Play all audios:
ABSTRACT CDK4/6 inhibition in combination with endocrine therapy is the standard of care for estrogen receptor (ER+) breast cancer, and although cytostasis is frequently observed, new
treatment strategies that enhance efficacy are required. Here, we perform two independent genome-wide CRISPR screens to identify genetic determinants of CDK4/6 and endocrine therapy
sensitivity. Genes involved in oxidative stress and ferroptosis modulate sensitivity, with GPX4 as the top sensitiser in both screens. Depletion or inhibition of GPX4 increases sensitivity
to palbociclib and giredestrant, and their combination, in ER+ breast cancer models, with GPX4 null xenografts being highly sensitive to palbociclib. GPX4 perturbation additionally
sensitises triple negative breast cancer (TNBC) models to palbociclib. Palbociclib and giredestrant induced oxidative stress and disordered lipid metabolism, leading to a
ferroptosis-sensitive state. Lipid peroxidation is promoted by a peroxisome AGPAT3-dependent pathway in ER+ breast cancer models, rather than the classical ACSL4 pathway. Our data
demonstrate that CDK4/6 and ER inhibition creates vulnerability to ferroptosis induction, that could be exploited through combination with GPX4 inhibitors, to enhance sensitivity to the
current therapies in breast cancer. SIMILAR CONTENT BEING VIEWED BY OTHERS OXIDATIVE PHOSPHORYLATION IS A METABOLIC VULNERABILITY OF ENDOCRINE THERAPY AND PALBOCICLIB RESISTANT METASTATIC
BREAST CANCERS Article Open access 14 July 2023 EPIGENETIC DEREPRESSION CONVERTS PPARΓ INTO A DRUGGABLE TARGET IN TRIPLE-NEGATIVE AND ENDOCRINE-RESISTANT BREAST CANCERS Article Open access
27 September 2021 N6-METHYLADENOSINE REGULATED FGFR4 ATTENUATES FERROPTOTIC CELL DEATH IN RECALCITRANT HER2-POSITIVE BREAST CANCER Article Open access 13 May 2022 INTRODUCTION Cyclin
D-dependent kinases 4 and 6 inhibitors (CDK4/6i) including palbociclib, ribociclib and abemaciclib in combination with endocrine therapy are the standard of care for patients with estrogen
receptor-positive (ER + ) and human epidermal growth factor receptor 2-negative breast cancer1,2,3,4,5. Inhibition of CDK4/6 and suppression of ER signalling each attenuate cell
proliferation by arresting cells in G1 of the cell cycle, and their combination enhances cell cycle arrest6. Despite the success of these treatments, the vast majority of patients progress
in the advanced setting, and many patients relapse despite adjuvant treatment. Therefore, novel approaches are required to overcome resistance and enhance sensitivity. Investigational oral
estrogen receptor antagonist and degraders, such as giredestrant, promote more effective suppression of ER and may be more effective than current endocrine therapies, especially in the
presence of _ESR1_ mutations7,8,9. Moreover, CDK4/6i have demonstrated limited activity in other types of breast cancer, such as triple negative breast cancer (TNBC), and therapies that
sensitise these cancers to CDK4/6i could have substantial utility. Ferroptosis has been identified as an iron dependent non-apoptotic mechanism of cell death, mediated by accumulation of
lipid peroxides that cause a rapid and unrepairable damage of the plasma membrane10. Phospholipids (PL) that contain polyunsaturated fatty acids (PUFAs) are highly susceptible to
peroxidation, and glutathione peroxidase 4 (GPX4) protects against ferroptosis by catalysing the reduction of phospholipid and cholesterol hydroperoxides11,12,13. Various pathways regulate
the membrane abundance of PUFA-PLs that are susceptible to peroxidation, with ACSL4 and LPCAT3 generally being the key enzymes involved in this process14. Recently, peroxisomes have emerged
as an additional regulator of ferroptosis, involved in the synthesis PUFA ether-linked phospholipids (PUFA-ePLs) independently of ACSL4, with AGPS, FAR1 and AGPAT3 as key enzymes in this
pathway15. Peroxidation of lipids can result from a non-enzymatic reaction with alkoxyl radicals or hydroxyl radicals, or in an iron dependent Fenton-type reaction16. To identify potential
targets that enhance the efficacy of CDK4/6 inhibitors and endocrine therapy, we performed whole genome CRISPR/Cas9 suppressor screens, which unexpectedly identified GPX4 depletion as the
top hit sensitising palbociclib and giredestrant treated cells, with multiple other genes involved in ferroptosis modulating sensitivity. Here we show that GPX4 depletion sensitises multiple
breast cancer models to CDK4/6 and ER inhibitors and investigate how these agents generate a ferroptosis vulnerable state. RESULTS A PALBOCICLIB WHOLE GENOME CRIPSR SCREEN REVEALS
MODULATORS OF FERROPTOSIS AND OXIDATIVE STRESS THAT SENSITISE CDK4/6 INHIBITORS To identify mechanisms of sensitivity and resistance to CDK4/6 inhibitors in ER+ breast cancer, we performed a
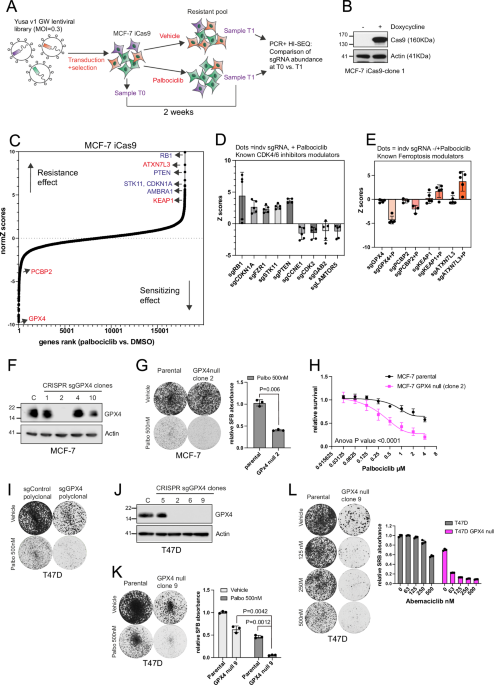
negative selection genome-wide CRISPR/Cas9 mutagenesis screen using a human sgRNA library containing 87,897 sgRNAs17 and doxycycline Cas9 inducible MCF-7 cell (MCF-7 iCas9; Fig. 1A).
Following the sgRNA library transduction at a low MOI (MOI 0.3), cells were exposed to doxycycline to induce Cas9 expression and gene mutagenesis, and then exposed to vehicle (DMSO) or
palbociclib 500 nM (SF50) for two weeks. Deep sequencing was used to compare sgRNA abundance in pre-treated (T0) and post-treated (T1) cell populations to identity genes that modified the
response to palbociclib. In the vehicle controls we observed a significant depletion of sgRNAs targeting commonly accepted “core essential genes”, suggesting the screen had sensitivity
detecting genetic perturbations (Supplementary Fig. 1A). Moreover, the screen confirmed previous findings. Hence, CRISPR-Cas9 targeting of the cell cycle inhibitors _RB1_18,19,20, _CDKN1A_
(p21), and _FZR1_21, induced resistance to palbociclib (Fig. 1C, D). CRISPR-Cas9 targeting of PI3K/AKT/mTOR growth pathway inhibitors _PTEN_19 and _STK11_ induced resistance, while loss of
pathway activators _GAB2_ and _LAMTOR5_ increased sensitivity (Fig. 1C, D), supporting prior data showing that activation of this pathway results in resistance to CDK4/6
inhibitors18,19,22,23,24. Our screen also confirmed that depleting cell cycle progression activators such as _CDK2_ and _CCNE1_ increased sensitivity to palbociclib18,25,26. AMBRA1 that has
been shown to lead to cyclin D1 stabilization and CDK4/6i resistance in a similar CRISPR screen27, was also a resistant hit in our CRISPR screen (Fig. 1C), demonstrating the robustness of
the screen. The top sensitising hit was _GPX4_, a lipid peroxidase that protect cells against oxidative damage and ferroptosis. Similarly, CRISPR-Cas9 targeting of _PCBP2_, an iron chaperone
that prevents ferroptosis by delivering Fe2+ to ferroportin, also increased sensitivity to palbociclib (Fig. 1C, E and Supplementary Fig. 1B). One of the top resistant hits was _AXTN7L3_,
which has been recently identified as a regulator of ferroptosis15. In addition, CRISPR-Cas9 targeting of _KEAP1_, an inhibitor of the NRF2 antioxidant pathway, lead to resistance to
palbociclib and has been shown to induce resistance to ferroptosis28,29,30. TARGETING GPX4 INCREASES SENSITIVITY TO PALBOCICLIB IN ER+ BREAST CANCER CELL LINES The screen data suggested that
CDK4/6 inhibition may create a GPX4-dependent state where cells rely on GPX4 to avoid ferroptosis. In the screen, five independent sgRNA targeting GPX4 increased sensitivity to palbociclib
(Fig. 1E). Next, we targeted _GPX4_ using the CRISPR Edit-R System with predesigned sgGPX4 (CM-011676). Three sgGPX4 led to greater sensitivity to palbociclib in MCF-7 cells (Supplementary
Fig. 1C, D). We then developed single-cell knockout clones from sgGPX4-02 transfected MCF-7 cells (Fig. 1F). The MCF-7 _GPX4_ null clone grew to a similar rate to parental MCF-7, but showed
greater sensitivity to palbociclib in clonogenic assays (Fig. 1G) and cell survival assays (Fig. 1H). Poly-clonal sgGPX4-02 transfected T47D cells showed reduced growth compared to
sgControl, and substantial sensitivity to palbociclib (Fig. 1I). We next developed a T47D _GPX4_ null clone (Fig. 1J) in the presence of ferrostatin-1 (an inhibitor of ferroptosis), due to
the higher sensitivity of T47D to GPX4 loss, which was highly sensitive to palbociclib when ferrostatin-1 was removed (Fig. 1K). The T47D GPX4 null clone was also highly sensitive to the
CDK4/6 inhibitor abemaciclib (Fig. 1L). Therefore, ER+ cancer models showed heterogenous sensitivity to depletion of GPX4, but with consistent sensitisation to CDK4/6 inhibition by GPX4
depletion. THE COMBINATION OF GPX4 AND CDK4/6 INHIBITORS IS MORE EFFECTIVE THAN THE SINGLE DRUGS IN ER+ AND TRIPLE NEGATIVE BREAST CANCER MODELS To further evaluate the potential of
targeting GPX4 to enhance CDK4/6 inhibitor sensitivity, we investigated the effect of combining GPX4 inhibitors with palbociclib in a panel of ER+ and _RB1_ wild type TNBC cell lines. To
inhibit GPX4, we used RSL3 and ML210 that covalently bind the selenocysteine residue (Sec46) of GPX4 leading to its inactivation and degradation31. There was a high degree of heterogeneity
in the response to the ferroptosis inducer RSL3 among breast cancer cell lines, with TNBC cell lines being more sensitive than ER+ cell lines as previously reported14 (Fig. 2A), and
conversely ER+ cells were more sensitive to palbociclib than TNBC lines (Fig. 2B). In ER+ models, MCF-7 cells were more resistant to RSL3 compared to T47D cells (Fig. 2A and Supplementary
Fig. 2A), mirroring the _GPX4_ CRISPR depletion experiments (Fig. 1G, K). The combination of RSL3 with palbociclib was highly synergistic (blue) in T47D and the TNBC cell line Cal51 (Fig.
