex Corp.) and responses recorded as median fluorescent intensity (MFI), in arbitrary units. ANALYSIS OF PLANAR ARRAY DATA
Planar array data were processed as previously described33. Proteome-wide array data were analyzed per subarray. Spots that were flagged in image acquisition, that were smaller than 30
pixels, or that had a green channel signal lower than 4 SD above the mean local background were removed. Duplicate spots were deduplicated by selecting the spot with highest signal in the
green channel. Finally, red channel data were _Z-_scored, and antigens with _Z_ ≥ 12 were classified as reactive. Secretome array data was analyzed per subarray. Microarray spots that were
flagged in image acquisition, that were smaller than 40 pixels, or that had a green channel signal at or below the local background level were removed. Duplicate spots were filtered if their
CV exceeded 50, and remaining spot pairs were deduplicated by taking the mean. Finally, local background was subtracted from red channel data and resulting values were _Z-_scored. Antigens
with _Z_ ≥ 8 were classified as reactive. _Z-_scored planar array data were used for the selection of antigens for further investigation in the full HCW and hospitalized patient cohorts.
First, antigens reactive in single samples on single arrays were selected. Second, lowered selection thresholds and detection in multiple samples were used to diversify the selection. The
following selection criteria were used: protein fragments meeting _Z_ ≥ 8 in multiple samples; full-length proteins meeting _Z_ ≥ 4 in multiple samples; protein fragments and full-length
proteins meeting _Z_ ≥ 4 in single or multiple samples on both array types. Third, antigens from the literature and in-house studies were selected: protein fragments noted in both the
literature and in-house studies; protein fragments meeting _Z_ ≥ 8 and noted in either the literature or in-house studies; full-length proteins noted in multiple publications; full-length
proteins meeting _Z_ ≥ 2 and noted in the literature; handpicked protein fragments and full-length proteins noted in either the literature or in-house studies. Selected antigens and their
matching selection criteria are listed in Supplementary Data 1. CLASSIFICATION OF NEW-ONSET AUTOANTIBODIES IN INDIVIDUALS WITH A SERONEGATIVE BASELINE SAMPLE New-onset autoantibodies were
classified by applying Partitioning Around Medoids (PAM) clustering60 to the bead array data of baseline seronegative individuals. The approach is documented in the companion R package
abtract61. The log2 FC of each autoantibody trajectory was computed relative to the most recent seronegative sample. Individuals with autoantibody data at seroconversion and the samplings
immediately before and after seroconversion were considered for PAM clustering (_n_ = 374). Principal component analysis (PCA) was conducted on these data to identify and exclude outlying
individuals with an increase in most autoantibody trajectories at seroconversion (_n_ = 5, Supplementary Fig. 7a). Outliers were defined as PC1 ≥ mean + 3 × SD of PC1 (PC1 ≥8.85). To reduce
the running time of the model, trajectories that never exceed background MFI levels were pre-classified as not new-onset. In each antigen class (protein fragments and full-length proteins),
background MFI level was defined as robust _Z-_score = 3 in the seronegative samples from September 2020 and January 2021 from the 40 individuals that seroconverted in May 2021 (MFIfragment
≥73, MFIfull-length ≥322; _n_ filtered trajectories = 619894/910041 (68%)). The PAM clustering was performed with the custom cosine × euclidean distance metric. To evaluate the parameter _k_
(number of clusters), the PAM clustering was run with _k_ ranging from 1 to 20, each with 10 random starts of the clustering algorithm. Using the silhouette method, the optimal cluster
number was determined to be 7. Based on a median log2 FC ≥2, clusters 5, 6, and 7 were classified as new-onset. CLASSIFICATION OF NEW-ONSET AUTOANTIBODIES IN INDIVIDUALS WITHOUT ANY
