Play all audios:
ABSTRACT Idiopathic hypersomnia (IH) is a rare, heterogeneous sleep disorder characterized by excessive daytime sleepiness. In contrast to narcolepsy type 1, which is a well-defined type of
central disorders of hypersomnolence, the etiology of IH is poorly understood. No susceptibility loci associated with IH have been clearly identified, despite the tendency for familial
aggregation of IH. We performed a variation screening of the _prepro-orexin/hypocretin_ and _orexin receptors_ genes and an association study for IH in a Japanese population, with
replication (598 patients and 9826 controls). We identified a rare missense variant (g.42184347T>C; p.Lys68Arg; rs537376938) in the cleavage site of _prepro-orexin_ that was associated
with IH (minor allele frequency of 1.67% in cases versus 0.32% in controls, _P_ = 2.7 × 10−8, odds ratio = 5.36). Two forms of orexin (orexin-A and -B) are generated from cleavage of one
precursor peptide, prepro-orexin. The difference in cleavage efficiency between wild-type (Gly-Lys-Arg; GKR) and mutant (Gly-Arg-Arg; GRR) peptides was examined by assays using proprotein
convertase subtilisin/kexin (PCSK) type 1 and PCSK type 2. In both PCSK1 and PCSK2 assays, the cleavage efficiency of the mutant peptide was lower than that of the wild-type peptide. We also
confirmed that the prepro-orexin peptides themselves transmitted less signaling through orexin receptors than mature orexin-A and orexin-B peptides. These results indicate that a subgroup
of IH is associated with decreased orexin signaling, which is believed to be a hallmark of narcolepsy type 1. SIMILAR CONTENT BEING VIEWED BY OTHERS SLEEP AND DIURNAL ALTERNATIVE
POLYADENYLATION SITES ASSOCIATED WITH HUMAN APA-LINKED BRAIN DISORDERS Article Open access 01 November 2024 EXOME SEQUENCING IDENTIFIES GENES ASSOCIATED WITH SLEEP-RELATED TRAITS Article 04
January 2024 SLEEP DISTURBANCES AS RISK FACTORS FOR NEURODEGENERATION LATER IN LIFE Article Open access 27 May 2025 INTRODUCTION Idiopathic hypersomnia (IH) is a rare heterogeneous disorder
characterized by prolonged and disabling excessive daytime sleepiness (intolerable sleepiness in the daytime)1. Patients with narcolepsy type 1, which is a well-recognized type of central
disorders of hypersomnolence, exhibit excessive daytime sleepiness, cataplexy (sudden loss of muscle tone in response to strong emotion), and abnormal rapid eye movement (REM) sleep.
However, patients with IH do not display cataplexy or REM sleep abnormalities. The onset of IH is most frequently during adolescence or young adulthood2. IH is a rare disease and is
estimated to affect approximately 0.005% of the general population3. However, the exact prevalence of IH remains unknown due to the absence of epidemiological studies. A high proportion of
IH patients (26.9–39.1%) reported a family member with excessive daytime sleepiness3,4,5. The human leukocyte antigen (HLA) class II region is strongly associated with narcolepsy type 1 and
type 2. Genome-wide association studies (GWAS) of narcolepsy type 1 have identified more than 10 genomic loci involved in immune and metabolic pathways6,7,8. When the 2009 influenza A (H1N1)
pandemic was declared, the AS03-adjuvanted vaccine Pandemrix was used in several European countries. A significant association between the onset of narcolepsy type 1 and exposure to
Pandemrix in children and adolescents was reported in Finland and Sweden9. A GWAS of Pandemrix-associated narcolepsy type 1 was performed, and _GDNF-AS1_ was identified as a novel genetic
factor10. Kleine-Levin syndrome (KLS) is a rare sleep disorder characterized by recurrent episodes of hypersomnia and is associated with cognitive disturbances and behavioral abnormalities.
A GWAS of KLS found a significant association with variants in _TRANK1_ region and links between KLS, circadian regulation, and bipolar disorder11. Meanwhile, no significant associations
between IH and HLA have been observed12, and no other genetic variants associated with IH have been clearly identified13. Thus, the underlying genetic contribution to IH is poorly
understood. Orexin, also known as hypocretin, is a neuropeptide that regulates sleep-wake cycles and REM sleep14,15,16. Narcolepsy type 1 is caused by the loss of orexin-producing neurons in
the hypothalamus, leading to low or undetectable levels of orexin-A in the cerebrospinal fluid (CSF)17,18. Rare missense mutations in _MOG_ and _P2RY11_ have been reported to be associated
with narcolepsy type 119,20. However, pathogenic mutations in _prepro-orexin_ and _orexin receptor-1_ (_OX1R_) and _-2_ (_OX2R_) were not identified in patients with narcolepsy type 1,
except for one rare severe case18,21. Previous studies have reported that postnatal cell death of orexin-producing neurons in narcolepsy is associated with cell type-specific autoimmune
targeting22,23. Unlike patients with narcolepsy type 1, patients with IH show normal levels of orexin-A in the CSF. Therefore, orexin signaling system in IH has been believed to be normal24.
Genetic variants in _prepro-orexin_, _OX1R_, and _OX2R_ have not been studied in IH. Orexin is a master regulator of sleep, and we hypothesized that genetic variants may exist that affect
the protein structure or its function. In this study, we focused on rare missense and loss-of-function variants in _prepro-orexin_, _OX1R_, and _OX2R_ to search for genetic factors of IH
because no significant associations with common variants in these gene regions were observed in our GWAS13. IH has been reported to be clinically heterogeneous25. To identify rare genetic
variants associated with IH, we conducted an association study between IH and genetic variants selected by variation screening in exons of these genes in the Japanese population, which is
relatively homogeneous. RESULTS A RARE VARIANT ASSOCIATED WITH IDIOPATHIC HYPERSOMNIA A flow diagram of the study design is shown in Supplementary Fig. 1. For _prepro-orexin_, _OX1R_, and
_OX2R_, variation screening of 195 patients with IH identified only one rare missense variant in _prepro-orexin_ through the filtering process (see Methods) (g.42184347T>C; p.Lys68Arg;
rs537376938). Then, we performed an association study between IH and p.Lys68Arg in 8380 controls and 440 patients with IH including the 195 patients with IH analyzed in the variation
screening. The minor allele frequency (MAF) of p.Lys68Arg was 1.59% in the IH group and 0.30% in the control group, showing a significant association (_P_ = 2.5 × 10−6, odds ratio = 5.40)
(Table 1). We next assessed p.Lys68Arg in an independent set of 158 patients with IH and 1446 controls as a replication study. As a result, the p.Lys68Arg variant was replicated successfully
(_P_ = 6.0 × 10−3, odds ratio = 4.62) (Table 1). In the combined sample, the MAF of p.Lys68Arg was significantly higher in patients with IH than in controls, demonstrating a strong
susceptibility to IH (MAF of 1.67% in cases versus 0.32% in controls, _P_ = 2.7 × 10−8, odds ratio = 5.36) (Table 1). In contrast, no significant association between narcolepsy (type 1 and
type 2) and p.Lys68Arg was observed (Supplementary Table 1). All patients with this mutation were heterozygous carriers. Clinical polysomnography (PSG) and multiple sleep latency test (MSLT)
variables were available in a subset of our IH patients, and therefore, we compared these clinical data to find characteristic changes in patients with this orexin mutation (Table 2).