2C). In MCF-7 cells, combination synergy was only achieved at higher concentrations of RSL3 (Fig. 2C), which possibly were required to fully inhibit GPX4 as synergy was more evident in fully
depleted GPX4 -MCF-7 clones (Fig. 1C, H). Abemaciclib and ribociclib showed similar synergy when combined with RSL3 (Fig. 2D and Supplementary Fig. 2B). Propidium Iodide (PI) staining
showed higher percentage of cell death with the combination of palbociclib plus RSL3 in MCF-7 and T47D cells (Supplementary Fig. 2C). We further extended this observation in a panel of
breast cancer cell lines doing clonogenic assays with 12–14 days treatment. Across ER+ and TNBC cell line models there was combination efficacy with palbociclib plus RSL3 (Fig. 2E and
Supplementary Fig. 2D) independently of their degree of sensitivity to the single drugs. Some TNBC cells were highly sensitive to RSL3 alone such as Sum149, Sum159, Sum185PE and BT20 (Fig.
2E and Supplementary Fig. 2C). Therefore, inhibition of GPX4 enhanced the activity of CDK4/6 inhibition in ER+ and TNBC models. CYTOSTASIS INDUCED BY CDK4/6 INHIBITORS INCREASES LIPID
PEROXIDATION LEADING TO A GPX4 DEPENDENT STATE Ferroptosis can be triggered by iron dependent lipid peroxidation of PUFAs, which disrupts membrane integrity when is not prevented by
anti-ferroptosis defences such as GPX413,32. We investigated whether palbociclib induces lipid peroxidation as a marker of ferroptosis vulnerability. General, and mitochondrial, lipid
peroxidation was measured by C11-BODIPY (581/591) and Mito-PerOx (581/591) sensors, respectively, which shift fluorescence from red (590 nm) to green emission (510 nm) when they are
oxidised. Palbociclib led to BODIPY and Mito-PerOX oxidation in FACS and microscopy analysis in MCF-7, T47D and Cal51 cells (Fig. 3A–C and Supplementary Fig. 3A–C). The combination of
palbociclib with the GPX4 inhibitors RSL3 and ML210 lead to further increase in lipid peroxidation. Palbociclib did not induce lipid peroxidation in a palbociclib resistant cell line
T47DpR18 that fails to arrest due to RB1 loss (Fig. 3C). In addition, we did not observe combination efficacy of palbociclib plus RSL3 in clonogenic assays performed in T47DpR, T47DpR-B (RB1
loss) (Fig. 3D) and MCF-7pR (cyclin E1 amplification)18 (Supplementary Fig. 3D). These results suggest that the promotion of a ferroptosis-vulnerable stage may be dependent on cell cycle
arrest, rather than CDK4/6 inhibition per se. Anti-ferroptosis compounds such as TEMPO, liproxstatin-1, and ferrostatin-1 did not rescue the palbociclib effect of reduced clonogenic growth,
indicating that palbociclib alone doesn’t induce ferroptosis (Fig. 3E and Supplementary Fig. 3E and Fig. 3F). Anti-ferroptosis compounds did rescue the effects of inhibiting or depleting
GPX4, and rescued the combination to levels similar to what palbociclib treatment achieves as a single agent. Similarly, ferrostatin-1 prevented cell death induced by RSL3 alone and in
combination with palbociclib (Fig. 3F). These experiments confirmed that ferroptosis was the mechanism of cell death induced by the addition of GPX4 inhibition to palbociclib, which resulted
in greater cell death than the single treatments. In summary, our results suggest that palbociclib induces lipid peroxidation sensitising cells toward ferroptosis, which only occurs when
the protection exerted by GPX4 is inhibited. Proteomics analysis showed that palbociclib treatment led to reduced GPX4 levels and we further explored this observation (Supplementary Fig.
4A). ER+ cells had substantially higher GPX4 levels than the RSL3-highly sensitive TNBC cell lines Sum149, Sum159, and BT20 (Fig. 3G), potentially consistent with the general resistance of
naïve ER+ cell lines to ferroptosis induction14. In accordance, ER + MCF-7 cells that are innately more resistant to the ferroptosis inductor RSL3, had higher levels of GPX4 compared to T47D
(Fig. 3G, H). CDK4/6 inhibition with palbociclib substantially reduced GPX4 and ferritin levels (Fig. 3H–K). Depletion of GPX4 has been shown to substantially sensitise to ferroptosis
induction11, and degradation of ferritin may increase free Fe2+ production leading to ferroptosis susceptibility33,34. The combination of palbociclib with RSL3 further suppressed GPX4
expression (Fig. 3I and Supplementary Fig. 4B). Palbociclib did not reduce GPX4 in MCF-7pR and T47DpR (Fig. 3J and Supplementary Fig. 4C), and antioxidants such as ferrostatin-1,
liproxstatin-1 and TEMPO did not rescue GPX4 reduction induced by palbociclib. (Fig. 3K), pointing cell cycle arrest as the main mechanism leading to reduced GPX4 levels. Noteworthy,
palbociclib treatment did not affect _GPX4_ transcription, but accelerated GPX4 degradation in a cycloheximide chase experiment (Supplementary Fig. 4D–F). This suggested that the observed
reductions in GPX4 protein levels were likely due to a post-translational mechanism. Dihydroorotate dehydrogenase (DHODH) protects against mitochondrial ferroptosis independently of GPX435.
However, treatment of MCF-7 and T47D cells with the DHODH activator DHO did not rescue palbociclib, RSL3, or combination-induced cell death. Hence, the observed vulnerability to ferroptosis
in the context of CDK4/6 inhibition appeared to be independent of DHODH activation (Supplementary Fig. 4G). WHOLE GENOME CRISPR SCREEN REVEALS THAT GPX4 LOSS INCREASES SENSITIVITY TO THE ER
ANTAGONIST AND DEGRADER GIREDESTRANT Clinically, palbociclib and other CDK4/6 inhibitors are used as standard of care in combination with endocrine therapies. Therefore, we expanded our
CRISPR screening efforts to identify determinants of giredestrant response (Supplementary Fig. 5A). We observed striking parallels in both resistance and sensitivity hits across the
palbociclib and giredestrant screens, consistent with the convergence of ER and CDK4/6 signalling on cell cycle regulation. In particular, loss of function _PTEN_, _CDKN1A_, _STK11_, _RB1_
and _KEAP1_ were identified as mediating resistance to giredestrant (Fig. 4A, Supplementary Fig. 5B). These resistance-associated genes also overlapped with those identified in an additional
genetic screen seeking to identify drivers of estrogen-independence - with _CSK_ also as a shared top hit – suggesting that the identified genes are relevant for general endocrine
resistance (and perhaps G1 arrest), rather than representing a giredestrant-specific resistance program36. Notably, loss of function _GPX4_ was the top hit that enhances sensitivity to
giredestrant, in line with the palbociclib screen. _PLAA_, encoding the phospholipase A2 activating protein, was an additional gene whose loss of function had a pronounced effect on
enhancing sensitivity to giredestrant. Like GPX4, phospholipase A2 (PLA2) family members have been implicated in modulating ferroptosis sensitivity. PLA2 proteins act to hydrolyse the ester
bonds of phospholipids to release free fatty acids, including arachidonic acid, a key substrate for phospholipid peroxidation37,38. Next, we sought to address if giredestrant increased
dependence on GPX4 in additional ER+ breast cancer cell lines. We thus conducted a matrixed dose response of giredestrant and RSL3 in ER+ breast cancer cell lines that had been pre-treated
with either vehicle (DMSO) or giredestrant for 7 days. In DMSO pre-treated T47D cells, single agent giredestrant achieved a maximal saturating inhibition of approximately 40% (Fig. 4B).
Co-treatment with RSL3 improved the inhibitory effect of giredestrant, with the combination being synergistic, similar to the observations made for the combined activity of palbociclib and
RSL3 (Fig. 4B, see Supplementary Data 1 for full matrixed dose response and BLISS synergy scores). Moreover, giredestrant reduced colony formation in MCF-7-GPX4 null cells treated with
vehicle or palbociclib (Supplementary Fig. 5C) The activity of RSL3, both as a single agent and in combination with giredestrant, was inhibited by the synthetic antioxidant Ferrostatin-1
(Fer-1), while single agent giredestrant activity was maintained, supporting that RSL3 pushes vulnerable cells toward ferroptosis. Pre-treatment of T47D cells with giredestrant profoundly
sensitized T47D cells to single agent RSL3, driving down the concentration of RSL3 required to impact cell viability (Fig. 4C, Supplementary Data 1). CAMA1 and EFM-19 cells display
differential sensitivity to single agent RSL3 and the RSL3 plus giredestrant combination in DMSO pre-treated cells, but both were sensitized to RSL3 following giredestrant pre-treatment. In
HCC1500 cells, the combination activity of RSL3 with giredestrant, both in DMSO and giredestrant pre-treatment conditions, was additive rather than synergistic (Fig. 4C). Together, these
data suggested that the combined action of giredestrant with GPX4 inhibition is highly active across multiple ER+ cell lines, strikingly resembling the palbociclib-GPX4 inhibition synergy.
GIREDESTRANT INCREASES PUFA-CONTAINING PHOSPHOLIPIDS AT THE EXPENSE OF MUFA-PHOSPHOLIPIDS We investigated the mechanism of GPX4 dependency in giredestrant treated cells, with transcriptional
profiling of MCF-7 cells that had been transduced with either control sgRNAs (targeting olfactory receptor genes OR51Q1 and OR5M9), or GPX4 sgRNAs. Cells were harvested 10 days following
either vehicle (DMSO) or giredestrant treatment. Perturbation of _GPX4_ in the DMSO-treated condition had little impact on the transcriptome, while its perturbation had a profound effect on
cells in the presence of giredestrant (Fig. 4D, E). In particular, genes reflecting an oxidative stress response, for example _SLC7A11_ and _HMOX1_, were profoundly upregulated upon _GPX4_
perturbation in cells treated with giredestrant, and only minimally in DMSO treated cells. HALLMARK gene set enrichment analysis (GSEA) identified suppression of Estrogen Response genes, E2F
targets and other proliferation-associated gene sets as being suppressed by giredestrant, consistent with its anticipated effects as an ER antagonist (Supplementary Fig. 5D). Perturbation
of _GPX4_ in cells that were simultaneously treated with giredestrant revealed a distinct transcriptional response, with induction of “Reactive Oxygen Species” and “Fatty Acid Metabolism”
gene sets, among others. These gene sets were not enriched in _GPX4_ perturbed cells in the absence of giredestrant, consistent with the screen results that identified GPX4 as a
giredestrant-dependent vulnerability (Supplementary Fig. 5D). Sensitivity to ferroptosis results from peroxidation of PUFAs-PLs. We thus investigated whether giredestrant altered PL
homoeostasis with a time course analysis of MCF-7 cell lipid profile. Giredestrant had little impact on the relative abundance of PL species 48 h after treatment. However, cells displayed a
marked change in fatty acid saturation in PLs and glycerolipids after 7 days of exposure to giredestrant, that was maintained 14 days following treatment (Fig. 4F). Specifically, MUFA-PLs
decreased upon giredestrant treatment while both saturated-PLs and PUFA-PLs were elevated, relative to vehicle control. Increased PUFA-PLs include species that are major substrates for
ferroptosis, for example phosphatidylethanolamine-linked arachidonic acid [PE(FA20:4)]39. Arachidonic acid (AA)-linked ePL (aka plasmalogen) species, for example PE(P-16:0/20:4), are also
increased upon giredestrant treatment, as are PUFA-triacylglycerols (TAGs) (Fig. 4F, Supplementary Fig. 5E). Giredestrant thus appeared to provoke, in MCF-7 cells, a broad remodelling of
lipids towards PUFA-containing species, specifically after longer duration (7 and 14 day) treatment. In addition to changes in the saturation of ePLs and TAGs, giredestrant treatment also
resulted in a global decrease in diacylglycerols (DAGs) and hexosylceramide (HCER), and an elevation of sphingomyelin (SM) (Supplementary Fig. 5F). PALBOCICLIB COMBINATION WITH GIREDESTRANT
ENHANCES THE ACCUMULATION OF PUFA-PHOSPHOLIPIDS COMPARED TO THE SINGLE TREATMENTS With the converging discoveries that GPX4 inhibition sensitised cells to both palbociclib and to
giredestrant, we investigated the role of GPX4 in the giredestrant-palbociclib combination setting. We first assessed the impact of giredestrant and a sub-saturating (200 nM) concentration
of palbociclib, intended to model the clinical scenario, as single agents and in combination, on the lipid profile of both MCF-7 and T47D cells 7 days after treatment. Principal component
analysis of the global lipidome showed that approximately 80% of the variance across samples can be attributed to the distinct lipid profiles of the two cell lines at baseline (Fig. 5A).