SERONEGATIVE BASELINE SAMPLE New-onset autoantibodies were assessed in individuals without seronegative baseline samples using multinomial linear regression (MNL)62,63. The MNL model was
built using classifications and autoantibody trajectories of the 22 prevalent new-onset autoantibodies in the baseline seronegative individuals that had follow-up samples at 4 and 8 months
after seroconversion. The MNL model specification was \({\bf{New}}{\boldsymbol{\hbox{-}}}{\bf{onset\; type}}\, \sim \,{\log
}_{10}{{\bf{MFI}}}_{t\left(0\right)}+{{\bf{FC}}}_{t\left(4\right)}+{{\bf{FC}}}_{t\left(8\right)}\), where _t_ is months from seroconversion, and FC is fold change relative to time of
seroconversion (_t_(0)). The trajectories were split into training (0.8) and testing (0.2) sets using randomized stratified sampling on the trajectory outcome (target antigen and simplified
new-onset classification; acute, delayed, or not new-onset autoantibody). The MNL model was trained using repeated stratified _K-_fold cross-validation (repeats = 10, _K_ = 5, stratification
on trajectory outcome). Grid search was used to optimize the penalizing decay parameter, which was set to 0 based on model accuracy. Individuals that did not have seronegative baseline
samples, but that did have autoantibody data at seroconversion and the two samplings immediately after seroconversion, were considered for MNL classification (_n_ = 135). PCA was used to
exclude outlying individuals with an increase in most autoantibody trajectories in the considered time points (_n_ = 2, Supplementary Fig. 7b). Outliers were defined as PC1 ≥ mean + 3 × SD
of PC1 (PC1 ≥2.1). Trajectories of the 22 prevalent new-onset autoantibodies in the remaining 133 individuals were classified using the MNL model (_n_ trajectories = 2926). ANNOTATION OF
ANTIGEN LOCATION Antigen location was determined based on Cellular Component Gene Ontology (GO) terms for each corresponding gene64,65. The GO terms were simplified using the Generic GO
subset66 and further subdivided into broad location categories as detailed in Supplementary Table 3. For antigens with multiple locations, the annotation was selected in the following order:
Extracellular > Plasma membrane > Nuclear > Intracellular. Antigens may lack annotation of location if no parent terms are included in the GO subset. ASSOCIATION OF NEW-ONSET
AUTOANTIBODIES WITH SYMPTOMS POST-COVID-19 Association of new-onset autoantibodies with symptoms post-COVID-19 was performed using data from HCW displaying one or more of the 22 prevalent
new-onset autoantibodies, or none (_n_ = 403). Among these, neuropsychiatric symptoms were defined as reporting anxiety, brain fatigue, difficulties concentrating, depressive disorders, or
impaired memory that lasted for at least 2 months after COVID-19 (_n_ = 384). The highest reported severity was used (severe = 20 (5%); moderate = 58 (15%); mild = 23 (6%); no
neuropsychiatric sx = 283 (74%)). For other symptoms post-COVID-19, reported mild symptoms were excluded (Supplementary Table 4). IDENTIFICATION OF MAIN EPITOPES The peptide bead array was
used for epitope mapping. Data were acquired from 142 baseline samples and 150 samples after autoantibody onset in the 142 individuals who had one or more new-onset autoantibodies whose
antigens were represented on the peptide bead array. (Eight individuals had both acute and delayed new-onset autoantibodies, increasing the number of samples after autoantibody onset.) The
FC was computed relative to baseline, both for beads with coupled peptides as well as the negative control bead with coupled biotin. To adjust for individual background levels, the FC of
biotin was subtracted from the FC of the peptides, and the median was set to 1. Any resulting negative values (_n_ = 2) were imputed with the smallest positive value present (0.07). Main
epitopes were defined in individuals having the corresponding new-onset autoantibody by taking the mean FC of each peptide. Peptides, where the mean exceeded the background FC cutoff, were