Subjective sleepiness as evaluated with the Japanese version of the Epworth Sleepiness Scale (JESS) scores in unmedicated conditions and arousal index were nominally higher in the orexin
mutation-positive patients (_P_ = 0.049 and _P_ = 0.046, respectively), suggesting that mutation-positive IH patients suffered more sleepiness and sleep instability similar to
orexin-deficient narcolepsy type 1. Then, excluding patients who had periodic limb movement index (PLMI) ≥ 15 events per hour in adults, PLMI ≥ 5 events per hour in children, or apnea
hypopnea index ≥5 events per hour, which may be associated with sleep problems at night, the remaining 317 patients with IH were analyzed for association with p.Lys68Arg. The variant also
showed a significant association with these 317 patients (_P_ = 8.5 × 10−8, odds ratio = 7.13) (Supplementary Table 2). We searched for MAFs of p.Lys68Arg in populations other than the
Japanese population using whole-genome sequencing data of the Han Chinese Genomes Database (PGG.Han) and Genome Aggregation Database (gnomAD)26,27. The MAFs in the Han Chinese, European
(non-Finnish), African, and European (Finnish) populations were 0.34%, 0.013%, 0%, and 0%, respectively. Next, we performed a principal component analysis (PCA) to determine whether
population stratification or differences in the genetic background are present between the orexin mutation-positive and -negative IH groups. Genome-wide single nucleotide polymorphism (SNP)
data in patients with IH (13 orexin mutation-positive patients and 116 orexin mutation-negative patients) and HapMap samples were utilized for the analysis. Our samples clustered in the JPT
(Japanese in Tokyo)/CHB (Han Chinese in Beijing) cluster and separately from the CEU (Utah residents with Northern and Western European ancestry) and YRI (Nigeria Yoruba in Ibadan) clusters
(Supplementary Fig. 2). When a PCA was conducted using our samples and JPT/CHB samples, the orexin mutation-positive and -negative IH samples seemed to be equally distributed in the JPT
cluster (Supplementary Fig. 3). The result suggested that an effect of population stratification would be negligible in the samples. We also confirmed the absence of familial relationships
among the mutation-positive patients using identity-by-descent (PIHAT) values (<0.1) calculated from the genome-wide SNP data. Large-scale GWASs using data from the UK biobank and 23andMe
have identified that a common missense variant (g.55277539A>G; p.Ile308Val; rs2653349) in _OX2R_ is significantly associated with self-reported napping28 (the Sleep Disorder Knowledge
Portal website (http://www.sleepdisordergenetics.org)). The A allele of rs2653349 shows an association with more frequent napping during the day. Given the possibility of accumulation of
genetic variants associated with daytime napping in the orexin pathway, we genotyped rs2653349 in patients with IH and controls. As a result, the A allele frequency of rs2653349 in the
orexin mutation-positive IH group (15.0%) was significantly higher than that in the orexin mutation-negative IH group (4.7%) or control group (4.8%) (Table 3). Two missense variants,
p.Arg168Trp (rs141639071) and p.Arg213His (rs200068306), were detected in _OX2R_ by variation screening in patients with IH (Supplementary Table 3). These missense variants were registered
in the Japanese Multi Omics Reference Panel (jMorp) (MAF of p.Arg168Trp = 0.11%, MAF of p.Arg213His = 0.03%). Only one IH patient each carried these missense variants, and thus, the two
variants were excluded from further analyzes according to the work flow of this study. However, all in silico analyses estimated that p.Arg168Trp was damaging or deleterious, and therefore,
we genotyped this mutation in IH patients not analyzed in the variation screening. No additional IH patients carried this variant, suggesting that p.Arg168Trp is not associated with IH, or
the effect size of this variant may be weak even if an association between IH and p.Arg168Trp exists. FUNCTIONAL ANALYSIS OF THE IDENTIFIED VARIANT Orexin-A and orexin-B are cleaved from
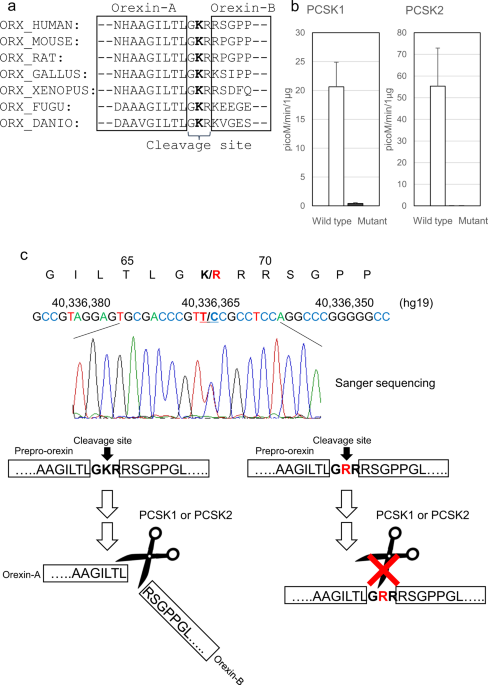
prepro-orexin. p.Lys68Arg is located in the cleavage site of prepro-orexin. GKR (Gly-Lys-Arg), which is the wild-type amino acid sequence of this cleavage site, is substituted with GRR
(Gly-Arg-Arg) by the mutation. The Lys residue is conserved across almost all species that express prepro-orexin (Fig. 1a). This amino acid substitution was predicted to be damaging or
deleterious by all in silico analyses (Supplementary Table 3). We hypothesized that the mutant amino acid sequence, GRR, affects the proteolytic cleavage efficiency of prepro-orexin. Two
peptides, pLTLGKR-AMC (aminomethylcoumarin) (wild-type prepro-orexin) and pLTLGRR-AMC (mutant prepro-orexin), were prepared for an enzyme activity analysis. We examined proprotein convertase
subtilisin/kexin (PCSK) type 1 and PCSK2 activity, which may process prepro-orexin29. In both the PCSK1 and PCSK2 assays, the mutant peptide was significantly less processed than the
wild-type peptide (50-fold difference in PCSK1; 1100-fold difference in PCSK2) (Fig. 1b). The result suggested that large amounts of the mutant precursor prepro-orexin remain uncleaved in IH
patients that carry p.Lys68Arg (Fig. 1c). Next, we investigated whether the prepro-orexin peptides have pharmacological effects on orexin signaling through OX1R and OX2R. The effects of
orexin-A, orexin-B, wild-type prepro-orexin, and mutant prepro-orexin were evaluated using the ligand-induced β-arrestin recruitment system (Fig. 2a, b). Orexin-A induced the highest
β-arrestin recruitment in the OX1R assay. The recruitment levels of the other three peptides were lower and similar to each other. In the OX2R assay, recruitment levels of both wild-type and
mutant prepro-orexins were lower than those of orexin-A and orexin-B. These results indicated that the pharmacological effects of the prepro-orexin peptides on OX1R and OX2R were weak in
terms of orexin signaling. HPLC ANALYSIS OF CSF OREXIN-A We collected a CSF sample from one of the 20 patients with IH who carried the mutant allele of p.Lys68Arg. Radioimmunoassay (RIA)
measurement using a polyclonal antibody against orexin-A showed that the orexin-A level in CSF was normal at 227 pg/mL. Because degraded orexin-A fragments are largely found in the CSF and
orexin-A production is partly perturbed by the mutation at the cleavage site of prepro-orexin, the degree of degradation of orexin-A peptide in the CSF was evaluated using high performance
liquid chromatography (HPLC) followed by RIA, a method that has been recently developed. We compared the CSF orexin-A degradation pattern in this IH patient with the orexin mutation and
another IH patient without this mutation. We detected two peaks in both CSF samples that are expected to be orexin-A fragments according to previous studies30,31. However, no significant
differences in degradation in terms of the amount or separation pattern were observed (Supplementary Fig. 4). DISCUSSION The orexin pathway was not believed to contribute to the development
of IH because the CSF orexin-A levels are normal in patients with IH24. However, considering the heterogeneity of IH and the importance of orexin in sleep–wake regulation, we hypothesized