Despite their differences under basal conditions, drug treatments altered the lipid profiles of MCF7 and T47D cells in a similar manner, with 200 nM palbociclib at the 7 day time point
having a less pronounced effect than giredestrant, and with the greatest lipid changes occurring with the combination of palbociclib plus giredestrant (Fig. 5A). This experiment reproduced
our prior observation that giredestrant elevates the levels of AA-linked phosphatidylcholine [PC(FA20:4)] and PE(FA20:4), and further extends this finding to T47D cells, while also
suggesting that a giredestrant-palbociclib combination enhances this effect (Fig. 5B). PUFA-ePLs were likewise elevated by giredestrant and/or palbociclib in both cell lines, with the
combined action of both drugs generally driving greatest increases in individual PUFA-linked ePL species (Fig. 5C). Treatment of MCF-7 and T47D cells with giredestrant and palbociclib
altered the PUFA/MUFA ratio of both PC and PE, favouring PUFAs (Fig. 5D, E). TRIPLET COMBINATION OF GIREDESTRANT, PALBOCICLIB, AND GPX4 INHIBITION PREVENTS CLONAL OUTGROWTH EVEN AFTER SHORT
EXPOSURE TO TREATMENT The combination of CDK4/6 inhibition with endocrine therapies has been transformative for patients with ER+ breast cancer. However, both targeted therapies converge on
a G1 cell cycle arrest and drive limited cell death, suggesting that a sustained drug exposure is required to maintain tumour control. Given our observations that giredestrant and
palbociclib sensitize to regulators of ferroptosis, a cell death pathway, we tested whether RSL3 short exposure treatment combinations would eradicate tumour cells. T47D cells were
pre-treated with either DMSO, giredestrant, palbociclib or their combination for 7 days. Pre-treated cells were then re-seeded and treated either continuously for 18 days or for only 3 days
followed by a washout of drugs; cell confluency was then analysed at the same 18-day endpoint (Fig. 5F–M). For continuous treatment, the doublet combinations of giredestrant-RSL3,
giredestrant-palbociclib, and the triplet giredestrant-palbociclib-RSL3, ablated colony formation. In contrast, a short exposure to giredestrant or the giredestrant-palbociclib combination
did not prevent colony formation following washout (Fig. 5G, K, right side). Importantly however, RSL3-containing combinations displayed growth control even after drug washout, with the
triple combination being most effective and independent of the pre-treatment regime, consistent with having a cell lethal effect (Fig. 5K). Notably, cells pre-treated with giredestrant alone
or the giredestrant-palbociclib combo were particularly sensitive to a short-term exposure of the RSL3 single agent (Fig. 5H, L). Taken together, following a pre-treatment of cells with
giredestrant plus palbociclib, cell outgrowth was sustainably inhibited via short-duration GPX4 blockade, consistent with an induced ferroptosis phenotype. LIPID PEROXIDATION AND FERROPTOSIS
IN ER+ BREAST CANCER CELL LINES IS NOT DEPENDENT ON ACSL4, BUT PROMOTED BY FAR1 AND AGPAT3 We next investigated in more detail the potential molecular mechanisms of ferroptosis
vulnerability induced by palbociclib in ER+ cell lines, by performing transcriptional and global proteomic profiling in MCF-7 cells treated with vehicle or palbociclib. RNA sequencing
analysis revealed that palbociclib treatment induces features previously reported in senescence cells40, with top upregulated Enricher terms being “senescence and autophagy”, “cytokines and
inflammatory response”, “PI3K-AKT-mTOR survival pathway” and “TGF-beta regulation of extracellular matrix” (Fig. 6A Supplementary Fig. 6A, B). We also observed enrichment of the terms
“biological oxidation”,“oxidative stress”, and “ferroptosis” that included genes encoding arachidonate 5-lipoxygenase (ALOX5) and cytochrome P450 monooxygenases (CYPs family), both of which
can promote lipid peroxidation10,39,41,42 (Fig. 6A and Supplementary Fig. 6A, B). In relation, proteins involved in peroxisome beta-oxidation of fatty acids, such as acetyl-coenzyme oxidases
(ACOXs) and CROT, were upregulated in the proteomic data (Fig. 6B and Supplementary Fig. 6C, D)16,39. Genes involved in antioxidant or detoxification defence were also enriched in the terms
“NRF2 pathway” and “glutathione metabolism”10, confirming a redox unbalance upon palbociclib treatment (Fig. 6A and Supplementary Fig. 6A, B). As reported previously we also observed
reduction of GPX4 levels in the proteomics data (Fig. 6D). In addition, palbociclib led to the upregulation of genes and proteins involved in lipid metabolism, including “glycerophospholipid
and triacylglycerol (TAG) biosynthesis”, “sphingolipid synthesis”, “arachidonic acid metabolism”, and “peroxisomal lipid metabolism” correlating with the lipidomic findings (Supplemental
Fig. 6A–D). Specifically, palbociclib treatment resulted in the upregulation of the proteins DGAT1 and AGPAT3, which regulate the synthesis of TAG43, the major constituents of glycerolipids
that can lead to PUFA-TAGs or PUFA-PLs accumulation (Fig. 6B, C). FAR1 and AGPAT3 have been shown to regulate the synthesis of plasmalogen (ether-glycerophospholipids) containing PUFA
(PUFA-ePL) in a peroxisome dependent pathway that promote ferroptosis15. PUFA content in phospholipids (PUFA-PL) are classically catalysed by the ACSL4 pathway10,14. However, our panel of
ER+ cell lines have low levels of ACSL4 compared to the TNBC cells lines (Fig. 6E). Moreover, palbociclib further reduced ACSL4 in MCF-7 and T47D (Fig. 6F). Silencing ACSL4 (Fig. 6I) rescued
clonogenic growth (Fig. 6G) and lipid peroxidation (Fig. 6H) induced by RSL3 or the combination in the TNBC cell line Cal120, but had little impact in the ER+ cell lines T47D. Therefore,
the ACSL4-PUFA pathway is likely not involved in lipid peroxidation in ER+ cells lines, whereas it is in TNBC cell lines with higher ACSL4 levels. The transcription and proteomic data
suggested that the generation of PUFA-PLs in ER+ cells may be catalysed by a peroxisome dependent pathway (Fig. 6B). In this pathway, peroxisome enzymes including FAR1 act to synthesize an
ether lipid precursor, and endoplasmic reticulum enzymes such as AGPAT3 catalyse the synthesis of PUFA-ePLs (or PUFA-plasmalogens)15. To study their role in ferroptosis in ER+ breast cancer
models, we depleted AGPAT3 and FAR1 using siRNA SMARTpools or a CRISPR sgRNA approach and treated MCF-7 and T47D cells with palbocicilib, RSL3, or the combination in clonogenic assays (Fig.
7A and Supplementary Fig. 7C) and cell survival assays (Supplementary Fig. 7A, B). Depleting AGPAT3 desensitised the ER+ cell lines MCF-7 and T47D to RSL3 (Fig. 7A, and Supplementary Fig.
7A), however did not affect the TNBC cell line Cal51 (Supplementary Fig. 7B). Targeting FAR1 likewise induced resistance to RSL3 in T47D cells, however with no effect in MCF-7 cells (Fig. 7A
and Supplementary Fig. 7C). Moreover, RSL3 induced lipid peroxidation was reduced by depletion of AGPAT3 in MCF-7 cells, and depletion of AGPAT3 and FAR1 in T47D cells, in BODIPY-C11
fluorescence assays (Fig. 7B). Therefore, lipid peroxidation and ferroptosis was dependent on AGPAT3 in MCF-7 cells, and FAR1-AGPAT3 in T47D cells, implicating the peroxisome pathway, and
not the classical ACSL4 pathway, in ferroptosis vulnerability. ATXN7L3 was a top resistant hit in the palbociclib MCF-7 CRISPR screen (Fig. 1B). AXTN7L3 has been shown to modulate global
transcription as part of histone acetylation (HAT) complex SAGA, which remodels chromatin and mediates histone acetylation and deubiquitination44,45. PSDM4, a component of the proteasome
complex, also modulated sensitivity to palbociclib (Supplementary Fig. 7D). Depletion of ATXN7L3 induced resistance to RSL3 and palbociclib, reduced lipid peroxidation induced by RSL3, and
led to upregulation of GPX4 and ferritin (Supplementary Fig. 7E–G). These data suggests that ATXN7L3 is a pro-ferroptosis modulator in breast cancer cells, linking additional screen hits to
ferroptosis regulation. MCF-7 GPX4 NULL XENOGRAFT TUMOURS SHOW HIGHER SENSITIVITY TO PALBOCICLIB Finally, we investigated whether depletion of GPX4 would increase palbociclib sensitivity in
MCF-7 xenografts. MCF-7 and MCF-7 GPX4 null cells were injected subcutaneously into the mouse flanks, and tumours that reached 150–200 mm3 were randomised to vehicle or palbociclib
treatment. We used a palbociclib dose of 25 mg/kg that had a moderate effect on MCF-7 growth but led to a significant reduction of tumour size in MCF-7 GPX4 null xenografts compared to those
treated with vehicle (Fig. 7F, G), confirming that palbociclib induced dependency on GPX4 in vivo. Western blots of residual tumour samples after 34 days treatment (Fig. 7H), revealed that
the combination of palbociclib with GPX4 depletion profoundly reduced phospho-RB1, phospho-CDK2, p21 and cyclin D1 compared to palbociclib alone, indicating an acute arrest. The combination
of palbociclib with GPX4 depletion also repressed SLC7A11 and increased ALOX5, as markers of ferroptosis33,46. SLC7A11 is an essential components of the glutathione (GSH)-glutathione
peroxidase 4 (GPX4) antioxidant systems that prevents ferroptosis, while ALOX5 is a member of the lipoxygenase family that is closely related to lipid peroxidation and induction of
ferroptosis10,33. Apoptosis markers such as cleavage PARP and Caspase 3 were reduced with the combination47,48. DISCUSSION The introduction of CDK4/6 combination therapies in ER+ breast
cancer has been enormously beneficial for patients but requires sustained treatment to maintain tumor control. The limited cell death that occurs in the face of ER and CDK4/6 inhibition may
also allow for cell adaptation and acquisition of resistance. Here, we show that giredestrant, palbociclib, and their combination, induced a ferroptosis vulnerable state, with enhanced PUFA
synthesis and lipid peroxidation. Combinations with GPX4 inhibition leads to ferroptosis in ER+ breast cancer models, and the triplet combination of palbociclib, giredestrant, and GPX4
inhibition was highly effective in eradicating cancer cells and preventing clonal outgrowth. In addition, combination of CDK4/6 and GPX4 inhibition sensitised multiple TNBC models,
suggesting that this approach may extend the types of breast cancer targetable with CDK4/6 inhibitors. Combining GPX4 and CDK4/6 inhibition leads to the collapse of the glutathione
(GSH)-glutathione peroxidase 4 (GPX4) antioxidant systems that fails to protect against ferroptosis, and it is accompanied by a deep reduction of cell cycle and apoptosis markers33,47,48.