defined as main epitopes that were common across individuals. The background FC cutoff was defined as mean + 5 × SD of the FC of the negative biotin control (FC ≥ 2.52). SEQUENCE ALIGNMENT
Sequence alignment was performed using NCBI BLASTP adjusted for short input sequences: _E_ < 20 000, word size ≥3, PAM30 substitution matrix, gap costs 7/2, no compositional adjustment,
and no filters or masks. STATISTICAL ANALYSIS Correlations were performed using Spearman correlation. Group differences of continuous variables were investigated using the Mann–Whitney
_U_-test. Binary variables were investigated using logistic regression, categorical variables using multinomial logistic regression, and ordinal variables using proportional odds logistic
regression. Regression models used correction for age and sex in group comparisons. False discovery rate correction was performed using the Benjamini–Hochberg procedure, and resulting values
are reported as _q_ values. All statistical tests were two-sided. All measurements were taken from distinct samples. All data analysis and statistical analysis was performed in RStudio with
R 4.1.2 and 4.2.1 and R packages tidyverse, lubridate, rlang, scales, knitr, pander, httr2, readxl, rstatix, proxy, cluster, caret, MLmetrics, MASS, broom, Omixer, ggpubr, ragg, patchwork,
cowplot, GGally, ggsignif, ggfortify, ggdist, ggbeeswarm, ggrepel, ComplexHeatmap, and circlize. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio
Reporting Summary linked to this article. DATA AVAILABILITY The antibody repertoire data generated in this study have been registered in the SciLifeLab Data Repository under accession code
26318929. The antibody repertoire data are available under restricted access due to data privacy laws. Access can be obtained by researchers who meet the specified criteria by submitting a
request through the Data Repository67. The processed antibody repertoire data are provided in the Source Data file. The self-reported post-COVID-19 symptom data are protected and are not
available due to data privacy laws. Access requests can be initiated by email to the corresponding author with an approximate timeframe to reply of 4 weeks. The processed post-COVID-19
symptom data were provided in the Supplementary Information and Source Data file. Source data are provided with this paper. CODE AVAILABILITY The abtract package for antibody trajectory
clustering, implemented in R 4.4.0, is available on GitHub at https://github.com/jernbom/abtract. The version relevant to this work, v0.1.0, is archived on Zenodo61. REFERENCES * Bastard, P.
et al. Autoantibodies neutralizing type I IFNs are present in ~4% of uninfected individuals over 70 years old and account for ~20% of COVID-19 deaths. _Sci. Immunol._ 6, eabl4340 (2021). *
Zhang, Q. et al. Autoantibodies against type I IFNs in patients with critical influenza pneumonia. _J. Exp. Med._ 219, e20220514 (2022). * Knight, J. S. et al. The intersection of COVID-19
and autoimmunity. _J. Clin. Invest._ 131, e154886 (2021). * Puel, A., Bastard, P., Bustamante, J. & Casanova, J.-L. Human autoantibodies underlying infectious diseases. _J. Exp. Med._
219, e20211387 (2022). * Franke, C. et al. High frequency of cerebrospinal fluid autoantibodies in COVID-19 patients with neurological symptoms. _Brain Behav. Immun._ 93, 415–419 (2021).
Article PubMed CAS Google Scholar * Baiocchi, G. C. et al. Cross‐sectional analysis reveals autoantibody signatures associated with COVID‐19 severity. _J. Med. Virol._ 95, e28538 (2023).
* Vasilevska, V. et al. Molecular mimicry of NMDA receptors may contribute to neuropsychiatric symptoms in severe COVID-19 cases. _J. Neuroinflammation_ 18, 245 (2021). * Emmenegger, M. et
al. Anti-prothrombin autoantibodies enriched after infection with SARS-CoV-2 and influenced by strength of antibody response against SARS-CoV-2 proteins. _PLoS Pathog._ 17, e1010118 (2021).