that some IH patients may carry variants associated with a dysfunction in the orexin signaling pathway and that detailed examination of orexin-related genes would be important. In this
study, we found a significant association between IH and p.Lys68Arg, which is located in the cleavage site of prepro-orexin. Low cleavage efficiency of prepro-orexin with this mutation was
confirmed. In addition, this form of prepro-orexin exhibited weak pharmacological effects on orexin signaling. MAFs of p.Lys68Arg in populations other than Asian populations are very low,
suggesting that the effect size of p.Lys68Arg would be low in other populations. A further replication analysis to confirm the association between IH and p.Lys68Arg should be performed in
Asian populations. Rare variants show typically stronger stratification than common variants32. Although we performed a PCA, a family-based association study for replication would be more
appropriate to exclude the possibility of rare variant stratification in a future study. In addition, the MAF of p.Lys68Arg in the Japanese controls was 0.32%. The sleep phenotype of these
healthy individuals, not patients, with this mutation should be analyzed to determine whether they have specific sleep characteristics. In this study, sleep phenotype data for 398 healthy
individuals were collected including the MSLT and the JESS. However, no individuals in this group have this mutation, and thus, we cannot currently address this issue. MSLT, the gold
standard for the diagnosis of central disorders of hypersomnolence, is criticized for poor precision in the differentiation of IH and narcolepsy type 233. In this study, no significant
association between narcolepsy type 2 and p.Lys68Arg was observed (Supplementary Table 1). Larger sample sizes of narcolepsy type 2 are required to provide a more reliable result about the
association between them. In the future, prospective studies should be carried out to examine whether subjects with this orexin mutation tend to have a change in their diagnosis following
MSLT. Whole-exome sequencing is an efficient method to discover rare coding variants in all genes. However, according to the gnomAD, the sequence depth of p.Lys68Arg in the whole-exome
sequencing is low because 36.9% of variant carriers have sequence depth in the 0–15 range. No variant carriers have sequence depth in the 0–15 range for whole-genome sequencing in the
gnomAD. The GC content of the coding sequence of _prepro-orexin_ is 73%. A high GC content region can affect sequence depth for exome sequencing34. GC content has less influence on sequence
depth in whole-genome sequencing than whole-exome sequencing35. These observations suggest that Sanger sequencing or whole-genome sequencing is an appropriate method to search for coding
variants in _prepro-orexin_. GWASs in individuals of European ancestry of the UK Biobank and 23andMe have identified genetic loci associated with sleep duration, chronotype, or daytime
sleepiness36,37,38. Because MAFs of p.Lys68Arg in populations other than Asian populations are extremely low, these GWASs did not identify p.Lys68Arg in _prepro-orexin_ as a locus associated
with sleep phenotypes. Therefore, p.Lys68Arg may be a population-specific variant associated with IH. In addition, our study suggested that intensive studies in populations with
non-European ancestry may be useful for identification of rare variants with larger effect sizes that cannot be found in studies of populations with European ancestry. GWAS for napping
identified a common missense variant (p.Ile308Val; rs2653349) in _OX2R_28. No significant association of this variant with IH in general was found, but when we examined the association of
this _OX2R_ mutation in orexin mutation-positive IH patients, we found a significantly higher risk for IH in the population (Table 3). This result indicated that a combination effect of
p.Ile308Val in _OX2R_ and p.Lys68Arg in _prepro-orexin_ may contribute to a higher predisposition to IH. In addition, a significant difference was found in the frequency of rs2653349 in
_OX2R_ between orexin mutation-positive IH and -negative IH groups. The result suggested that mutations in the orexin signaling pathway can be used as a marker for the IH subclass. In this
analysis, all patients with this mutation were heterozygous carriers. Because they have one normal allele (Lys), the phenotypes are considered to be different from those with narcolepsy.
Decreased efficiency of prepro-orexin cleavage may lead to a decreased amount of the mature form of orexin A and B, resulting in difficulty in maintenance of wakefulness. A mutation in
_OX2R_ causes autosomal recessive canine narcolepsy in Dobermans16. Although heterozygotes do not show spontaneous narcoleptic symptoms, they exhibit unambiguous cataplexy-like symptoms
after administration of drugs that act on cholinergic and monoaminergic systems39. This result suggests that these drugs can induce side effects in people with the p.Lys68Arg mutant allele.
On the other hand, because nonpeptide orexin receptor agonists have been developed for the treatment of narcolepsy40, orexin receptor agonists may have a treatment effect in IH patients that
carry p.Lys68Arg. We assessed the pharmacological effects of four orexin peptides on orexin signaling (Fig. 2a, b). Orexin-B induced β-arrestin recruitment that was weaker than orexin-A for
OX1R, whereas similar levels of orexin-A and -B were observed for OX2R. The results were consistent with a previous report14. In the OX1R analysis, both wild-type and mutant prepro-orexin
peptides induced β-arrestin recruitment that was much lower than that of orexin-A for OX1R and that was similar to that of orexin-B. Both prepro-orexin peptides induced β-arrestin
recruitment that was also lower compared with that of orexin-A and orexin-B. These results indicated that prepro-orexin peptides that are not appropriately processed cannot bind to or cannot
function as ligands for OX1R or OX2R (Fig. 2c). Orexin-A levels are generally measured with an RIA using a polyclonal antibody against orexin-A. Therefore, patients with IH carrying one
mutant allele of the p.Lys68Arg substitution are expected to show a normal level of orexin-A in the CSF. In fact, we confirmed that the CSF orexin-A level of a patient with the orexin
mutation was normal, although only one patient was analyzed. Degradation of orexin-A in one patient with the orexin mutation and another patient without the mutation was also evaluated with
recently developed technology that utilizes HPLC and RIA; no significant differences between the samples were found (Supplementary Fig. 4). In this analysis, two peaks were detected in the
CSF samples. Integrated analysis of the CSF peptidome and proteome found two peptide variants (residues 34–47 and 34–49) derived from the N-terminal part of orexin-A30. Both peptides contain
cysteine moieties that form disulfide bonds (Cys39-Cys45 and Cys40-Cys47), providing stability to the peptide. Taken together, we suggest that either or both of the two peaks detected in
our study are two peptides containing cysteine moieties in the N-terminal part of orexin-A (Supplementary Fig. 5). No cysteine moieties are present in orexin-B or in other regions of
orexin-A. These would be mostly degraded in the CSF by other factors. Therefore, we assume that no abnormality is present in the degradation process in the patient with the mutant allele.