Our results suggest that the promotion of a ferroptosis-vulnerable stage induced by CDK4/6 and ER inhibitors, is dependent on cell cycle arrest, which leads to a redox-lipid imbalance with
increased PUFA generation, reduced GPX4 and ferritin levels, and increased lipid peroxidation (Fig. 7E). Peroxisomes, in conjunction with endoplasmic reticulum enzymes, have recently been
implicated in the synthesis of ether-liked glycerophospholipids known as plasmalogens that can bind to PUFAs (PUFA-ePL), which are susceptible of peroxidation at the cell membrane15,49.
Lipidomics analysis revealed that giredestrant and palbociclib remodel the lipidome in favour of PUFA-phospholipids, including PUFA-ePL species. In relation, proteomic data demonstrate that
palbociclib upregulates enzymes involved in PUFA-ePL synthesis such as FAR1 and AGPAT3 in ER+ models. AGPAT3 promoted RSL3 induced lipid peroxidation and ferroptosis in the ER+ cell lines
T47D and MCF-7, although was dispensable in the TNBC Cal51 cells line that have higher levels of ACSL4. The ACSL4 pathway has been known as the classical way to enhance PUFA content in cell
membranes, being more predominant in TNBC as shown by us and others, which was thought to explain their higher sensitivity to ferroptosis14. Here we show that, in ER+ breast cancer cell
lines, lipid peroxidation is ACSL4 independent and required the action of AGPAT3, suggesting that there may be multiple, potentially redundant mechanisms that regulate PUFA-lipid content and
ferroptosis susceptibility in breast cancer cells. The lipidomic data highlights that innate resistance to ferroptosis, as observed in specific cell lines such as MCF-7, could be attributed
to a distinct lipid profile-metabolism. We noticed that MCF-7 cells are more effective at accumulating PUFA-TAGs (Fig. 5A, B) and upregulate DGAT1 (Fig. 6B). It has been shown that cell
cycle arrest-induced DGAT leads to the sequestration of PUFAs into TAG-lipid droplets, which protects against ferroptosis50. Therefore, a balance toward PUFA-TAG may have a protective effect
in MCF-7 cells. However, treatment with CDK4/6 and ER inhibitors, and specifically their combination, seems to turn the balance toward PUFA-PL accumulation and ferroptosis sensitivity.
Lipid peroxidation seems to be promoted by cell cycle arrest, as it was not observed in palbociclib resistant cell lines with an _RB1_ loss or _CCNE1_ amplification. Indeed, recent work has
likewise implicated cell cycle arrest, induced either by p53 activation or CDK4/6 inhibition, in sensitizing fibrosarcoma cells to GPX4 inhibition51. Mechanistically, it was proposed that
cell cycle arrest resulted in suppression of MBOAT1 and EMP2, leading to elevated PUFA-ePLs and thus a ferroptosis-susceptible state. MBOAT1 was also identified in a recent genetic screen
for ferroptosis suppressors, and was shown to be positively regulated by ER signalling in ER+ breast cancer cells52. Similar to what we report, treatment of ER+ breast cancer cells with the
ER antagonist and degrader fulvestrant sensitized to GPX4 inhibition. The induction of a ferroptosis vulnerable state with PUFA accumulation/lipid peroxidation was more evident after 5-14
days of treatment, correlating with the time required to establish a quiescent/senescent phenotype53,54. In relation, oxidative stress and lipid remodelling is a known feature of senescence
cells55. The current clinical schedule for palbociclib allows enough time for the cancer cells to reach this ferroptosis vulnerable state, as it is based on three weeks of administration
followed by a one week drug holiday56. Our study observed potential differences between giredestrant and palbociclib with respect to PUFA-PL production. Giredestrant treatment over 7 days
resulted in greater induction of PUFA-ePLs than palbociclib (Fig. 5A–E), and pre-sensitised cells to GPX4 inhibition to a greater extent than palbociclib (Fig. 5G–N). However, these
differences may reflect the distinct timing and extent to which palbociclib and giredestrant achieve cell cycle arrest and senescence, due to the kinetics of drug action, or drug
concentrations used. Our data identifies a number of potential avenues for future investigation. RNA sequencing of giredestrant and palbociclib treated cells, and proteomic analysis of
palbociclib treated cells identified pathways consistent with oxidative stress and increased lipid peroxidation (Fig. 6A). MCF-7 cells showed increased expression of ALOX5 with palbociclib,
an iron-dependent enzyme that can promote lipid peroxidation and ferroptosis10,39,41,42. Oxidating pathways regulated by cytochrome P450 monoxygenases (CYPs) and aldehyde dehydrogenases
(ALDH) were also upregulated (Fig. 6A). Moreover, proteomic analysis revealed upregulation of peroxisome beta-oxidation of fatty acids by acetyl-coenzyme oxidases (ACOXs), which through the
generation of free radicals, can trigger lipid peroxidation in a non-enzymatic or Fenton reaction10,16,39,41,42. In addition, palbociclib led to a reduction of GPX4 and ferritin levels that
likely contributed to increase ferroptosis vulnerability. Down-regulation of GPX4 has been shown to sensitise to GPX4 inhibitors in prior studies11. GPX4 reduction was promoted by cell cycle
arrest and likely through a post-transcriptional mechanism. We have identified a strategy to enhance the efficacy of CDK4/6 and ER inhibition and extend the potential role of promoting
ferroptosis as an anti-cancer strategy in ER+ and potentially TN breast cancers. In the context of ER+ breast cancer treatment, the triplet combination of CDK4/6 inhibition with palbociclib,
ER inhibition with giredestrant and GPX4 inhibition was highly efficacious, inducing cell lethality and preventing clonal outgrowth. Our work emphasises the importance of developing
drug-like GPX4 inhibitors, to investigate exploiting this vulnerability in the clinic. MATERIALS AND METHODS CELL LINES MCF-7 (HTB-22), T47-D (HTB-133), MDA-MB175VII (HTB-25), HCC1428
(CRL-2327), and BT-483 (HTB-121) cell lines were obtained from American Type Culture Collection (ATCC), MFM-223 (ACC 422), Cal-519 (ACC 302), and Cal-120 (ACC 459) cell lines were from
Leibniz Institute DSMZ, and Sum149PT (HUMANSUM-0003004), Sum159PT (HUMANSUM-0003006) cell lines were from Bioivit. All cell lines maintained according to the supplier’s instructions. Cell
lines were banked in multiple aliquots on receipt to reduce risk of phenotypic drift, and identity confirmed by STR profiling with the PowerPlex 1.2 System (Promega). STR profiling at
Genentech was performed using the PowerPlex 16 System (Promega). All stocks are tested for mycoplasma prior to and after cells are cryopreserved. Two methods are used to avoid false
positive/negative results: Lonza Mycoalert and Stratagene Mycosensor. COMPOUND SOURCES AND TREATMENT CONDITIONS Palbociclib (PD0332991)-Isethionate (S1579), RSL3 (S8155), ML210 (S0788) and
Liproxstatin-1 (S7699) were obtained from Selleckchem. Ferrostatin-1 (SML-0583), L-Dihydroorotic acid (DHO, D7128) and TEMPO (2564-83-2) were from Sigma-Aldrich. C11-BODIPY581/591 dye
(D3861) was from ThermoFisher Scientific and MitoPerOx dye (ab146820) was obtained from Abcam. Giredestrant was synthesized as previously described7. ANTIBODIES The following primary
antibodies were used: anti-GPX4 (#52455), anti-SLC7A11 (#12691), anti-phosphoSirT1 (Ser47) (#2314), anti-phosphoCDK2 (Thr160) (#2561), anti-PhosphoRB1 S807/811 (#8516), anti-cleaved PARP
(#5625), anti-cleaved Casp3 (#9664), anti- Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (#4370), Anti- 5-Lipoxygenase (ALOX5) (#3289) from Cell Signaling Technology at a 1:1000 dilution.
Anti-FAR1 (Novus Biologicals, LLC, NBP1–89847, 1:1000 dilution), anti-AGPAT3 (abcam, ab211435, 1:500), monoclonal Anti-β-Actin antibody (Sigma, A5441, 1:5000), anti-ACSL4 (Santa Cruz
Biotechnology, sc-365230, 1:1000 dilution), anti-ferritin (abcam, ab75973, 1:2000), anti-ATXN7L3 (abcam, ab99947, 1:1000). The following secondary antibodies were used: HRP-linked
anti-rabbit IgG (Sigma A99169,1:10000 dilution), HRP-linked anti-mouse IgG (Sigma A9044,1:10000 dilution). GENOME-WIDE CRISPR/CAS9 SCREEN: PALBOCICLIB SENSITIZATION AND RESISTANCE A
doxycycline-inducible Cas9-expressing MCF-7 iCas9 clone was generated by transduction of MCF-7 cells with the Edit-R Inducible Lentiviral hEF1a-Blast-Cas9 Nuclease (Dharmacon) and selected
in 10 μg/ml blasticidin for 5 days (MCF-7iCas9). Cas9 expression was confirmed in western blot after treatment with vehicle or 2 μg/ml doxycycline. For the screen, 300 × 106 MCF-7 iCas9
cells were infected at a multiplicity of infection (MOI) of 0.3, aiming for 1000 cells per individual sgRNA, with a previously published and validated genome-wide human lentiviral CRISPR
library17. Transduced cells were selected with 2 µg/mL puromycin initially for 2 days, followed by 4 days of puromycin and doxycycline, at that point a T0 sample was taken. 100 × 106 cells
were exposed to palbociclib 500 nM or vehicle for 2 weeks, before T1 was taken. DNA was extracted from samples T0 and T1, and CRISPR guide regions amplified by PCR, and sgRNA in each sample
were sequenced using a U6 custom primer57 on the HiSeq (Illumina) to generate gRNA count data. Pre-processing and quantification of CRISPR screens data was performed as previously
described57. For prioritisation of genes, we used normZ which aggregates sgRNA z-scores to gene level by normalising to the number of sgRNAs as previously explained58. GENOME-WIDE
CRISPR/CAS9 SCREEN: GIREDESTRANT SENSITIZATION AND RESISTANCE MCF-7 cells were transduced with lentiviral vector pLenti6.3 encoding _S. pyogenes_ Cas9 and BlastR genes, selected with 10
µg/ml blasticidin, and single-cell cloned to isolate a clone showing 96% CD81-negative cells by flow cytometry after transduction with lentiviral CD81 sgRNA. A sgRNA library targeting all
protein-coding genes in the human genome at a complexity of 8 sgRNAs per gene was designed and produced in 8 plasmid pools, as described by Callow et al. 59, for an analogous mouse sgRNA
library. The sgRNA sequences are available in Supplementary Data 2. Library sgRNA plasmid pools were packaged into lentivirus and titered in the screening cell line as described by Callow et
al. 59. For the screen, lentiviral stocks of the 8 sub-library pools were combined in proportion to their viral titers. MCF-7 Cas9-expressing cells were transduced in independent triplicate
samples of 1.73 × 108 cells each with the lentiviral whole-genome sgRNA library at a multiplicity of infection of 0.25-0.29, as measured by puromycin resistance, to achieve a representation
of 252–287 cells per sgRNA. All cell culture and passaging was performed on the CompacT SelecT (Sartorius Stedim Biotech) automation system in barcoded, triple-layer T500 flasks (132925,
ThermoFisher Scientific). Two days after transduction (day 2), 1.60 × 108 cells were collected for a reference time point from each replicate, and 1.73 × 108 cells per replicate were
passaged for selection with 2 µg/ml puromycin starting on day 3. 8.05 × 107 cells per replicate were then passaged without puromycin on days 6 and 10. For each replicate on day 13, 8.00 ×
107 cells were collected for a baseline time point, and three sets of 8.05 × 107 cells were plated for treatment the next day (treatment day 0), two sets with 0.6 nM giredestrant and one set
with an equivalent amount of diluent DMSO. For giredestrant treatments, 8.05 × 107 cells were passaged on treatment day 3 into fresh 0.6 nM giredestrant, compound and medium were
replenished every 3–4 days, and one set of cells for each replicate was collected on treatment days 10 and 17 (7.00 × 107 to 8.00 × 107 cells per sample). For DMSO control treatments, 8.05 ×
107 cells were passaged on treatment days 3, 6, 10, and 13. In addition, 8.00 × 107 cells were collected on treatment days 10 and 17. The mean growth inhibition since treatment day 0
achieved for the replicates with giredestrant was 56% at treatment day 10 and 83% at day 17. For all cell collections, genomic DNA isolation, PCR amplification of the sgRNA sequences,
amplicon purification, and next-generation paired-end sequencing were performed according to Callow et al. 59. Sufficient genomic DNA was used for PCR amplification to maintain a
representation of 280-511 cells per sgRNA for all baseline and treated cell samples (191-455 cells per sgRNA for reference cell samples). For each read pair, both read sequences were
searched for an exact match of any one of the sgRNA barcode sequences, using the ScreenCounter R package available from the crisprVerse60. The number of reads matched to each guide in each
sample was used to obtain a guide-by-sample count matrix for further analysis. The raw count data were stored in a standard Bioconductor SummarizedExperiment object61. Normalisation is
needed to adjust for the difference in sequencing depth between samples, and to account for potential compositional biases due to the competitive nature of pooled genetic screening. We
estimated normalisation factors for each sample by applying the TMM method62 on the count data for sgRNAs targeting a gold-standard set of non-essential genes63. The sequence reads across
all samples achieved a mean sgRNA count of at least 546, with less than 2.2% of sgRNAs showing complete depletion (Supplementary Data 2). We performed a differential abundance analysis at
the sgRNA level using the popular limma-voom approach64. Specifically, we fitted a linear model to the log-CPM values for each sgRNA, using voom-derived observation and quality weights. We
performed robust empirical Bayes shrinkage to obtain shrunken variance estimates for each sgRNA, and we used moderated F-tests to compute p-values for each of the two-group comparisons of
interest. To control the FDR in each comparison, we applied the standard Benjamini-Hochberg method to obtain an adjusted p-value for each sgRNA (Supplementary Data 2). We used sgRNA-level
statistics to obtain gene-level summaries using two complementary statistical approaches. The first approach was to aggregate sgRNA statistics by reusing the “fry” gene-set enrichment
analysis method implemented in limma, and considering sgRNAs targeting a given gene as a “gene set” (Supplementary Data 3). This allows the detection of genes that are consistently enriched
or depleted for the majority of the sgRNAs designed for each gene. The second approach uses Simes’ method (Simes, 1986) to obtain a combined p-value for each gene based on the p-values for
all associated sgRNAs (Supplementary Data 3). This allows the detection of differentially abundant genes in cases where only a small proportion of the sgRNAs show a strong signal. Gene-level
p-values were corrected for multiple comparisons using the Benjamini-Hochberg method. In addition, DrugZ v1.1.0.258 was run using default parameters on the raw sgRNA read counts for each
comparison of interest across the three available replicates. DrugZ computes Z-scores of each sgRNA for the fold change between the replicate-matched giredestrant and DMSO treatments.