Article MathSciNet PubMed PubMed Central CAS Google Scholar * Liu, Y. et al. Paradoxical sex-specific patterns of autoantibody response to SARS-CoV-2 infection. _J. Transl. Med._ 19,
524 (2021). * Fonseca, D. L. M. et al. Severe COVID-19 patients exhibit elevated levels of autoantibodies targeting cardiolipin and platelet glycoprotein with age: a systems biology
approach. _npj Aging_ 9, 21 (2023). Article PubMed PubMed Central CAS Google Scholar * Anaya, J.-M. et al. Latent rheumatic, thyroid and phospholipid autoimmunity in hospitalized
patients with COVID-19. _J. Transl. Autoimmun._ 4, 100091 (2021). Article PubMed PubMed Central CAS Google Scholar * Wang, E. Y. et al. Diverse functional autoantibodies in patients
with COVID-19. _Nature_ 595, 283–288 (2021). Article ADS PubMed CAS Google Scholar * Bondt, A. et al. Human plasma IgG1 repertoires are simple, unique, and dynamic. _Cell Syst._ 12,
1131–1143.e1135 (2021). Article PubMed PubMed Central CAS Google Scholar * Neiman, M. et al. Individual and stable autoantibody repertoires in healthy individuals. _Autoimmunity_ 52,
1–11 (2019). Article PubMed CAS Google Scholar * Chang, S. E. et al. New-onset IgG autoantibodies in hospitalized patients with COVID-19. _Nat. Commun._ 12, 5417 (2021). * Jaycox, J. R.
et al. SARS-CoV-2 mRNA vaccines decouple anti-viral immunity from humoral autoimmunity. _Nat. Commun._ 14, 1299 (2023). Article ADS PubMed PubMed Central CAS Google Scholar * Soriano,
J. B., Murthy, S., Marshall, J. C., Relan, P. & Diaz, J. V. A clinical case definition of post-COVID-19 condition by a Delphi consensus. _Lancet Infect. Dis._ 22, e102–e107 (2022).
Article PubMed CAS Google Scholar * Wulf Hanson, S. et al. Estimated global proportions of individuals with persistent fatigue, cognitive, and respiratory symptom clusters following
symptomatic COVID-19 in 2020 and 2021. _JAMA_ 328, 1604 (2022). Article PubMed PubMed Central Google Scholar * Chen, C. et al. Risk surveillance and mitigation: autoantibodies as
triggers and inhibitors of severe reactions to SARS-CoV-2 infection. _Mol. Med._ 27, 160 (2021). * Damoiseaux, J. et al. Autoantibodies and SARS-CoV2 infection: the spectrum from association
to clinical implication: report of the 15th Dresden Symposium on Autoantibodies. _Autoimmun. Rev._ 21, 103012 (2022). Article PubMed CAS Google Scholar * Mehandru, S. & Merad, M.
Pathological sequelae of long-haul COVID. _Nat. Immunol._ 23, 194–202 (2022). * Latorre, D. Autoimmunity and SARS‐CoV‐2 infection: unraveling the link in neurological disorders. _Eur. J.
Immunol._ 52, 1561–1571 (2022). * Ariño, H. et al. Neuroimmune disorders in COVID-19. _J. Neurol._ 269, 2827–2839 (2022). * Frontera, J. A. & Simon, N. M. Bridging knowledge gaps in the
diagnosis and management of neuropsychiatric sequelae of COVID-19. _JAMA Psychiatry_ 79, 811 (2022). Article PubMed Google Scholar * Needham, E. J. et al. Brain injury in COVID-19 is
associated with dysregulated innate and adaptive immune responses. _Brain_ 145, 4097–4107 (2022). * Prüss, H. Autoantibodies in neurological disease. _Nat. Rev. Immunol._ 21, 798–813 (2021).
* Klein, J. et al. Distinguishing features of long COVID identified through immune profiling. _Nature_ 623, 139–148 (2023). Article ADS PubMed PubMed Central CAS Google Scholar *
Hober, S. et al. Systematic evaluation of SARS‐CoV‐2 antigens enables a highly specific and sensitive multiplex serological COVID‐19 assay. _Clin. Transl. Immunol._ 10, e1312 (2021). *
Rudberg, A.-S. et al. SARS-CoV-2 exposure, symptoms and seroprevalence in healthcare workers in Sweden. _Nat. Commun._ 11, 5064 (2020). * Havervall, S. et al. SARS-CoV-2 induces a durable
and antigen specific humoral immunity after asymptomatic to mild COVID-19 infection. _PLoS ONE_ 17, e0262169 (2022). Article PubMed PubMed Central CAS Google Scholar * Havervall, S. et
al. Robust humoral and cellular immune responses and low risk for reinfection at least 8 months following asymptomatic to mild COVID‐19. _J. Intern. Med._ 291, 72–80 (2021). * Uhlén, M. et
al. The human secretome. _Sci. Signal._ 12, eaaz0274 (2019). Article PubMed Google Scholar * Jernbom Falk, A. et al. Autoantibody profiles associated with clinical features in psychotic
disorders. _Transl. Psychiatry_ 11, 474 (2021). * Wu, F. et al. A new coronavirus associated with human respiratory disease in China. _Nature_ 579, 265–269 (2020). Article ADS PubMed
PubMed Central CAS Google Scholar * Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. _Nucleic Acids Res._ 25, 3389–3402 (1997).