However, considering that mutant prepro-orexin is less processed by PCSK1 and PCSK2 and that the regular RIA method largely detects degenerated fragments, the orexinergic system may be
partially impaired in IH patients with the mutant allele newly identified in this study (Fig. 2c). In the present study, we found that a portion of IH patients was associated with p.Lys68Arg
in _prepro-orexin_ and that this variant is an East Asian-specific mutation. Further association studies in other populations are needed to test whether other rare genetic variants in
_prepro-orexin_, _OX1R_, and _OX2R_ affect the development of IH. IH is probably a heterogeneous disease, suggesting that associations between IH and genes other than those associated with
the orexin pathway may exist. Therefore, much effort, such as greatly increasing the sample sizes and whole-genome analyses, is required to identify more genetic variants associated with IH.
International collaboration is necessary to collect sufficient DNA samples for future studies. METHODS SUBJECTS The sample set was composed of 598 patients with IH and 9826 healthy controls
in a Japanese population (initial set: 440 patients and 8380 controls; replication set: 158 patients and 1446 controls). Physician sleep specialists diagnosed the patients with IH according
to the International Classification of Sleep Disorders third edition (ICSD-3)1. The statistical power of the replication study was calculated at the significance level of 0.05. We set the
frequency of the susceptibility allele and the odds ratio to be 0.005 and 5.5, respectively, which were estimated from the initial stage41. The power was estimated to be ~0.8. Additional
clinical details (PSG and MSLT) for a subset of IH patients for whom additional data were available were provided by several collaborative institutions. The reliability and validity of the
Japanese version of the ESS have been confirmed in the Japanese population, and the JESS is used as a subjective measure of daytime sleepiness of patients42. We obtained scores of the JESS
from 313 IH patients in unmedicated conditions. Genotyping for the _HLA-DQB1_ locus in 592 patients with IH was conducted with a Luminex Multi-Analyte Profiling system with WAKFlow HLA
typing kits (Wakunaga Pharmaceutical, Wakunaga, Hiroshima, Japan). Clinical and demographic characteristics of the patients are shown in Table 2 and Supplementary Table 4, respectively.
Regarding the 8380 controls in the initial sample set, we utilized data from subjects provided by the jMorp (https://jmorp.megabank.tohoku.ac.jp/201911/)43,44. The previous study provided
demographic characteristics of the jMorp subjects44. We also studied 235 patients with narcolepsy type 2 and 514 patients with narcolepsy type 1. Narcolepsy type 2 exhibits REM sleep
abnormalities and has the same symptoms as type 1 except for cataplexy. Because narcolepsy type 2 is a central disorder of hypersomnolence as defined in the ICSD-31, we included narcolepsy
type 2 as a case group in the present study. All subjects provided written informed consent. This study was approved by the Human Genome, Gene Analysis Research Ethics Committee of the
University of Tokyo (G0910-(32)) and the Research Ethics Committee of Tokyo Metropolitan Institute of Medical Science (21-10). VARIATION SCREENING, ASSOCIATION STUDIES, AND STATISTICAL
ANALYSES PCR and Sanger sequencing were used to screen the exons for genetic variants in _prepro-orexin_, _OX1R_, and _OX2R_. Sequences of amplification and sequencing primers are shown in
Supplementary Table 5. This variation screening was performed in 195 patients with IH. Rare missense and loss-of-function variants with a MAF of <5% were selected. When two or more
patients carried a specific variant, we conducted association studies between the variant and IH. The variant was genotyped with the Taqman method in 598 patients with IH (initial set: _n_ =
440; replication set: _n_ = 158), 235 patients with narcolepsy type 2, 514 patients with narcolepsy type 1, and 1446 healthy controls. Genetic data from 8380 subjects of the jMorp
(whole-genome sequence) were included as controls. For in silico analyses, SIFT45, PolyPhen-246, LRT47, Mutation Taster48, Mutation Assessor49, and PROVEAN50 were used to predict the
pathogenicity of a variant by referring to a previous study51. SNPs (rs2653349 and rs141639071) in _OX2R_ were genotyped with the Genome-Wide Human SNP Array 6.0, described later, and the
Taqman method. Comparisons of frequencies between two groups were done using the Fisher exact test. Clinical characteristics were compared between mutation-positive IH patients and -negative
IH patients using the t test or Fisher exact test, because the distributions of age, sex, and body mass index were not significantly different between the two groups. The significance level
was set at _P_ < 0.05 for all statistical analyses. Genomic positions refer to GRCh38/hg38. GENOME-WIDE SNP TYPING Genome-wide SNP typing was performed in 13 orexin mutation-positive IH
patients, 116 orexin mutation-negative IH patients, and 420 healthy controls using the Genome-Wide Human SNP Array 6.0 according to the manufacturer’s protocols (Affymetrix Inc., Santa
Clara, CA). Data generated by the array were analyzed with GeneChip Operating Software and Genotyping Console 4.0 (Affymetrix Inc). We included SNPs that showed genotyping call rates of
>97%, MAFs of >5%, and _P_ values of more than the threshold of the Hardy–Weinberg equilibrium (_P_ > 0.001), which was evaluated using the Chi-2 test. We checked unknown familial
relationships between subjects in this study with PIHAT values as calculated by PLINK 1.952. When calculating PIHAT values, linkage disequilibrium-based SNP pruning was conducted (_r_2 <
0.5). PCA was performed using the EIGENSTRAT program53 and PLINK 1.9 to evaluate population stratification or differences in genetic background among the study subjects genotyped with the
SNP array. In the PCA, 45 JPT (Japanese in Tokyo, Japan), 90 CEU (Utah residents with Northern and Western European ancestry from the CEPH collection), 90 YRI (Nigeria Yoruba in Ibadan,
Nigeria), and 45 CHB (Han Chinese in Beijing, China) were utilized that were derived from the International HapMap project. PCSK1 AND PCSK2 ENZYME ACTIVITY ASSAY Human orexin-A and orexin-B
peptides were purchased from the PEPTIDE Institute (Osaka, Japan). Human wild-type orexin-A cleavage site substrate pLTLGKR-AMC
(pyroleucine–threonine–leucine–glutamine–lysine–arginine–aminomethylcoumarin) and human mutant orexin-A cleavage site substrate pLTLGRR-AMC
(pyroleucine–threonine–leucine–glutamine–arginine–arginine–aminomethylcoumarin) were synthesized by the PEPTIDE Institute. Recombinant human PCSK1 and PCSK2 were purchased from R & D
Systems (Minneapolis, MN). Enzyme activity studies were performed according to the manufacturer’s protocols. In brief, PCSK1 activity was measured as the rate of cleavage of 200 μM synthetic
fluorogenic substrates, pLTLGKR-AMC or pLTLGRR-AMC, in 50 μL assay buffer (pH 6.0) including 25 mM MES, 5 mM CaCl2, 1% Brij-35, and 4 μg/mL recombinant human PCSK1 at 37 °C for 60 min.