Z-scores are then aggregated at the gene-level into a normalised Z-score (normZ; Supplementary Data 358;). NormZ scores, fry method p-values, and Simes’ method p-values were used to identify
top screen hits for sgRNA depletion and enrichments. CELLULAR VIABILITY ASSAYS Cells were seeded in 384-well plates at an approximate number of 600–1000 cells per well. Drugs were added 24
h after seeding at the indicated concentrations and plates were incubated at 37 C for 6 days. Viability was estimated using CellTiter-Glo luminescence reagent (CTG, Promega). Final
luminescence intensity value was normalised to DMSO median and surviving fractions of cells were plotted where lines of best fit were drawn using a four-parameter nonlinear regression.
Comparisons of dose–response curves were performed using two-way ANOVA testing. Combenefit 2.021 was used to quantify the synergy using the 384-well plate dose-response data. Combenefit
assesses synergy/antagonism from dose-response data using three classical models, namely the Loewe, the Bliss, and the Highest Single Agent (HAS). We used the Bliss synergy model to generate
a surface map, which represents the percentage of synergy and antagonism between RSL3 and Palbociclib. A score >0% indicates synergy (represented as blue), while a score <0% indicates
antagonism (represented as orange or yellow). For CTG assays encompassing giredestrant, a “pre-treatment phase” was also included. Cells were grown in RPMI medium in T175 flasks with DMSO
(as vehicle control), 1 nM giredestrant, 200 nM palbociclib, or 1 nM giredestrant plus 200 nM palbociclib for 7 days, with one round of cell passaging conducted on day 4. For the “treatment
phase”, 3000 cells (pre-treated as above) were seeded in 384 well plates in 54 μl overnight. The following day, cells were treated simultaneously with 6 μl compounds in a 10 (giredestrant) x
8 (RSL3) dose matrix design. The highest concentration for giredestrant was 450 nM, and 10 μM for RSL3. 3-fold dilutions were conducted for each drug, with DMSO as vehicle control, in
quadruplicate wells in the 384 well plates in the presence or absence of 1 μM ferrostatin-1 for 7 days. ATP was quantified as a surrogate for cellular viability using the CellTiter-Glo
Luminescent Cell Viability Assay (Catalog No. G7573, Promega), according to manufacturer’s instructions, using an EnVision plate reader (Catalog No. 2104-0010 A, PerkinElmer). Curve fitting
and half maximal inhibitory concentration (IC50) calculations were carried out using GraphPad Prism 9 (GraphPad Software). Combination effect of Giredestrant and RSL3 was determined by Bliss
independence analyses (Borisy, A. A. et al., Systematic discovery of multicomponent therapeutics. Proc Natl Acad Sci U S A 2003 100(13): p. 7977-7982.). A heatmap was generated to represent
the percent inhibition and Delta. Bliss scores using an in-house R script. A Bliss expectation for the combined response (_C_) was calculated by the equation: _C_ = (_A_ + _B_) − (_A_ ×
_B_) where _A_ and _B_ are the fractional growth inhibitions of Giredestrant and RSL3 at a given dose. Values greater than 1 reflect synergy. INCUCYTE IMAGING FOR GROWTH QUANTIFICATION WITH
PREDESIGNED CRISPR EDIT-R CRGPX4 We targeted GPX4 using the CRISPR Edit-R System and three predesigned crGPX4 (CM-011676) from Dharmacon. Cells were transfected for 48 h, collected, and
plated in 12 well plates in triplicates for treatment with vehicle (DMSO) or 500 nM palbociclib. Cells were imaged on an Incucyte at 4x magnification every 12 h for 12 days. Cell confluence
was analysed using the Incucyte software. CELL DEATH ASSAY USING PROPIDIUM IODIDE (PI) Cells were seeded at a density of 600–1000 cells per well in 384-well plates. Drugs were added 24 h
after seeding at the indicated concentrations and plates were incubated at 37 C for 6 days. Next, cells were stained with Hoechst 33342 (10 mg/mL, ThermoFisher) at a final concentration of 1
μg/mL, and propidium iodide (PI, 1 mg/mL, Sigma-Aldrich) at a final concentration of 0.4 μg/mL. The plates were incubated at 37 C and 5% CO2 for 30 min and images acquired with the Celigo S
high content microscope for cytometric analysis (% of cell death). Two fluorescent filters were used: red 531/629 (PI-positive dead cells) and blue 377/447 (Hoechst-positive live cells).
CLONOGENICS All clonogenic assays were conducted 6-well plates. 6000 to 15,000 cells/well were seeded per well 24 h prior to exposure to the indicated drug concentrations, or vehicle. Wells
were treated continuously for at least 2 weeks replacing media/drug every 3–4 days. 10% trichloroacetic acid (TCA) was added for 1 h to fix the cells, rinsed with water, and stained with
sulforhodamine B Solution (SFB) (0.4% in 1% acetic acid). Stained clonogenics were washed with 1% acetic acid prior image acquisition. To determine clonogenics confluency, bound SFB was
solubilised using 1 ml of 10 mM Tris pH = 10.5 for 5 min at room temperature and 200 µl was used to measure absorbance at a wavelength of 490 nm. For Incucyte and clonogenic assays that
encompassed giredestrant, a “pre-treatment phase” was included. T47D cells were grown in RPMI medium in T175 flasks with DMSO, 1 nM giredestrant, 200 nM palbociclib, or 1 nM giredestrant
plus 200 nM palbociclib for 7 days, with one time cell passaging conducted on day 4. For the “treatment phase”, 30,000 cells (pre-treated as above) were seeded in 12 well plates. The
following day they were treated with DMSO, 1 nM giredestrant, or 1 nM giredestrant plus 200 nM palbociclib, in the presence or absence of RSL3, in duplicates within each of two plates. Cell
growth was tracked with a 10x objective and analyzed using the Incucyte S3 live-cell analysis system (Sartorius). After 3 days, the plates were washed, and compounds in fresh media were
added back to one of the duplicate wells on each plate (sustained treatment), while the other duplicates received only fresh media (washout). The plates were loaded back into the Incucyte
and tracked for 15 more days with media changes in every 3 to 4 days. After the final confluency reading, cells were stained in 0.5% crystal violet solution with 20% methanol for 30 min.
After 4–20 min washes in PBS, the plates were air dried and scanned using Epson Perfection 4870 Photo Scanner (Epson). FLOW CYTOMETRY ANALYSIS FOR LIPID PEROXIDATION MCF-7 and T47D cells
were treated with DMSO, RSL3, ML210, palbociclib or combinations for 6 days. For the last 30 min cells were also treated with 5 μM of C11-BODIPY581/591 dye (D3861, ThermoFisher Scientific)
or MitoPerOx (ab146820, Abcam) resuspended in culture medium. Cells were then washed with ice-cold PBS twice, trypsinized and collected in PBS. Flow cytometry analysis was performed on a BD
Symphony A5, using B530_30A (FITC) filter for oxidised BODIPY-C11, and YG610_20-A (PE-Texas Red) filter for reduced BODIPY-C11. A minimum of 10,000 cells were analyzed for each condition.
Data analysis was performed using the FlowJo 10 software. The gating performed is showed in Supplementary Fig. 3G. Histograms represent gated live cells with similar sizes. LIPIDOMICS SAMPLE
PREPARATION: MCF-7 GIREDESTRANT TIME COURSE EXPERIMENT MCF7 were seeded at 4 × 106 cells in 17 T175 flasks in RPMI medium overnight. On day 0, the cells from 4 untreated flasks were
trypsinized and washed; 2 × 106 cells were collected and frozen; 6–7 flasks of cells were treated with DMSO, 0.6 nM giredestrant respectively. On day 2, the cells from four flasks of each
treatment were trypsinized and washed, 2 × 106 cells were collected and frozen. On day 4, the rest of flasks were passaged and 4 × 106 cells were reseeded in T175 flasks: 6 flasks for
continuous DMSO treatment, 8 flasks for continuous giredestrant treatment. On day 7, four flasks of each treatment cells were trypsinized and washed, 2 × 106 cell were collected and frozen;
the cells from 2 DMSO treatment flasks were passaged and 4 × 106 cells were reseeded in 2 T175 flasks; The medium in 4 giredestrant treatment flasks were replenished as the treatment
continued. On day 11, the cells in 2 DMSO treatment flasks were passaged and 4 × 106 cells were reseeded in 4 T175 flasks; and the medium in 4 giredestrant treatment flasks were replenished
again. On day 14, the cells were trypsinized and washed, 2 × 106 cells from each flask were collected and frozen for lipid analysis. _SAMPLE PREPARATION:_ MCF-7 AND T47D CELLS WITH
GIREDESTRANT AND/OR PALBOCICLIB MCF-7 and T47D cells were seeded at same density, 4 × 106 cells in RPMI medium in T175 flask (16 flasks each for each cell line) overnight. On day 0, the
cells were treated with DMSO, 1 nM giredestrant, 200 nM palbociclib, or 1 nM giredestrant plus 200 nM palbociclib in quadruplicates. On day 4, after the cells were passaged, and 4 × 106
cells from each flask were reseeded into another T175, and continued with the same treatment for 3 more days. On day 7, the cells were trypsinized and counted, and 1.5–2 × 106 cells from
each flask were collected and frozen for lipid analysis. EXTRACTION AND ANALYSIS Cells were homogenised in dichloromethane (DCM):methanol (1:1, v:v). After centrifuging, homogenate
containing the same amount of proteins was transferred into a v-bottom glass tube. 0.5 ml water, 0.45 ml DCM and 1.0 ml methanol were added to the supernatant to form a single phase. After
30 min, isotope labelled internal standards were added to the mixture, followed by 0.45 ml DCM and 0.5 ml water. The mixture was centrifuged at 1000 × _g_ for 20 min. Phase separation was
achieved after centrifuge. The bottom layer was then collected into a clean glass tube, and the upper layer was re-extracted by adding 1.8 ml of DCM. The bottom layer was combined and dried
under a gentle stream of nitrogen. The residue was reconstituted in 300 μl of DCM:Methanol (1:1), 10 mM ammonium acetate for LipidyzerTM Platform direct infusion analysis [ref:Zhijun Cao,
Thomas C. Schmitt, Vijayalakshmi Varma et al. Evaluation of the Performance of Lipidyzer Platform and Its Application in the Lipidomics Analysis in Mouse Heart and Liver; J. Proteome Res.