Article PubMed PubMed Central CAS Google Scholar * Pisetsky, D. S. & Lipsky, P. E. New insights into the role of antinuclear antibodies in systemic lupus erythematosus. _Nat. Rev.
Rheumatol._ 16, 565–579 (2020). Article PubMed PubMed Central CAS Google Scholar * Feng, A. et al. Autoantibodies are highly prevalent in non–SARS-CoV-2 respiratory infections and
critical illness. _JCI Insight_ 8, e163150 (2023). * Zandian, A. et al. Untargeted screening for novel autoantibodies with prognostic value in first-episode psychosis. _Transl. Psychiatry_
7, e1177 (2017). Article PubMed PubMed Central CAS Google Scholar * Shaw, E. R. et al. Temporal dynamics of anti-type 1 interferon autoantibodies in COVID-19 patients. _Clin. Infect.
Dis._ 75, e1192–e1194 (2021). * Patel, K. G., Liu, C., Cameron, P. L. & Cameron, R. S. Myr 8, a novel unconventional myosin expressed during brain development associates with the protein
phosphatase catalytic subunits 1α and 1γ1. _J. Neurosci._ 21, 7954–7968 (2001). Article PubMed PubMed Central CAS Google Scholar * Low, J. S. et al. ACE2-binding exposes the SARS-CoV-2
fusion peptide to broadly neutralizing coronavirus antibodies. _Science_ 377, 735–742 (2022). Article ADS PubMed CAS Google Scholar * Dacon, C. et al. Broadly neutralizing antibodies
target the coronavirus fusion peptide. _Science_ 377, 728–735 (2022). Article ADS PubMed CAS Google Scholar * Bodansky, A. et al. Molecular mimicry in multisystem inflammatory syndrome
in children. _Nature_ 632, 622–629 (2024). Article PubMed PubMed Central CAS Google Scholar * Greene, C. et al. Blood–brain barrier disruption and sustained systemic inflammation in
individuals with long COVID-associated cognitive impairment. _Nat. Neurosci._ 27, 421–432 (2024). Article PubMed PubMed Central CAS Google Scholar * Vanderlugt, C. L. & Miller, S.
D. Epitope spreading in immune-mediated diseases: implications for immunotherapy. _Nat. Rev. Immunol._ 2, 85–95 (2002). Article PubMed CAS Google Scholar * Arunachalam, P. S. et al.
Systems vaccinology of the BNT162b2 mRNA vaccine in humans. _Nature_ 596, 410–416 (2021). Article ADS PubMed PubMed Central CAS Google Scholar * Sarin, K. Y. et al. Impaired innate and
adaptive immune responses to BNT162b2 SARS-CoV-2 vaccination in systemic lupus erythematosus. _JCI Insight_ 9, e176556 (2024). * Pedersen, I. B. et al. Thyroid peroxidase and thyroglobulin
autoantibodies in a large survey of populations with mild and moderate iodine deficiency. _Clin. Endocrinol._ 58, 36–42 (2003). Article Google Scholar * Lim, S. H. et al. Autoimmune and
autoinflammatory connective tissue disorders following COVID-19. _JAMA Netw. Open_ 6, e2336120–e2336120 (2023). Article PubMed PubMed Central Google Scholar * Tesch, F. et al. Incident
autoimmune diseases in association with SARS-CoV-2 infection: a matched cohort study. _Clin. Rheumatol._ 42, 2905–2914 (2023). Article PubMed PubMed Central Google Scholar * Daniloski,
Z. et al. Identification of required host factors for SARS-CoV-2 infection in human cells. _Cell_ 184, 92–105.e116 (2021). Article PubMed CAS Google Scholar * García-Dorival, I. et al.