PCSK2 activity was measured as the rate of cleavage of 100 μM synthetic fluorogenic substrates, pLTLGKR-AMC or pLTLGRR-AMC, in 50 μL assay buffer (pH 5.0) including 50 mM NaOAc, 100 mM NaCl,
0.5% Brij-35, and 0.4 μg/mL recombinant human PCSK2 at 37 °C for 30 min. Cleaved fluorogenic substrate measurements were obtained eight times at 380-nm excitation and 460-nm emission using
the endopoint mode in an EnSpire Workstation (PerkinElmer Co., Ltd., Waltham, MA). PATHHUNTER Β-ARRESTIN ASSAY To test the effect of prepro-orexin peptides against OX1R and OX2R, we used the
ligand-induced β-arrestin recruitment system. β-arrestins are ubiquitously expressed in all cell types and function in the desensitization of G-protein-coupled receptors (GPCR), control of
GPCR intracellular trafficking, and activation of GPCRs in multiple signaling pathways54,55,56,57. PathHunter eXpress β-arrestin GPCR cells are engineered to co-express ProLink (PL)-tagged
GPCR and Enzyme Acceptor (EA)-tagged β-arrestin. Activation of GPCR-PL induces β-arrestin-EA recruitment, resulting in complementation of the two β-galactosidase enzyme fragments (EA and
PL). The resulting functional enzyme hydrolyzes substrate to generate a chemiluminescent signal. PathHunter eXpress β-arrestin human OX1R or human OX2R CHO-K1 cells (DiscoverX, Fremont, CA)
were seeded in AssayComplete Cell Plating Reagent (DiscoverX) and incubated for 48 h at 37 °C and 5% CO2. Then, cells were treated with designated concentrations of human orexin-A, orexin-B,
wild-type prepro-orexin, or mutant prepro-orexin for 90 min at 37 °C and 5% CO2. The resulting active enzymes hydrolyze the detection substrate (DiscoverX) to generate light, which was
measured sequentially in duplicate using an EnSpire plate reader (PerkinElmer Co., Ltd.). MEASUREMENT OF CSF OREXIN Orexin-A levels in CSF were measured with a commercially available 125I
RIA kit using a polyclonal antibody (RK-003-30, Phoenix Pharmaceuticals, Burlingame, CA). The orexin-A levels were defined as low (≤110 pg/mL), intermediate (>110 to ≤200 pg/mL), or
normal (>200 pg/mL). To assess degradation of the orexin-A peptide in the CSF, 1-mL CSF samples were injected onto HPLC (Model 526 HPLC pump, Alltech) at a flow rate of 1 ml/min and
linear gradient of 10–60% acetonitrile/0.1% trifluoroacetic acid over 40 min. Samples were separated by μBondapak C18 column 3.9 × 300 mm, 10 μM 125 A, (Waters Corporation, Milford, MA). The
fractions were collected every minute. The orexin-A levels in each fraction were dried up using a vacuum centrifuge system (SpeedVac, Savant). After resuspension in 250 µl of deionized
water, 100 µl was used in duplicates to measure orexin-A immunoreactive peaks in RIA. An in-house orexin-A antibody (1:200) and 125I-labeled orexin-A isotope (T-003-30, Phoenix
Pharmaceuticals) were used. The detailed protocol was described in the methods section of a previous study31. REPORTING SUMMARY Further information on research design is available in the
Nature Research Reporting Summary linked to this article. DATA AVAILABILITY The genome-wide data can be accessed upon application to NBDC Human Database
(https://humandbs.biosciencedbc.jp/en/) (NBDC research ID: hum0264, Japanese Genotype-phenotype Archive (JGA) accession number: JGAS000508). All remaining data are within the manuscript and
its Supporting Information files. REFERENCES * ICSD-3. International Classification of Sleep Disorders. 3rd edn, (American Academy of Sleep Medicine, 2014). * Billiard, M. & Sonka, K.
Idiopathic hypersomnia. _Sleep. Med. Rev._ 29, 23–33 (2016). Article PubMed Google Scholar * Billiard, M. & Dauvilliers, Y. Idiopathic hypersomnia. _Sleep. Med. Rev._ 5, 349–358
(2001). Article CAS PubMed Google Scholar * Ali, M., Auger, R. R., Slocumb, N. L. & Morgenthaler, T. I. Idiopathic hypersomnia: clinical features and response to treatment. _J. Clin.
Sleep. Med._ 5, 562–568 (2009). Article PubMed PubMed Central Google Scholar * Roth, B. Idiopathic hypersomnia: a study of 187 personally observed cases. _Int. J. Neurol._ 15, 108–118
(1981). CAS PubMed Google Scholar * Miyagawa, T. & Tokunaga, K. Genetics of narcolepsy. _Hum. Genome Var._ 6, 4 (2019). Article PubMed PubMed Central Google Scholar * Mignot, E.
et al. Complex HLA-DR and -DQ interactions confer risk of narcolepsy-cataplexy in three ethnic groups. _Am. J. Hum. Genet._ 68, 686–699 (2001). Article CAS PubMed PubMed Central Google
Scholar * Juji, T., Satake, M., Honda, Y. & Doi, Y. HLA antigens in Japanese patients with narcolepsy. All the patients were DR2 positive. _Tissue Antigens_ 24, 316–319 (1984). Article
CAS PubMed Google Scholar * Partinen, M. et al. Increased incidence and clinical picture of childhood narcolepsy following the 2009 H1N1 pandemic vaccination campaign in Finland. _PLoS
One_ 7, e33723 (2012). Article CAS PubMed PubMed Central Google Scholar * Hallberg, P. et al. Pandemrix-induced narcolepsy is associated with genes related to immunity and neuronal
survival. _EBioMedicine_ 40, 595–604 (2019). Article PubMed PubMed Central Google Scholar * Ambati, A. et al. Kleine-Levin syndrome is associated with birth difficulties and genetic
variants in the TRANK1 gene loci. _Proc. Natl Acad. Sci. USA._ 118, https://doi.org/10.1073/pnas.2005753118 (2021). * Miyagawa, T. et al. An association analysis of HLA-DQB1 with narcolepsy
without cataplexy and idiopathic hypersomnia with/without long sleep time in a Japanese population. _Hum. Genome Var._ 2, 15031 (2015). Article PubMed PubMed Central Google Scholar *
Tanida, K. et al. Genome-wide association study of idiopathic hypersomnia in a Japanese population. _Sleep. Biol. Rhythms_ 20, 137–148 (2022). Article Google Scholar * Sakurai, T. et al.
Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. _Cell_ 92, 1 page following 696 (1998). * Chemelli, R. M.
et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. _Cell_ 98, 437–451 (1999). Article CAS PubMed Google Scholar * Lin, L. et al. The sleep disorder
canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. _Cell_ 98, 365–376 (1999). Article CAS PubMed Google Scholar * Nishino, S., Ripley, B., Overeem, S.,
Lammers, G. J. & Mignot, E. Hypocretin (orexin) deficiency in human narcolepsy. _Lancet_ 355, 39–40 (2000). Article CAS PubMed Google Scholar * Peyron, C. et al. A mutation in a
case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. _Nat. Med._ 6, 991–997 (2000). Article CAS PubMed Google Scholar * Hor, H. et
al. A missense mutation in myelin oligodendrocyte glycoprotein as a cause of familial narcolepsy with cataplexy. _Am. J. Hum. Genet._ 89, 474–479 (2011). Article CAS PubMed PubMed
Central Google Scholar * Degn, M. et al. Rare missense mutations in P2RY11 in narcolepsy with cataplexy. _Brain_ 140, 1657–1668 (2017). Article PubMed Google Scholar * Hungs, M., Lin,
L., Okun, M. & Mignot, E. Polymorphisms in the vicinity of the hypocretin/orexin are not associated with human narcolepsy. _Neurology_ 57, 1893–1895 (2001). Article CAS PubMed Google
Scholar * Latorre, D. et al. T cells in patients with narcolepsy target self-antigens of hypocretin neurons. _Nature_ 562, 63–68 (2018). Article CAS PubMed Google Scholar * Luo, G. et
al. Autoimmunity to hypocretin and molecular mimicry to flu in type 1 narcolepsy. _Proc. Natl Acad. Sci. USA_ 115, E12323–E12332 (2018). Article CAS PubMed PubMed Central Google Scholar
* Mignot, E. et al. The role of cerebrospinal fluid hypocretin measurement in the diagnosis of narcolepsy and other hypersomnias. _Arch. Neurol._ 59, 1553–1562 (2002). Article PubMed
Google Scholar * Anderson, K. N., Pilsworth, S., Sharples, L. D., Smith, I. E. & Shneerson, J. M. Idiopathic hypersomnia: a study of 77 cases. _Sleep_ 30, 1274–1281 (2007). Article
PubMed PubMed Central Google Scholar * Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. _Nature_ 536, 285–291 (2016). Article CAS PubMed PubMed Central
Google Scholar * Gao, Y. et al. PGG.Han: the Han Chinese genome database and analysis platform. _Nucleic Acids Res._ 48, D971–D976 (2020). Article CAS PubMed Google Scholar * Dashti, H.
S. et al. Genetic determinants of daytime napping and effects on cardiometabolic health. _Nat_. _Commun._ 12, 900 (2021). https://doi.org/10.1038/s41467-020-20585-3. * Helwig, M. et al.
PC1/3 and PC2 gene expression and post-translational endoproteolytic pro-opiomelanocortin processing is regulated by photoperiod in the seasonal Siberian hamster (Phodopus sungorus). _J.
Neuroendocrinol._ 18, 413–425 (2006). Article CAS PubMed Google Scholar * Zougman, A. et al. Integrated analysis of the cerebrospinal fluid peptidome and proteome. _J. Proteome Res._ 7,
386–399 (2008). Article CAS PubMed Google Scholar * Sakai, N., Matsumura, M., Lin, L., Mignot, E. & Nishino, S. HPLC analysis of CSF hypocretin-1 in type 1 and 2 narcolepsy. _Sci.
Rep._ 9, 477 (2019). Article PubMed PubMed Central Google Scholar * Mathieson, I. & McVean, G. Differential confounding of rare and common variants in spatially structured
populations. _Nat. Genet._ 44, 243–246 (2012). Article CAS PubMed PubMed Central Google Scholar * Trotti, L. M., Staab, B. A. & Rye, D. B. Test-retest reliability of the multiple
sleep latency test in narcolepsy without cataplexy and idiopathic hypersomnia. _J. Clin. Sleep. Med._ 9, 789–795 (2013). Article PubMed PubMed Central Google Scholar * Clark, M. J. et
al. Performance comparison of exome DNA sequencing technologies. _Nat. Biotechnol._ 29, 908–914 (2011). Article CAS PubMed PubMed Central Google Scholar * Wang, Q., Shashikant, C. S.,
Jensen, M., Altman, N. S. & Girirajan, S. Novel metrics to measure coverage in whole exome sequencing datasets reveal local and global non-uniformity. _Sci. Rep._ 7, 885 (2017). Article
CAS PubMed PubMed Central Google Scholar * Jones, S. E. et al. Genome-wide association analyses of chronotype in 697,828 individuals provides insights into circadian rhythms. _Nat.
Commun._ 10, 343 (2019). Article CAS PubMed PubMed Central Google Scholar * Lane, J. M. et al. Genome-wide association analyses of sleep disturbance traits identify new loci and
highlight shared genetics with neuropsychiatric and metabolic traits. _Nat. Genet._ 49, 274–281 (2017). Article CAS PubMed Google Scholar * Dashti, H. S. et al. Genome-wide association
study identifies genetic loci for self-reported habitual sleep duration supported by accelerometer-derived estimates. _Nat. Commun._ 10, 1100 (2019). Article PubMed PubMed Central Google
Scholar * Mignot, E. et al. Heterozygosity at the canarc-1 locus can confer susceptibility for narcolepsy: induction of cataplexy in heterozygous asymptomatic dogs after administration of a
combination of drugs acting on monoaminergic and cholinergic systems. _J. Neurosci._ 13, 1057–1064 (1993). Article CAS PubMed PubMed Central Google Scholar * Irukayama-Tomobe, Y. et
al. Nonpeptide orexin type-2 receptor agonist ameliorates narcolepsy-cataplexy symptoms in mouse models. _Proc. Natl Acad. Sci. USA_ 114, 5731–5736 (2017). Article CAS PubMed PubMed
Central Google Scholar * Ohashi, J. et al. Comparison of statistical power between 2 * 2 allele frequency and allele positivity tables in case-control studies of complex disease genes.