2020, 19, 7, 2742–2749] on AB Sciex 6500 + LC-MS/MS. Flow rate is set at 7 μl/min. The injection volume is 50 μl. The autosampler temperature was kept at 15 °C. Buffer A and B are the same
as the reconstitution buffer [DCM:Methanol (1:1), 10 mM ammonium acetate]. Lipids concentrations were calculated by the LipidyzerTM platform based on the known concentrations of spiked
internal standards. Heatmap was generated using R. SIRNA TRANSFECTION KNOCKDOWN EXPERIMENTS Reverse transfections using the Non-Targeting Control siRNA (siCON) 1 and 2, or specific siGENOME
human SMARTpools (Dharmacon) against UBB (positive control, M-013382-01), ATXN7L3 (M-023237-01), AGPAT3 (M-008620-00), FAR1 (M-014806-01), and ACSL4 (M-009364-00) were carried out in
384-well plates or 6-well plates using 20 nmol/L of siRNA. At 48 h post-transfection cells were collected and plated for clonogenics or cell viability assays in triplicates, BODIPY-FACs
assays, or western blot assays, and treated with the indicated drugs. All viability assays were performed using CellTiter-Glo luminescence reagent (Promega). RNA PREPARATION, SEQUENCING, AND
DATA ANALYSIS MCF-7 cells were treated with 500 nM palbociclib or vehicle for 12 days and subjected to RNA sequencing. Total RNA was extracted from cancer cells in culture using the RNeasy
Mini Kit (Qiagen) following manufacturer’s instructions. Total RNA samples were tested for contamination and degradation. After the QC procedures, RNA sequencing library was prepared using
Novogene NGS RNA Library Prep Set (PT042) for 250–300 bp insert cDNA library. Messenger RNA was purified from total RNA using poly-T oligo-attached magnetic beads. After fragmentation, the
first strand cDNA was synthesized using random hexamer primers followed by the second strand cDNA synthesis. The library was ready after end repair, A-tailing, adapter ligation, size
selection, amplification, and purification. The sequencing was performed on Novaseq 6000 machine and S4 flow cell with PE150 sequencing strategy. Differential expression analysis was carried
using the edgeR v3.36.0 R package65. Transcripts were linked to the genes using the tximport v1.22.0 package66. Scaled counts were obtained and library sizes were computed from the scaled
counts to account for composition biases between samples. The effective library sizes were combined with the length factors to calculate the offsets for a general linear model. The samples
were filtered to remove low expression genes using min.count = 100 and min.total.count = 300 for the medium stringency set and using min.count = 150. Filtered counts were normalised with the
calcNormFactors function. For each comparison of interest, the glmTreat function was used to identify differentially expressed genes using a threshold of log2 fold change >= 1, _p_-value
<= 0.05. The gene names, gene symbols and ENTREZID were obtained for each transcript using the AnnotationDbi v1.56.2 R package. Gene set enrichment analysis was carried out to identify
pathways of interest using both fgsea v1.20.0 on the unfiltered gene list and enrichR v3.167 on the differentially expressed genes with a threshold of log2 fold change >= 1.5, and p-value
<= 0.05. GO-terms obtained with enrichR v3.1 and p-values are shown. Genes associated with the terms “lipid metabolism” and “biological oxidation” were further analysed and shown in
networks obtained with STRING. A medium-confidence threshold was set for mapping the network, using a minimum required interaction score of 0.4 for connecting nodes. Single unconnected nodes
were excluded from the network plots. MASS SPECTROMETRY AND DATA ANALYSIS MCF7 or T47D cells were seeded in T25 flasks and treated with vehicle or 1 μM palbociclib for 7 days. Media and
drugs were changed every 72 h. Cells were rinsed in PBS, harvested, and washed twice in PBS. The cell pellets were frozen at −80 °C and used for downstream proteomic analysis. For sample
preparation and TMT labelling, cell pellets were dissolved in 150 μL lysis buffer of 1% sodium deoxycholate (SDC), 100 mM triethylammonium bicarbonate (TEAB), 10% isopropanol, 50 mM NaCl and
Halt protease and phosphatase inhibitor cocktail (100X) (Thermo, #78442) on ice with pulsed probe sonication for 15 s followed by boiling at 90 °C for 5 min and re-sonication for 5 s.
Protein concentration was measured with the Coomassie Plus Bradford Protein Assay (Pierce) according to manufacturer’s instructions. Protein aliquots of 60 μg were reduced with 5 mM
tris-2-carboxyethyl phosphine (TCEP) for 1 h at 60 °C and alkylated with 10 mM iodoacetamide (IAA) for 30 min in the dark. Proteins were digested with trypsin (Pierce) at 75 ng/μL overnight.
The peptides were labelled with the TMTpro reagents (Thermo) according to manufacturer’s instructions. For High-pH Reversed-Phase Peptide Fractionation and LC-MS Analysis, peptides were
fractionated with the XBridge C18 column (2.1 ×150 mm, 3.5 μm, Waters) on a Dionex UltiMate 3000 HPLC system at high-pH. Mobile phase A was 0.1% (v/v) ammonium hydroxide and mobile phase B
was acetonitrile, 0.1% (v/v) ammonium hydroxide. The TMTpro labelled peptide mixture was fractionated using a multi-step gradient elution at 0.2 mL/min. The separation method was: for 5 min
isocratic at 5% B, for 35 min gradient to 35% B, gradient to 80% B in 5 min, isocratic for 5 min and re-equilibration to 5% B. Fractions were collected every 42 s and vacuum dried. LC-MS
analysis was performed on the Dionex UltiMate 3000 system coupled with the Orbitrap Lumos Mass Spectrometer (Thermo Scientific). Peptide fractions were reconstituted in 40 μL 0.1% formic
acid and 10 μL were loaded to the Acclaim PepMap 100, 100 μm × 2 cm C18, 5 μm, trapping column at 10 μL/min flow rate. The samples were then analysed with the Acclaim PepMap RSLC (75 μm × 50
cm, 2 μm) C18 capillary column at 45 °C. Mobile phase A was 0.1% formic acid and mobile phase B was 80% acetonitrile, 0.1% formic acid. The gradient method at flow rate 300 nL/min was: for
90 min gradient from 5% to 38% B, for 10 min up to 95% B, for 5 min isocratic at 95% B, re-equilibration to 5% B in 5 min, for 10 min isocratic at 5% B. Precursor ions within 375–1500 m/z
were selected at mass resolution of 120 K in top speed mode (3 s cycle) and were isolated for CID fragmentation with quadrupole isolation width 0.7 Th, collision energy 35% and max IT 35 ms.
MS3 spectra were obtained with further HCD fragmentation of the top 5 most abundant CID fragments isolated with Synchronous Precursor Selection (SPS). Collision energy was applied at 55%
with 86 ms IT and 50 K resolution. Targeted precursors were dynamically excluded for further activation for 45 s with 7 ppm mass tolerance. For protein identification and quantification, the
mass spectra were submitted to SequestHT for database search in Proteome Discoverer 2.4 (Thermo Scientific) using reviewed UniProt homo sapiens protein entries (downloaded on 12 June 2020).
The precursor mass tolerance was set at 20 ppm and the fragment ion mass tolerance at 0.5 Da for fully tryptic peptides with a minimum length of 6 amino acids. TMTpro at N-terminus/K and
Carbamidomethyl at C were selected as static modifications. Dynamic modifications were oxidation of M and Deamidation of N/Q. Peptide confidence was estimated with the Percolator node and
peptides were filtered for q-value < 0.01 based on target-decoy database search. Proteins were identified with at least one high confidence peptide. The reporter ion quantifier node
included a TMTpro quantification method with an integration window of 15 ppm at the MS3 level. Only unique peptides were used for quantification, considering protein groups for peptide
uniqueness. Peptides with average reporter signal-to-noise > 3 were used for protein quantification. Analysis was carried out to identify pathways of interest using enrichR v3.167 on the
differentially expressed proteins. GO-terms obtained with enrichR v3.1 and p-values are shown in Fig. 4. Proteins associated with the terms “lipid metabolism” and “biological oxidation” were
further analysed and shown in networks obtained with STRING. A medium-confidence threshold was set for mapping the network, using a minimum required interaction score of 0.4 for connecting
nodes. Single unconnected nodes were excluded from the network plots. WESTERN BLOTTING Cell lines were grown on 6 plates, treated as indicated, and lysed in NP40 lysis buffer, (1% v/v NP40,
10 mM Tris.Cl pH8, 150 mM NaCl, 1 mM EDTA, 1 mM DTT) supplemented with Protease/Phosphatase Inhibitor Cocktail (100X) (Cell Signalling Technology #5872). Western blots were carried out with
precast TA or Bis-Tris gels (Life Technologies). Unprocessed blots showing the molecular size are supplied in the Source data file. TUMOUR XENOGRAFTS All animals were bred on a mixed genetic
background. All mouse work was carried out in accordance with the Institute of Cancer Research (ICR) guidelines and with the UK Animals (Scientific Procedures) Act 1986. The dark/light
cycle were 7am to 7 pm light and 7 pm to 7am dark, ambient temperature was 22 C + /- 2 C and humidity was 45–65%. Twenty female NSG mice were used for the study, strain NOD.Cg-Prkdc scid
Il2rg tm1Wjl /SzJ (NOD SCID gamma mice), they were 7 weeks old at implantation. The study had ethical approval by the ICR Animal Welfare and Ethical Review Body. One estrogen pellet (NE-121,
Innovative Research of American) was implanted into each female NSG mouse 3 days before cell injection. 10 million MCF-7 or MCF-7 GPX4 null cells were mixed with Matrigel (Corning, Matrigel
Matrix, 354230) at 1:1 ratio and injected subcutaneously into the mouse flanks. Once tumours reached a mean volume of about 200 mm3, mice with similarly sized tumours were distributed into
treatment cohorts. Tumour growth was very slow, with seven mice (14 tumours) not being allocated as their tumour size didn’t reach the desired criteria. One flank was not injected by
mistake, leaving a total of 25 tumours that were randomly allocated to treatment cohorts. Mice were dosed daily and orally, with vehicle (0.5% methylcellulose/0.2% tween-80) or 25 mg/kg
palbociclib. Length (l) and width (W) of each tumour were measured using digital calipers and tumour volumes were calculated based on the following formula (WxWxI)/2. The maximum tumour mean
diameter permitted was 15 mm with no single diameter greater than 18 mm. Three tumours reached over the mean diameter permitted in this study and animals were culled within 24 h of the
measurement taken. All other tumours did not exceed the maximal tumour size covered by the home office licence under which the animal experiment was performed and permitted by the ICR Animal
Welfare and Ethical Review Body. Tumours growth was limited to the 90 days-live of the estrogen pellet, with five tumours having a shorter treatment period as they took longer to rich the
desired criteria for treatment. We have harvested residual tumours at the end of the experiment (34 days of treatment). 50 mg of frozen tissue was used to obtain protein lysates. Samples
were homogenised in NP40 lysis buffer using the Precellys tissue homogeniser, centrifuge, and analysed by western blotting. STATISTICS AND REPRODUCIBILITY Statistical differences between two
experimental groups were determined using paired and unpaired Student’s _t_ test. In the case of three or more experimental groups, a multiple unpaired t-test (two-stage-step-up) with a
False Discovery Rate (FDR) of 1% was performed. To test whether two independent drug concentrations affect cell survival, in combination, we used two-way ANOVA (Ordinary). A two-sided value
of _p_ < 0.05 was considered statistically significant, and a 95% confident level was used. All statistical analyses were performed using PRISM 10.3.0 software (GraphPad, San Diego, CA).