Identification of Niemann-Pick C1 protein as a potential novel SARS-CoV-2 intracellular target. _Antiviral Res._ 194, 105167 (2021). Article PubMed PubMed Central Google Scholar * Khan,
I. et al. Tubeimosides are pan-coronavirus and filovirus inhibitors that can block their fusion protein binding to Niemann-Pick C1. _Nat. Commun._ 15, 162 (2024). Article ADS PubMed
PubMed Central Google Scholar * Bodansky, A. et al. Autoantigen profiling reveals a shared post-COVID signature in fully recovered and long COVID patients. _JCI Insight_ 8, e169515 (2023).
* Just, D. et al. Autoantibodies against the C-terminus of lipopolysaccharide binding protein are elevated in young adults with psychiatric disease. _Psychoneuroendocrinology_ 126, 105162
(2021). Article PubMed CAS Google Scholar * Corman, V. M. et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. _Euro Surveill._ 25, 2000045 (2020). * Virhammar, J.
et al. Biomarkers for central nervous system injury in cerebrospinal fluid are elevated in COVID-19 and associated with neurological symptoms and disease severity. _Eur. J. Neurol._ 28,
3324–3331 (2021). Article PubMed Google Scholar * Berglund, L. et al. A whole-genome bioinformatics approach to selection of antigens for systematic antibody generation. _Proteomics_ 8,
2832–2839 (2008). Article PubMed CAS Google Scholar * Pin, E. et al. in _Cerebrospinal Fluid (CSF) Proteomics: Methods and Protocols_ (eds Santamaría, E. & Fernández-Irigoyen, J.) ch
19 (Springer, 2019). * Schubert, E. & Rousseeuw, P. J. Fast and eager k-medoids clustering: O(k) runtime improvement of the PAM, CLARA, and CLARANS algorithms. _Inform. Syst._ 101,
101804 (2021). Article Google Scholar * Jernbom, A. F. abtract v0.1.0. _Zenodo_ https://doi.org/10.5281/zenodo.13882631 (2024). * Venables, W. N. & Ripley, B. D. _Modern Applied
Statistics with S-Plus_ 4th edn. (Springer, 2002). * Kuhn, M. Building predictive models in R using the caret package. _J. Stat. Softw._ 28, 1–26 (2008). Article Google Scholar * Thomas,
P. D. et al. PANTHER: making genome-scale phylogenetics accessible to all. _Protein Sci._ 31, 8–22 (2022). Article PubMed CAS Google Scholar * Carbon, S. et al. AmiGO: online access to
ontology and annotation data. _Bioinformatics_ 25, 288–289 (2008). Article PubMed PubMed Central Google Scholar * Carbon, S. & Mungall, C. Gene ontology data archive. _Zenodo_
https://doi.org/10.5281/zenodo.10536401 (2024). * Jernbom, A. F. et al. Prevalent and persistent new-onset autoantibodies in mild to severe COVID-19. _SciLifeLab Data Repository_
https://doi.org/10.17044/scilifelab.26318929.v1 (2024). Download references ACKNOWLEDGEMENTS This research was funded by the Swedish Research Council (2022-03063, P.N.); Region Stockholm (RS
2020-0754, S. Hober); The Mental Health Fund (Fonden för Psykisk Hälsa, 2022), the Swedish Research Council (2022-06091), and ALF Funds from Uppsala University Hospital (J.L.C.); and
SciLife-U COVID-19, KAW-KTH SciLifeLab, and SLS Covid-19 (E.R.). The funding agencies had no influence on study design; the collection, analysis, or interpretation of data; the writing of
the report; or in the decision to submit the article for publication. Open access funding is provided by the KTH Royal Institute of Technology. The authors thank Eva Kumlien and Johan
Virhammar for providing control samples; the KTH node of Protein Production Sweden (PPS), a national research infrastructure funded by the Swedish Research Council, for providing antigens;
Eni Andersson, Ceke Hellström, and Jennie Olofsson at the SciLifeLab Autoimmunity and Serology Profiling unit, for assistance with experimental and computational work; Sára Mravinacová,
Sofia Bergström, and Jochen Schwenk for fruitful discussions; and all study participants for their participation. FUNDING Open access funding provided by Royal Institute of Technology.
AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Division of Affinity Proteomics, Department of Protein Science, SciLifeLab, KTH Royal Institute of Technology, Stockholm, Sweden August F.
Jernbom, Lovisa Skoglund, Elisa Pin, Ronald Sjöberg, Anna Månberg & Peter Nilsson * Division of Protein Technology, Department of Protein Science, KTH Royal Institute of Technology,
Stockholm, Sweden Hanna Tegel & Sophia Hober * Section of Neurosurgery, Department of Medical Sciences, Uppsala University Hospital, Uppsala, Sweden Elham Rostami * Department of
Neuroscience, Karolinska Institutet, Stockholm, Sweden Elham Rostami * Department of Medical Sciences, Psychiatry, Uppsala University, Uppsala, Sweden Annica Rasmusson & Janet L.
Cunningham * Department of Clinical Sciences, Karolinska Institutet, Danderyd Hospital, Stockholm, Sweden Sebastian Havervall & Charlotte Thålin Authors * August F. Jernbom View author
publications You can also search for this author inPubMed Google Scholar * Lovisa Skoglund View author publications You can also search for this author inPubMed Google Scholar * Elisa Pin
View author publications You can also search for this author inPubMed Google Scholar * Ronald Sjöberg View author publications You can also search for this author inPubMed Google Scholar *
Hanna Tegel View author publications You can also search for this author inPubMed Google Scholar * Sophia Hober View author publications You can also search for this author inPubMed Google
Scholar * Elham Rostami View author publications You can also search for this author inPubMed Google Scholar * Annica Rasmusson View author publications You can also search for this author
inPubMed Google Scholar * Janet L. Cunningham View author publications You can also search for this author inPubMed Google Scholar * Sebastian Havervall View author publications You can also
search for this author inPubMed Google Scholar * Charlotte Thålin View author publications You can also search for this author inPubMed Google Scholar * Anna Månberg View author
publications You can also search for this author inPubMed Google Scholar * Peter Nilsson View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS
A.F.J., P.N., A.M., S. Havervall, and C.T. conceptualized and designed the experiments and study. A.F.J., P.N., A.M., and E.P. interpreted the data and wrote the manuscript. A.F.J. and L.S.
performed experimental work. A.F.J. designed and performed analysis and visualization of the data. S. Hober, H.T., R.S., and P.N. produced the antigens and developed the arrays. C.T. and S.
Havervall collected and curated the COMMUNITY cohort. J.L.C., A.R., and E.R. provided the validation cohorts. The manuscript was edited and approved by all authors. CORRESPONDING AUTHOR
Correspondence to August F. Jernbom. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare the following competing interests: J.L.C. has received lecturing fees from Otsuka Pharma
Scandinavia, Janssen-Cilag AB, and H. Lundbeck AB. The remaining authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Carlo Perricone,
who co-reviewed with Giacomo Cafaro, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION
PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION
DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 SUPPLEMENTARY DATA 3 REPORTING SUMMARY SOURCE DATA SOURCE DATA TRANSPARENT PEER REVIEW FILE RIGHTS AND
PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any
medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The
images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not
included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly
from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Jernbom, A.F.,
Skoglund, L., Pin, E. _et al._ Prevalent and persistent new-onset autoantibodies in mild to severe COVID-19. _Nat Commun_ 15, 8941 (2024). https://doi.org/10.1038/s41467-024-53356-5 Download
citation * Received: 07 May 2024 * Accepted: 08 October 2024 * Published: 17 October 2024 * DOI: https://doi.org/10.1038/s41467-024-53356-5 SHARE THIS ARTICLE Anyone you share the following
link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature
SharedIt content-sharing initiative