_Ann. Hum. Genet._ 65, 197–206 (2001). Article CAS PubMed Google Scholar * Takegami, M. et al. Development of a Japanese version of the Epworth Sleepiness Scale (JESS) based on item
response theory. _Sleep. Med._ 10, 556–565 (2009). Article PubMed Google Scholar * Yamaguchi-Kabata, Y. et al. iJGVD: an integrative Japanese genome variation database based on
whole-genome sequencing. _Hum. Genome Var._ 2, 15050 (2015). Article PubMed PubMed Central Google Scholar * Tadaka, S. et al. 3.5KJPNv2: an allele frequency panel of 3552 Japanese
individuals including the X chromosome. _Hum. Genome Var._ 6, 28 (2019). Article PubMed PubMed Central Google Scholar * Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of
coding non-synonymous variants on protein function using the SIFT algorithm. _Nat. Protoc._ 4, 1073–1081 (2009). Article CAS PubMed Google Scholar * Adzhubei, I. A. et al. A method and
server for predicting damaging missense mutations. _Nat. Methods_ 7, 248–249 (2010). Article CAS PubMed PubMed Central Google Scholar * Chun, S. & Fay, J. C. Identification of
deleterious mutations within three human genomes. _Genome Res._ 19, 1553–1561 (2009). Article CAS PubMed PubMed Central Google Scholar * Schwarz, J. M., Rodelsperger, C., Schuelke, M.
& Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. _Nat. Methods_ 7, 575–576 (2010). Article CAS PubMed Google Scholar * Reva, B., Antipin, Y.
& Sander, C. Predicting the functional impact of protein mutations: application to cancer genomics. _Nucleic Acids Res._ 39, e118 (2011). Article CAS PubMed PubMed Central Google
Scholar * Choi, Y., Sims, G. E., Murphy, S., Miller, J. R. & Chan, A. P. Predicting the functional effect of amino acid substitutions and indels. _PLoS One_ 7, e46688 (2012). Article
CAS PubMed PubMed Central Google Scholar * Genovese, G. et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. _Nat. Neurosci._ 19,
1433–1441 (2016). Article CAS PubMed PubMed Central Google Scholar * Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. _Gigascience_ 4,
7 (2015). Article PubMed PubMed Central Google Scholar * Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. _PLoS Genet._ 2, e190 (2006). Article
PubMed PubMed Central Google Scholar * Gurevich, E. V. & Gurevich, V. V. Arrestins: ubiquitous regulators of cellular signaling pathways. _Genome Biol._ 7, 236 (2006). Article PubMed
PubMed Central Google Scholar * Lefkowitz, R. J. & Shenoy, S. K. Transduction of receptor signals by beta-arrestins. _Science_ 308, 512–517 (2005). Article CAS PubMed Google
Scholar * Moore, C. A., Milano, S. K. & Benovic, J. L. Regulation of receptor trafficking by GRKs and arrestins. _Annu. Rev. Physiol._ 69, 451–482 (2007). Article CAS PubMed Google
Scholar * Tian, X., Kang, D. S. & Benovic, J. L. beta-arrestins and G protein-coupled receptor trafficking. _Handb. Exp. Pharm._ 219, 173–186 (2014). Article CAS Google Scholar
Download references ACKNOWLEDGEMENTS The authors are deeply grateful to all participants in the present study. This study was supported by a Practical Research Project for Rare/Intractable
Diseases grant from the Japan Agency for Medical Research and Development (AMED), Grants-in-Aid for Scientific Research (B) from the Ministry of Education, Culture, Sports, Science and
Technology of Japan (15H04709 and 19H03588), Grants-in-Aid for Scientific Research (C) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (21K07534), and
Grants-in-Aid from the Takeda Science Foundation. The funders had no role in study design, data collection, analysis, the decision to publish, or preparation of the manuscript. AUTHOR
INFORMATION Author notes * These authors contributed equally: Taku Miyagawa, Susumu Tanaka. AUTHORS AND AFFILIATIONS * Sleep Disorders Project, Department of Psychiatry and Behavioral
Sciences, Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan Taku Miyagawa, Mihoko Shimada, Tohru Kodama & Makoto Honda * Department of Human Genetics, Graduate School of
Medicine, The University of Tokyo, Tokyo, Japan Taku Miyagawa, Mihoko Shimada, Kotomi Tanida, Kayoko Kato & Katsushi Tokunaga * Department of Anatomy, Faculty of Medicine, Kansai Medical
University, Osaka, Japan Susumu Tanaka & Masaaki Kitada * Genome Medical Science Project (Toyama), National Center for Global Health and Medicine, Tokyo, Japan Mihoko Shimada &
Katsushi Tokunaga * Sleep and Circadian Neurobiology Laboratory, School of Medicine, Stanford University, Stanford, CA, USA Noriaki Sakai & Seiji Nishino * Department of Neuropsychiatry,
Kurume University School of Medicine, Fukuoka, Japan Nozomu Kotorii, Yuji Hashizume, Kimihiro Ogi, Hiroshi Hiejima & Naohisa Uchimura * Kotorii Isahaya Hospital, Nagasaki, Japan Nozomu
Kotorii & Tatayu Kotorii * Ariyoshi Sleep Clinic, Fukuoka, Japan Yu Ariyoshi * International Institute for Integrative Sleep Medicine (WPI-IIIS), University of Tsukuba, Ibaraki, Japan
Takashi Kanbayashi, Hideaki Kondo & Kazuo Mishima * Ibaraki Prefectural Medical Center of Psychiatry, Ibaraki, Japan Takashi Kanbayashi * Department of Neuropsychiatry, Akita University
Graduate School of Medicine, Akita, Japan Aya Imanishi & Kazuo Mishima * Sleep Center, Kuwamizu Hospital, Kumamoto, Japan Azusa Ikegami & Kazuhiko Kume * Department of Laboratory
Medicine, National Center Hospital, National Center of Neurology and Psychiatry, Tokyo, Japan Yuichi Kamei * Kamisuwa Hospital, Nagano, Japan Yuichi Kamei * Department of Sleep-Wake
Disorders, National Institute of Mental Health, National Center of Neurology and Psychiatry, Tokyo, Japan Akiko Hida & Kazuo Mishima * Department of Psychiatry, Hannan Hospital, Osaka,
Japan Yamato Wada & Kenji Kuroda * Department of Neurology, Dokkyo Medical University, Tochigi, Japan Masayuki Miyamoto & Koichi Hirata * Department of Psychiatry, Shiga University
of Medical Science, Shiga, Japan Masanori Takami & Naoto Yamada * Department of Psychiatry and Neurology, Asahikawa Medical University, Hokkaido, Japan Yoshiyuki Tamura & Shigeru
Chiba * Department of Neurology, Junwakai Memorial Hospital, Miyazaki, Japan Yukari Taniyama & Kazuhito Tsuruta * Department of Nursing, Faculty of Health Science, Fukui Health Science
University, Fukui, Japan Naoto Omata * Department of Neuropsychiatry, Faculty of Medical Sciences, University of Fukui, Fukui, Japan Naoto Omata & Tomoyuki Mizuno * Department of
Psychiatry, Tokyo Women’s Medical University School of Medicine, Tokyo, Japan Shunpei Moriya * Department of Neurology, Neuro-Muscular Center, National Omuta Hospital, Fukuoka, Japan
Hirokazu Furuya * Department of Neurology, Kochi Medical School, Kochi University, Kochi, Japan Hirokazu Furuya * Department of Pediatrics, Yamagata University Faculty of Medicine, Yamagata,
Japan Mitsuhiro Kato * Department of Pediatrics, Showa University School of Medicine, Tokyo, Japan Mitsuhiro Kato * Institute of CNS Pharmacology, Tokyo, Japan Jun Ishigooka * Department of
Sleep Medicine, Shiga University of Medical Science, Shiga, Japan Masako Okawa * Japan Foundation for Neuroscience and Mental Health, Tokyo, Japan Masako Okawa * Department of Somnology,
Tokyo Medical University, Tokyo, Japan Masako Okawa & Yuichi Inoue * Department of Stem Cell Biology, Institute of Molecular Genetics and Embryology, Kumamoto University, Kumamoto, Japan
Kazuhiko Kume * Department of Neuropharmacology, Graduate School of Pharmaceutical Sciences, Nagoya City University, Aichi, Japan Kazuhiko Kume * Yoyogi Sleep Disorder Center,
Neuropsychiatric Research Institute, Tokyo, Japan Yuichi Inoue * Seiwa Hospital, Institute of Neuropsychiatry, Tokyo, Japan Makoto Honda Authors * Taku Miyagawa View author publications You
can also search for this author inPubMed Google Scholar * Susumu Tanaka View author publications You can also search for this author inPubMed Google Scholar * Mihoko Shimada View author
publications You can also search for this author inPubMed Google Scholar * Noriaki Sakai View author publications You can also search for this author inPubMed Google Scholar * Kotomi Tanida
View author publications You can also search for this author inPubMed Google Scholar * Nozomu Kotorii View author publications You can also search for this author inPubMed Google Scholar *
Tatayu Kotorii View author publications You can also search for this author inPubMed Google Scholar * Yu Ariyoshi View author publications You can also search for this author inPubMed Google
Scholar * Yuji Hashizume View author publications You can also search for this author inPubMed Google Scholar * Kimihiro Ogi View author publications You can also search for this author
inPubMed Google Scholar * Hiroshi Hiejima View author publications You can also search for this author inPubMed Google Scholar * Takashi Kanbayashi View author publications You can also
search for this author inPubMed Google Scholar * Aya Imanishi View author publications You can also search for this author inPubMed Google Scholar * Azusa Ikegami View author publications
You can also search for this author inPubMed Google Scholar * Yuichi Kamei View author publications You can also search for this author inPubMed Google Scholar * Akiko Hida View author
publications You can also search for this author inPubMed Google Scholar * Yamato Wada View author publications You can also search for this author inPubMed Google Scholar * Masayuki
Miyamoto View author publications You can also search for this author inPubMed Google Scholar * Masanori Takami View author publications You can also search for this author inPubMed Google
Scholar * Hideaki Kondo View author publications You can also search for this author inPubMed Google Scholar * Yoshiyuki Tamura View author publications You can also search for this author
inPubMed Google Scholar * Yukari Taniyama View author publications You can also search for this author inPubMed Google Scholar * Naoto Omata View author publications You can also search for
this author inPubMed Google Scholar * Tomoyuki Mizuno View author publications You can also search for this author inPubMed Google Scholar * Shunpei Moriya View author publications You can
also search for this author inPubMed Google Scholar * Hirokazu Furuya View author publications You can also search for this author inPubMed Google Scholar * Mitsuhiro Kato View author
publications You can also search for this author inPubMed Google Scholar * Kayoko Kato View author publications You can also search for this author inPubMed Google Scholar * Jun Ishigooka
View author publications You can also search for this author inPubMed Google Scholar * Kazuhito Tsuruta View author publications You can also search for this author inPubMed Google Scholar *
Shigeru Chiba View author publications You can also search for this author inPubMed Google Scholar * Naoto Yamada View author publications You can also search for this author inPubMed
Google Scholar * Masako Okawa View author publications You can also search for this author inPubMed Google Scholar * Koichi Hirata View author publications You can also search for this
author inPubMed Google Scholar * Kenji Kuroda View author publications You can also search for this author inPubMed Google Scholar * Kazuhiko Kume View author publications You can also
search for this author inPubMed Google Scholar * Naohisa Uchimura View author publications You can also search for this author inPubMed Google Scholar * Masaaki Kitada View author
publications You can also search for this author inPubMed Google Scholar * Tohru Kodama View author publications You can also search for this author inPubMed Google Scholar * Yuichi Inoue
View author publications You can also search for this author inPubMed Google Scholar * Seiji Nishino View author publications You can also search for this author inPubMed Google Scholar *
Kazuo Mishima View author publications You can also search for this author inPubMed Google Scholar * Katsushi Tokunaga View author publications You can also search for this author inPubMed
Google Scholar * Makoto Honda View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS T.M., S.T., M.S., K.T., and M.H. contributed to study concept
and design. T.M., S.T., M.S., N.S., and K.T. performed formal analyses. N.K., T.K., Y.A., Y.H., K.O., H.H., T.K., A.I., A.I., Y.K., A.H., Y.W., M.M., M.T., H.K., Y.T., Y.T., N.O., T.M.,
S.M., H.F., M.K., J.I., K.T., S.C., N.Y., M.O., K.H., K.K., K.K., N.U., Y.I., K.M., and M.H. recruited clinical cases and performed clinical examinations. T.M., S.T., M.S., N.S., K.T., K.K.,
M.K., T.K., and S.N. contributed to the experiments. S.N., K.M., K.T., and M.H. supervised the study. T.M. and M.S. performed validation. T.M., S.T., M.S., and M.H. participated in drafting
the article. All authors interpreted the data, provided conceptual comments, and approved the final manuscript. Taku Miyagawa and Susumu Tanaka contributed equally to this work.
CORRESPONDING AUTHOR Correspondence to Taku Miyagawa. ETHICS DECLARATIONS COMPETING INTERESTS Inoue Yuichi has received grants and payment for lectures including service on speakers’
bureaus, and has provided expert testimony for MSD K.K., Takeda Pharmaceutical Co. Ltd., and Eisai Co. Ltd. Makoto Honda has received consulting fees from Takeda Pharmaceutical Co. Ltd. The
other authors have no competing interests to declare. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and
institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTAL MATERIAL REPORTING SUMMARY CHECKLIST RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original
author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the
article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use
is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Miyagawa, T., Tanaka, S., Shimada, M. _et al._ A rare genetic variant in the
cleavage site of _prepro-orexin_ is associated with idiopathic hypersomnia. _npj Genom. Med._ 7, 29 (2022). https://doi.org/10.1038/s41525-022-00298-w Download citation * Received: 30
September 2021 * Accepted: 04 March 2022 * Published: 12 April 2022 * DOI: https://doi.org/10.1038/s41525-022-00298-w SHARE THIS ARTICLE Anyone you share the following link with will be able
to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing
initiative