Group and sample sizes were selected based on standard deviations and statistical assessments and/or prior studies conducted by our laboratories that yielded sufficient and reproducible
power to detect statistically significant differences. Investigators were not blinded to group allocation during data collection or analysis because both experimental setups and analyses
were generally performed by the same person(s). No data were excluded from the analyses. Western blots results were reproduced in at least two independent experiments. The expected molecular
weight band was observed with each antibody. The western blots band for FAR1, AGPAT3, ATXN7L3 and ACSL4 were loss upon specific siRNA treatments. The western blots band for GPX4 was loss
upon CRISPR-Cas9 against GPX4. An expected reduction of phospho-CDK2, phosphor-SirT1 and phosphor-RB1 was observed in cells treated with palbociclib. REPORTING SUMMARY Further information on
research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY The palbociclib RNA sequencing data have been deposited in the Sequence Read
Archive (SRA) under the accession code PRJNA989103. The Mass Spectrometry data have been deposited in the PRIDE Proteomics database under accession code PXD043133. The Giredestrant RNA
sequencing data have been deposited in the Gene Expression Omnibus database under accession code GSE260702. The Lipidomics Study 1 data Giredestrant Time Course Project have been deposited
into Metabolomics Workbench under the accession code PR002111. The Lipidomics Study 2 data Analysis of ER+ Breast Cancer Cells Treated with Giredestrant and Palbociclib Project have been
deposited in Metabolomics Workbench under the accession code PR002111. The remaining data are available within the Article, Supplementary Information or Source Data file. Source data are
provided with this paper. REFERENCES * Goetz, M. P. et al. MONARCH 3: Abemaciclib as initial therapy for advanced breast cancer. _J. Clin. Oncol._ 35, 3638–3646 (2017). Article CAS PubMed
Google Scholar * Hortobagyi, G. N. et al. Ribociclib as first-line therapy for HR-Positive, advanced breast cancer. _N. Engl. J. Med._ 375, 1738–1748 (2016). Article CAS PubMed Google
Scholar * Finn, R. S. et al. Palbociclib and Letrozole in advanced breast cancer. _N. Engl. J. Med._ 375, 1925–1936 (2016). Article CAS PubMed Google Scholar * Johnston, S. R. D. et al.
Abemaciclib combined with endocrine therapy for the adjuvant treatment of HR+, HER2-, node-positive, high-risk, early breast cancer (monarchE). _J. Clin. Oncol._ 38, 3987–3998 (2020).
Article CAS PubMed PubMed Central Google Scholar * Tolaney, S. M. et al. Abemaciclib plus trastuzumab with or without fulvestrant versus trastuzumab plus standard-of-care chemotherapy
in women with hormone receptor-positive, HER2-positive advanced breast cancer (monarcHER): a randomised, open-label, phase 2 trial. _Lancet Oncol._ 21, 763–775 (2020). Article CAS PubMed
Google Scholar * Finn, R. S. et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer
cell lines in vitro. _Breast Cancer Res._ 11, R77 (2009). Article PubMed PubMed Central Google Scholar * Liang, J. et al. GDC-9545 (Giredestrant): a potent and orally bioavailable
selective estrogen receptor antagonist and degrader with an exceptional preclinical profile for ER+ breast cancer. _J. Med. Chem._ 64, 11841–11856 (2021). Article ADS CAS PubMed Google
Scholar * Jhaveri, K. L. et al. Phase Ia/b Study of Giredestrant +/- Palbociclib and +/- luteinizing hormone-releasing hormone agonists in estrogen receptor-positive, HER2-Negative, locally
advanced/metastatic breast cancer. _Clin. Cancer Res._ 30, 754–766 (2024). Article CAS PubMed Google Scholar * Hurvitz, S. A. et al. Neoadjuvant palbociclib plus either giredestrant or
anastrozole in oestrogen receptor-positive, HER2-negative, early breast cancer (coopERA Breast Cancer): an open-label, randomised, controlled, phase 2 study. _Lancet Oncol._ 24, 1029–1041
(2023). Article CAS PubMed Google Scholar * Tang, D. et al. Ferroptosis: molecular mechanisms and health implications. _Cell Res._ 31, 107–125 (2021). Article CAS PubMed Google
Scholar * Yang, W. S. et al. Regulation of ferroptotic cancer cell death by GPX4. _Cell_ 156, 317–331 (2014). Article CAS PubMed PubMed Central Google Scholar * Jiang, X., Stockwell,
B. R. & Conrad, M. Ferroptosis: mechanisms, biology and role in disease. _Nat. Rev. Mol. Cell Biol._ 22, 266–282 (2021). Article PubMed PubMed Central Google Scholar * Dixon, S. J.
et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. _Cell_ 149, 1060–1072 (2012). Article CAS PubMed PubMed Central Google Scholar * Doll, S. et al. ACSL4 dictates
ferroptosis sensitivity by shaping cellular lipid composition. _Nat. Chem. Biol._ 13, 91–98 (2017). Article CAS PubMed Google Scholar * Zou, Y. et al. Plasticity of ether lipids promotes
ferroptosis susceptibility and evasion. _Nature_ 585, 603–608 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Porter, N. A., Caldwell, S. E. & Mills, K. A.
Mechanisms of free radical oxidation of unsaturated lipids. _Lipids_ 30, 277–290 (1995). Article CAS PubMed Google Scholar * Koike-Yusa, H. et al. Genome-wide recessive genetic screening
in mammalian cells with a lentiviral CRISPR-guide RNA library. _Nat. Biotechnol._ 32, 267–273 (2014). Article CAS PubMed Google Scholar * Herrera-Abreu, M. T. et al. Early adaptation
and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. _Cancer Res._ 76, 2301–2313 (2016). Article CAS PubMed PubMed Central Google Scholar * Costa,
C. et al. PTEN loss mediates clinical cross-resistance to CDK4/6 and PI3Kalpha inhibitors in breast cancer. _Cancer Discov._ 10, 72–85 (2020). Article CAS PubMed Google Scholar *
O’Leary, B. et al. The genetic landscape and clonal evolution of breast cancer resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. _Cancer Discov._ 8, 1390–1403 (2018). Article
PubMed PubMed Central Google Scholar * The, I. et al. Rb and FZR1/Cdh1 determine CDK4/6-cyclin D requirement in C. elegans and human cancer cells. _Nat. Commun._ 6, 5906 (2015). Article
ADS PubMed Google Scholar * Paul, M. R. et al. Genomic landscape of metastatic breast cancer identifies preferentially dysregulated pathways and targets. _J. Clin. Investig._ 130,
4252–4265 (2020). CAS PubMed PubMed Central Google Scholar * Asghar, U. S. et al. Single-cell dynamics determines response to CDK4/6 inhibition in triple-negative breast cancer. _Clin.
Cancer Res._ 23, 5561–5572 (2017). Article CAS PubMed PubMed Central Google Scholar * Palafox, M. et al. High p16 expression and heterozygous RB1 loss are biomarkers for CDK4/6
inhibitor resistance in ER(+) breast cancer. _Nat. Commun._ 13, 5258 (2022). Article ADS CAS PubMed PubMed Central Google Scholar * Al-Qasem, A. J. et al. Co-targeting CDK2 and CDK4/6
overcomes resistance to aromatase and CDK4/6 inhibitors in ER+ breast cancer. _NPJ Precis Oncol._ 6, 68 (2022). Article CAS PubMed PubMed Central Google Scholar * Freeman-Cook, K. et
al. Expanding control of the tumor cell cycle with a CDK2/4/6 inhibitor. _Cancer Cell_ 39, 1404–1421 e11 (2021). Article CAS PubMed Google Scholar * Carpintero-Fernandez, P. et al.
Genome wide CRISPR/Cas9 screen identifies the coagulation factor IX (F9) as a regulator of senescence. _Cell Death Dis._ 13, 163 (2022). Article CAS PubMed PubMed Central Google Scholar
* Fan, Z. et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. _Oncogenesis_ 6, e371 (2017). Article CAS PubMed PubMed Central Google Scholar *
Wohlhieter, C. A. et al. Concurrent mutations in STK11 and KEAP1 promote ferroptosis protection and SCD1 dependence in lung cancer. _Cell Rep._ 33, 108444 (2020). Article CAS PubMed
PubMed Central Google Scholar * Sun, X. et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. _Hepatology_ 63, 173–184 (2016).
Article CAS PubMed Google Scholar * Yang, W. S. et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. _Proc. Natl Acad. Sci. USA_ 113, E4966–E4975
(2016). Article CAS PubMed PubMed Central Google Scholar * Das, U. N. Saturated Fatty Acids, MUFAs and PUFAs Regulate Ferroptosis. _Cell Chem. Biol._ 26, 309–311 (2019). Article CAS
PubMed Google Scholar * Chen, X. et al. Iron Metabolism in Ferroptosis. _Front. Cell Dev. Biol._ 8, 590226 (2020). Article ADS PubMed PubMed Central Google Scholar * Torti, S. V.
& Torti, F. M. Winning the war with iron. _Nat. Nanotechnol._ 14, 499–500 (2019). Article ADS CAS PubMed PubMed Central Google Scholar * Mao, C. et al. DHODH-mediated ferroptosis
defence is a targetable vulnerability in cancer. _Nature_ 593, 586–590 (2021). Article ADS CAS PubMed PubMed Central Google Scholar * Xiao, T. et al. Estrogen-regulated feedback loop
limits the efficacy of estrogen receptor-targeted breast cancer therapy. _Proc. Natl Acad. Sci. USA_ 115, 7869–7878 (2018). Article ADS CAS PubMed PubMed Central Google Scholar * Sun,
W. Y. et al. Phospholipase iPLA(2)beta averts ferroptosis by eliminating a redox lipid death signal. _Nat. Chem. Biol._ 17, 465–476 (2021). Article CAS PubMed PubMed Central Google
Scholar * Chen, D. et al. iPLA2beta-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. _Nat. Commun._ 12, 3644 (2021). Article ADS CAS PubMed PubMed
Central Google Scholar * Kagan, V. E. et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. _Nat. Chem. Biol._ 13, 81–90 (2017). Article CAS PubMed Google Scholar
* Huang, W. et al. Cellular senescence: the good, the bad and the unknown. _Nat. Rev. Nephrol._ 18, 611–627 (2022). Article CAS PubMed PubMed Central Google Scholar * Ghosh, M. K.,
Mukhopadhyay, M. & Chatterjee, I. B. NADPH-initiated cytochrome P450-dependent free iron-independent microsomal lipid peroxidation: specific prevention by ascorbic acid. _Mol. Cell
Biochem._ 166, 35–44 (1997). Article CAS PubMed Google Scholar * Tang, Z. et al. Ferroptosis: the silver lining of cancer therapy. _Front. Cell Dev. Biol._ 9, 765859 (2021). Article
PubMed PubMed Central Google Scholar * Bhatt-Wessel, B. et al. Role of DGAT enzymes in triacylglycerol metabolism. _Arch. Biochem. Biophys._ 655, 1–11 (2018). Article CAS PubMed Google
Scholar * Atanassov, B. S. et al. ATXN7L3 and ENY2 coordinate activity of Multiple H2B Deubiquitinases Important for cellular proliferation and tumor growth. _Mol. Cell_ 62, 558–571
(2016). Article CAS PubMed PubMed Central Google Scholar * Bonnet, J. et al. The SAGA coactivator complex acts on the whole transcribed genome and is required for RNA polymerase II
transcription. _Genes Dev._ 28, 1999–2012 (2014). Article CAS PubMed PubMed Central Google Scholar * Chen, X. et al. Cellular degradation systems in ferroptosis. _Cell Death Differ._
28, 1135–1148 (2021). Article CAS PubMed PubMed Central Google Scholar * Hong, T. et al. PARP inhibition promotes ferroptosis via repressing SLC7A11 and synergizes with ferroptosis
inducers in BRCA-proficient ovarian cancer. _Redox Biol._ 42, 101928 (2021). Article CAS PubMed PubMed Central Google Scholar * Li, G. et al. A PARP1 PROTAC as a novel strategy against
PARP inhibitor resistance via promotion of ferroptosis in p53-positive breast cancer. _Biochem. Pharm._ 206, 115329 (2022). Article CAS PubMed Google Scholar * Cui, W. et al.
Peroxisome-driven ether-linked phospholipids biosynthesis is essential for ferroptosis. _Cell Death Differ._ 28, 2536–2551 (2021). Article CAS PubMed PubMed Central Google Scholar *
Lee, H. et al. Cell cycle arrest induces lipid droplet formation and confers ferroptosis resistance. _Nat. Commun._ 15, 79 (2024). Article ADS CAS PubMed PubMed Central Google Scholar
* Rodencal, J. et al. Sensitization of cancer cells to ferroptosis coincident with cell cycle arrest. _Cell Chem. Biol._ 31, 234–248 e13 (2024). Article CAS PubMed Google Scholar *
Liang, D. et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. _Cell_ 186, 2748–2764.e22 (2023). Article CAS PubMed PubMed Central Google
Scholar * Crozier, L. et al. CDK4/6 inhibitors induce replication stress to cause long-term cell cycle withdrawal. _EMBO J._ 41, e108599 (2022). Article CAS PubMed PubMed Central Google
Scholar * Leontieva, O. V. & Blagosklonny, M. V. CDK4/6-inhibiting drug substitutes for p21 and p16 in senescence: duration of cell cycle arrest and MTOR activity determine
geroconversion. _Cell Cycle_ 12, 3063–3069 (2013). Article CAS PubMed PubMed Central Google Scholar * Hamsanathan, S. & Gurkar, A. U. Lipids as regulators of cellular senescence.
_Front. Physiol._ 13, 796850 (2022). Article PubMed PubMed Central Google Scholar * Turner, N. C. et al. Overall survival with Palbociclib and Fulvestrant in advanced breast cancer. _N.
Engl. J. Med._ 379, 1926–1936 (2018). Article CAS PubMed Google Scholar * Llorca-Cardenosa, M. J. et al. SMG8/SMG9 Heterodimer Loss Modulates SMG1 Kinase to Drive ATR Inhibitor
Resistance. _Cancer Res._ 82, 3962–3973 (2022). Article CAS PubMed PubMed Central Google Scholar * Colic, M. et al. Identifying chemogenetic interactions from CRISPR screens with drugZ.
_Genome Med._ 11, 52 (2019). Article PubMed PubMed Central Google Scholar * Callow, M. G. et al. CRISPR whole-genome screening identifies new necroptosis regulators and RIPK1
alternative splicing. _Cell Death Dis._ 9, 261 (2018). Article PubMed PubMed Central Google Scholar * Hoberecht, L. et al. A comprehensive Bioconductor ecosystem for the design of CRISPR
guide RNAs across nucleases and technologies. _Nat. Commun._ 13, 6568 (2022). Article ADS CAS PubMed PubMed Central Google Scholar * Huber, W. et al. Orchestrating high-throughput
genomic analysis with Bioconductor. _Nat. Methods_ 12, 115–121 (2015). Article ADS CAS PubMed PubMed Central Google Scholar * Robinson, M. D. & Oshlack, A. A scaling normalization
method for differential expression analysis of RNA-seq data. _Genome Biol._ 11, R25 (2010). Article PubMed PubMed Central Google Scholar * Hart, T. et al. Measuring error rates in
genomic perturbation screens: gold standards for human functional genomics. _Mol. Syst. Biol._ 10, 733 (2014). Article PubMed PubMed Central Google Scholar * Law, C. W. et al. voom:
precision weights unlock linear model analysis tools for RNA-seq read counts. _Genome Biol._ 15, R29 (2014). Article PubMed PubMed Central Google Scholar * Robinson, M. D., McCarthy, D.
J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. _Bioinformatics_ 26, 139–140 (2010). Article CAS PubMed Google
Scholar * Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. _F1000Res_ 4, 1521 (2015). Article
PubMed Google Scholar * Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. _Nucleic Acids Res._ 44, W90–W97 (2016). Article CAS PubMed
PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS ICR BSU and Imperial College London CBS for supporting the in vivo study. David Vicente (Molecular and Cell Biology Team,
ICR) for his advice about in vivo studies. Kai Betteridge (Light Microscopy Facility Manager, ICR) for his support with microscopy. Honglin Chen, Scott Martin, Rajini Srinivasan, Jenille
Tan, Colin Watanabe for contributions to the giredestrant sensitization and resistance screen. This work has been funded by Breast Cancer Now grant with reference number CTR-Q5-Y3. The work
of T.I.R. and J.C. was funded by the CRUK Centre grant with reference number C309/A25144. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Breast Cancer Now Toby Robins Research Centre, The
Institute of Cancer Research, London, UK M. T. Herrera-Abreu, U. Khalid, R. Cutts, G. Wilson, J. Frankum, J. Alexander, O. Hickman, R. Brough, S. Haider, C. J. Lord & N. C. Turner *
Genentech, 1 DNA Way, South San Francisco, CA, USA J. Guan, M. R. Costa, J. Chan, Q. Li, J-P. Fortin, W. R. Wong, A. Biton, W. Sandoval, M. Hafner & C. Metcalfe * Tumour Modelling
Facility, Institute of Cancer Research, London, UK J. Ning & A. Swain * ProCogia Inc. under contract to Hoffmann-La Roche Limited, Toronto, ON, Canada P. Perampalam * Roche Informatics,
Mississauga, ON, Canada J. Vijay * CRUK Gene Function Laboratory, The Institute of Cancer Research, London, UK J. Frankum, R. Brough & C. J. Lord * Functional proteomics team, The
Institute of Cancer Research, London, UK T. I. Roumeliotis & J. Choudhary * Breast Unit, The Royal Marsden Hospital, London, UK N. C. Turner Authors * M. T. Herrera-Abreu View author
publications You can also search for this author inPubMed Google Scholar * J. Guan View author publications You can also search for this author inPubMed Google Scholar * U. Khalid View
author publications You can also search for this author inPubMed Google Scholar * J. Ning View author publications You can also search for this author inPubMed Google Scholar * M. R. Costa
View author publications You can also search for this author inPubMed Google Scholar * J. Chan View author publications You can also search for this author inPubMed Google Scholar * Q. Li
View author publications You can also search for this author inPubMed Google Scholar * J-P. Fortin View author publications You can also search for this author inPubMed Google Scholar * W.
R. Wong View author publications You can also search for this author inPubMed Google Scholar * P. Perampalam View author publications You can also search for this author inPubMed Google
Scholar * A. Biton View author publications You can also search for this author inPubMed Google Scholar * W. Sandoval View author publications You can also search for this author inPubMed
Google Scholar * J. Vijay View author publications You can also search for this author inPubMed Google Scholar * M. Hafner View author publications You can also search for this author
inPubMed Google Scholar * R. Cutts View author publications You can also search for this author inPubMed Google Scholar * G. Wilson View author publications You can also search for this
author inPubMed Google Scholar * J. Frankum View author publications You can also search for this author inPubMed Google Scholar * T. I. Roumeliotis View author publications You can also
search for this author inPubMed Google Scholar * J. Alexander View author publications You can also search for this author inPubMed Google Scholar * O. Hickman View author publications You
can also search for this author inPubMed Google Scholar * R. Brough View author publications You can also search for this author inPubMed Google Scholar * S. Haider View author publications
You can also search for this author inPubMed Google Scholar * J. Choudhary View author publications You can also search for this author inPubMed Google Scholar * C. J. Lord View author
publications You can also search for this author inPubMed Google Scholar * A. Swain View author publications You can also search for this author inPubMed Google Scholar * C. Metcalfe View
author publications You can also search for this author inPubMed Google Scholar * N. C. Turner View author publications You can also search for this author inPubMed Google Scholar
CONTRIBUTIONS N.T. and M.T.H.A. conceptualised the project and developed the methodology; M.T.H.A. performed most of the experiments and data analysis; U.K. performed most of the cell
survival, clonogenic and PI experiments in Figs. 2 and 3; G.W. performed a cell survival experiment; C.L., R.B., and J.F. developed the CRISPR screen methodology and provided the library and
material; M.T.H.A. performed the palbociclib CRISPR screen; R.C., J.A., and S.H. conceptualised the bioinformatic analysis; M.T.H.A. and R.C. did RNAseq data analysis; M.T.H.A., A.S., and
J.N. conceptualised the in vivo experiment, performed by M.T.H.A. and J.N.; O.H., J.C, and T.R. conceptualised the proteomics experiments, performed by O.H and T.R. and analysed by T.R. and
M.T.H.A.; MT.HA. prepared the manuscript. For the giredestrant-related portion of the manuscript, C.M., M.H., and M.R.C. conceptualised the work, wrote portions of the manuscript and/or
supervised experiments and analyses. J.C., J-P.F., P.P., A.B., and M.R.C. performed the giredestrant CRISPR screen and/or analyzed the screening data. J.G. performed giredestrant-related
viability experiments and generated samples for RNA-sequencing and lipidomics analysis. Q.L. and W.R.W. performed the lipidomics analysis and analyzed lipidomics data. W.S. supervised the
lipidomics experiments. J.V. performed the RNA-sequencing analyses, under the supervision of M.H. All authors reviewed the manuscript. CORRESPONDING AUTHORS Correspondence to C. Metcalfe or
N. C. Turner. ETHICS DECLARATIONS COMPETING INTERESTS J.C., J-P.F., P.P., A.B., M.R.C, J.G., Q.L., W.R.W., W.S., J.V., M.H., and C.M. are or were employees of Genentech/Roche and hold Roche
shares. All other Authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks the anonymous reviewers for their contribution to the peer review
of this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and
institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION REPORTING SUMMARY SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 SUPPLEMENTARY DATA 3 PEER REVIEW FILE DESCRIPTION
OF ADDITIONAL SUPPLEMENTARY FILES SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0
International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s)
and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material
derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a
credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted
use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions
ABOUT THIS ARTICLE CITE THIS ARTICLE Herrera-Abreu, M.T., Guan, J., Khalid, U. _et al._ Inhibition of GPX4 enhances CDK4/6 inhibitor and endocrine therapy activity in breast cancer. _Nat
Commun_ 15, 9550 (2024). https://doi.org/10.1038/s41467-024-53837-7 Download citation * Received: 25 May 2023 * Accepted: 18 October 2024 * Published: 05 November 2024 * DOI:
https://doi.org/10.1038/s41467-024-53837-7 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative