Play all audios:
ABSTRACT This study aimed to investigate four of the eight PFN-1 mutations that are located near the actin-binding domain and determine the structural changes due to each mutant and unravel
how these mutations alter protein structural behavior. Swapaa’s command in UCSF chimera for generating mutations, FTMAP were employed and the data was analyzed by RMSD, RMSF graphs, Rg,
hydrogen bonding analysis, and RRdisMaps utilizing Autodock4 and GROMACS. The functional changes and virtual screening, structural dynamics, and chemical bonding behavior changes, molecular
docking simulation with two current FDA-approved drugs for ALS were investigated. The highest reduction and increase in Rg were found to exist in the G117V and M113T mutants, respectively.
The RMSF data consistently shows changes nearby to this site. The _in silico_ data described indicate that each of the mutations is capable of altering the structure of PFN-1 _in vivo_. The
potential effect of riluzole and edaravone two FDA approved drugs for ALS, impacting the structural deviations and stabilization of the mutant PFN-1 is evaluated using _in silico_ tools.
Overall, the analysis of data collected reveals structural changes of mutant PFN-1 protein that may explain the neurotoxicity and the reason(s) for possible loss and gain of function of
PFN-1 in the neurotoxic model of ALS. SIMILAR CONTENT BEING VIEWED BY OTHERS CONFORMATIONAL DYNAMICS OF Α-SYNUCLEIN AND STUDY OF ITS INTRAMOLECULAR FORCES IN THE PRESENCE OF SELECTED
COMPOUNDS Article Open access 03 November 2023 IN SILICO ANALYSIS OF TUBA4A MUTATIONS IN AMYOTROPHIC LATERAL SCLEROSIS TO DEFINE MECHANISMS OF MICROTUBULE DISINTEGRATION Article Open access
06 February 2023 IDENTIFICATION OF ALLOSTERIC FINGERPRINTS OF ALPHA-SYNUCLEIN AGGREGATES IN MATRIX METALLOPROTEASE-1 AND SUBSTRATE-SPECIFIC VIRTUAL SCREENING WITH SINGLE MOLECULE INSIGHTS
Article Open access 06 April 2022 INTRODUCTION Amyotrophic lateral sclerosis (ALS) is an inexorable neurodegenerative disease and is the subject of multiple worldwide intense investigation
to determine the mechanisms for motor neuron death and treatment development to cure or slow the progression of the disease1. ALS (also known as Lou Gehrig’s disease) can progress quickly
with a life expectancy of only 3–5 years after diagnosis and is considered as one of the devastating diseases. Due to the selective loss of upper and lower motor neurons, ALS Patients become
progressively weak (mainly muscle weakness), and ultimately will cause death. ALS is largely sporadic (~ 90–95%) and only a small fraction of patients have the familial forms (fALS)
(5–10%), with an estimated overall incidence of 2–4 per 100,000, with the ratio of 1:1.4 women vs men2. Studies on the DNA samples from fALS patients led to the discovery of mutations in
numerous genes associated with the disease and involved in the mechanism of neurodegeneration3. In this study, we bring focus to a recent and important mutation in such a molecule with the
potential to unravel and shed light on the mechanism in axonal pathology, cytoskeletal importance in motor neurons in ALS. This molecule is the profilin-1 gene (_PFN-1_). Mutations in the
PFN-1 have been identified as one of the genetic causes for fALS4,5. The protein expressed from the _PFN-1_ gene is named PFN-1 and it is a multifunctional protein. PFN-1 is a critical
protein in the conversion of monomeric (G)-actin to filamentous (F)-actin6. The approach used in this study has the power and capability to generate critical data towards unraveling the
structural deviations and shed necessary light on the mechanisms underlying mutant PFN-1 neurotoxicity in ALS. The computational techniques on this approach are based on the report by
Potapov _et al_.7. These in silico tools enabled us to investigate the structure of PFN-1 in the wild-type and mutant forms, rapidly and cost-effectively. In contrast to conventional
approaches, the usable data generated and available from clinical trials at considerable expense and time and the pre-clinical studies that require the use of a large number of animals are
significantly burdensome that in silico studies have the edge8. Albeit none of these approaches can provide all the answers needed to swiftly produce FDA approved medicine to treat a disease
such as ALS. We posit that both approaches are needed and should be used with accuracy and precision. Systems biology utilizes modular bioinformatics platforms to enable studies of
molecular interactions at specific experimental setups and procedures that identify subcellular machinery responsible for specific functions in cells, tissues, and organ systems resulting in
physiological behaviors9,10. This bioinformatics-system integrated interface considers modularity in systems biology and uses the biophysics-based and molecular mechanics tools with the
choice on the selected functionally which becomes a vital component in the building and understanding of a circuit or a machine11. In this study, the perturbations caused by these four
mutations (C70G, M113T, E116G, and G117V) on the actin-binding area of the PFN-1 structure were the subject of investigation and we examined how they may impact its function that leads to a
toxic “gain/loss of function”. There is a sense of urgency to study for better understanding and facilitating the possible treatment for this disease. We report our investigations on mutant
PFN-1 using computational analytical tools under specific and pre-determined conditions, tested, and carried data analysis using computer-aided tools, data mining approaches, and systems
biology. It is of great interest and importance to investigate the effect of the other 4 mutations (A19T, T108M, R135W, Q138L) on the PLP domain of PFN1, which will be pursued in the future.
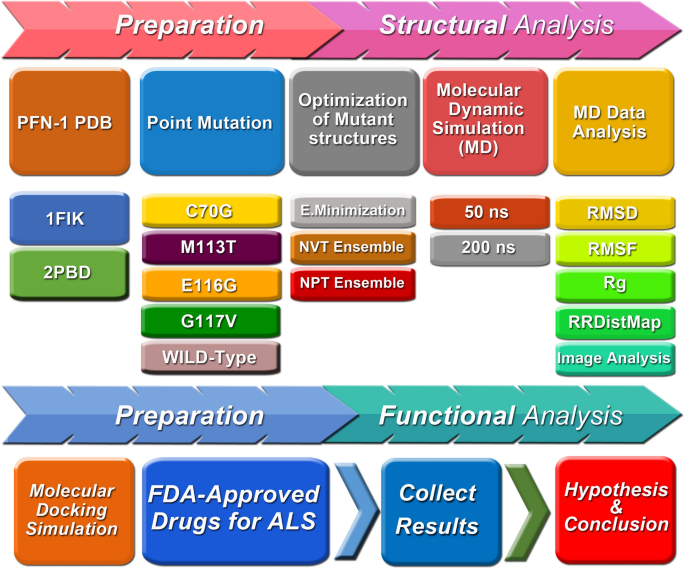
METHODS AND MATERIALS A comprehensive in silico analysis was employed for this study by utilizing a variety of bioinformatics tools including software and databases. The structure of
wild-type and mutant PFN-1 was examined, and the changes were explored at depth to shed light on potential reasons for the mutant PFN-1’s toxicities in ALS (Fig. 1). WILD-TYPE PFN-1 Protein
database searches in RCSB.org were performed using compiled crystal structure data for PFN-1. Amongst the 168,599 biological molecules’ crystal structures and macromolecules registered in
the protein data-bank to date, 15 of them belong to PFN-1 (Supplementary Table S1). These 15 PFN-1 crystal structures that are registered in the protein data-bank (Rcsb.org) includes two
structures with mutations (4X1M, 4X25), the other nine structures are linked to other proteins such as polyproline chain containing proteins (e.g. VASP) and actin (2PAV, 2PBD, 3CHW, 1AWI,
1CF0, 1CJF, 6NAS, 6NBW, 6NBE) binding proteins. The other four structures of PFN-1 unbound to any other protein or macromolecules, their amino acid chains crystal structure data are fully
resolved (1FIK, 1FIL, 1PFL and 4X1L). We have considered studying two of the crystal structures; 1FIK code from the unbounded set, contains one molecule in the asymmetric unit with
diffraction of at least 2.0 Å and 2PBD code from the bounded set as a profilin-actin complex structure (Fig. 1). PFN-1 PROTEIN DATA BASE IDENTIFICATION (PDBID): 1FIK The crystal structure of
PFN-1 coded 1FIK that is unique in the protein chain and has a phosphate ion and several water molecules bound to it and analyzed in this study. Our initial observation and analysis of the
structure using the University of California San Francisco (UCSF) Chimera program12, gave us the notion of removing excess molecules of phosphate and water. This approach enabled us to
create PFN-1 protein data that can be easily entered into the molecular dynamics (MD) simulation environment platform. The process of crystallography typically exerts stress on proteins due
to reduction in humidity, and temperature change may affect and cause bias to the actual structure of the protein13. To obtain a more realistic structure close to its de novo state, we used
a molecular dynamics simulation method and subsequently investigated the structural changes in PFN-1 by subjecting PFN-1 to 200 ns (ns) in MD simulations. MOLECULAR DYNAMICS SIMULATION All
MD simulations calculations were performed using GROMACS software (Ver.2018)14 and Ubuntu operating system (OS) Ver.1615. The polarizable simulations were carried out in a 3D cubic box as a
unit cell with a simple point charge (SPC) water model. The protonation state of ionizable amino acids with pKa of any given amino acid is assumed at pH 7. The positive and negative ions
were added to the solvent to neutralize the surface charge of the molecule and to establish normal conditions; for example, we added 2 negative ions of chlorine (Cl) to neutralize the total
of 9202 water molecules in the formed space as a solvent. The Optimized Potentials for Liquid Simulations for all atoms force field model also has been developed and used in MD simulation
series. The molecules were used as solvents in the aquatic environment with the SPC model. For the residue interactions, the thickness of this solvent layer is assumed to be fixed with a
threshold of at least 10 Å. We also ensured that no steric clashes or inappropriate geometry. in a solvated system of PFN-complexes exist. On the other hand, we were mindful of the fact that
performing simulations without the energy minimization step, may result in higher energy creation and disruption of structure. This phenomenon is due to the addition of hydrogen and
breakage of the hydrogen bond network in the water. Therefore, to eliminate these unwanted forces, the structure is relaxed through a process called energy minimization (EM). To avoid
(protein) position deviation during MD simulations, it was first performed during a 50,000 spatial limiting step as NVT and NPT ensembles equilibration where N is the number of molecules, V
is the volume of the system, T is the temperature and P is the pressure. These were used to stabilize the temperature, the pressure, and the density of the system, respectively.
Equilibration of pressure was conducted under an NPT ensemble, wherein the number of particles, pressure, and temperature were all kept constant16. Finally, softwares such as Qt Grace (is a
tool to make two-dimensional plots of numerical data, generate graphs and analyze the results)17 and UCSF Chimera were used to further visualize and analyze the data. All the simulations
were repeated three times for each set of parameters and in many independent runs. MODELING AND OPTIMIZATION, STRUCTURES The molecular structures required in this study were obtained from
the PubChem structural database18 with their unique chemical identification number (CID) and CAS number as numerical identifier assigned by chemical abstract service (CAS) as listed in Fig.
2. Then, we attempted to optimize the structural energy to obtain the proper spatial geometry structures. Further, the GaussView program (ver. 05)19 provided the necessary outputs for the
Gaussian quantum steering calculations (ver. 03) package, while making initial changes. In this program, we obtained the optimal structure by using the DFT/B3LYP protocol by command line20,
thereby generating the optimized model of structures (Fig. 2). MOLECULAR DOCKING SIMULATION Molecular docking simulation by Autodock (ver. 4.2)21 was used to investigate for virtual
screening of riluzole and edaravone and inspection of interaction with PFN-1 (at 200 ns) of molecular dynamic trajectories. This method is partly used to estimate the activity of various
molecules that accelerate the process before drug synthesis and during drug discovery. This technique is based on the atomic structure and the interaction of atoms relative to each other. In
this study, molecular structures were set for 250 × docking runs with an appropriately necessary algorithm to analyze the displacement at the PFN-1 surface. During these steps, the
molecular structures were flexible and found their ideal sites for binding at the PFN-1 molecule. Molecular mechanics are set to suit and calculate the ligand binding energy when interacting
with macromolecules. We used the Genetic algorithm-based method for docking, with the program running on a Linux operating system supercomputer system with a total of 16 cores, 32 logical
cores of CPU, and 1 compatible GPU. Ubuntu distribution (Linux operating system), (ver. 16). By the Autogrid4 section of Autodock4 Suite, (ver. 4.2), electrostatic interactions and
pre-calculations of grid maps, were obtained for each atom. To adjust the size of the box to cover the PFN-1, a grid map consisted of 110 × 100 × 80 Å points around the PFN-1WT before 200 ns
and 120 × 100 × 80 around PFN-1WT after MD simulation, and Å grid spacing of 0.375 Å (fourth of the carbon–carbon bond length) was used. The center of the grid was set to the coordinates of
the PFN-1 and for each mutation, the respective box sizes were determined. For flexible PFN-1-ligand docking, the Lamarckian Genetic Algorithm was examined for finding the optimized
condition for docking structural outcomes. The results were evaluated and analyzed by UCSF Chimera, Autodock suite, and Ligplot software program which automatically generates schematic 2-D
representations of protein–ligand complexes from standard protein data-bank file input used to generate schematic diagrams22. MUTANT PFN-1 PROTEINS To find the mechanism of PFN-1 toxicity
and to explain the harmful and deleterious effects of the point mutation-induced changes to the PFN-1 protein23, we created four of the mutant structures reported on the PFN-1 in ALS5,
primarily to investigate the structural changes of the protein and then examine the functional changes of protein due to mutations using UCSF-chimera command line to create point mutation.
The four mutations studied here are located near the actin-binding domain of the human PFN-1. Point mutation modifications were implemented by the Swapaa command code (one or more protein
residues to change can be specified in a single command) in the UCSF Chimera command line, ver. 1.13 (Supplementary Table S2). The main theme for this study was to focus on the structure of
PFN-1 including all point mutations in the actin-binding area. The post-mutational structure evaluation involves only a limited number of laboratory experiments2,24. Notably, the validation
of the point mutation on the protein crystal structure located in the PFN-1 is confirmed after molecular dynamics simulation. In the motion reconstruction and protein residual dynamics, an
equilibrium for mutant residue needed to be established in terms of energy and spatial geometry with other surroundings. It may be possible that mutation in an amino acid alters the
secondary structure of the protein or alters the active site or regulatory position of the protein25. MD simulations at 25, 50, and 100 million steps, which takes 50, 100, and 200 ns,
respectively, according to the method described in the previous section, was done by the Gromacs program which is a molecular dynamics package mainly designed for simulations of proteins,
lipids, and nucleic acids (2018) for four mutated structures similar to the PFN-1WT structure. In the first step, a detailed structural examination of the mutant PFN-1 was performed by
visual analysis with the help of structural display tools. The next step was to find changes in the PFN-1 function due to each mutation, using molecular docking simulation methods. To find
these changes, we collected data with 250 × docking runs for PFN-1 binding to each structure. RESULTS AND DISCUSSION MUTATION IMPACTS PFN-1 PROTEIN STRUCTURE ALS is a complex degenerative
disorder with genetic and environmental components, and it has been difficult to conceptualize and treat it. To evaluate the impact of each mutation, we analyzed the modified structural
characteristics of PFN-1. We have represented these by Root-Mean Square Deviation (RMSD)26,27, backbone (basic structure), and sidechain (showing amino acid residues), Root Mean Square
Fluctuation (RMSF), and evaluated based on the position of alpha carbons (Cα) using 50 ns and 200 ns MD simulations28. These are indicated as backbone data, the radius of gyration (Rg) and
were analyzed throughout the trajectory and followed up with visual analysis. To further demonstrate and better show the exact timing of the deviation in the molecule, we have produced short
movies (see Supplementary Section) on the structural changes where the largest deviations occur to help in the visualization of the graphical data presented in the figures and diagrams.
PFN1 POINT MUTATIONS ON THE ACTIN-BINDING DOMAIN The loss-of-function in PFN1 due to Cys 70 to Gly point mutation leads to the toxicity that kills motor neurons in ALS. Cys is a polar amino
acid with uncharged radical groups (R can be any group in which a carbon or hydrogen atom is attached to the rest of the amino acid residue) changed to a nonpolar amino acid with the
smallest R group. The 3D structure of PFN-1 shows that the location of Cys 70 is on the beta-sheet structure and changing it to Gly on this residue may not destabilize the beta-sheet but it
will cause a loss or inability to form any disulfide bond, which would cause a detrimental effect on the proper folding and interaction of other residues needed for the quaternary structure
and linking to other proteins such as actin. The Met 113 to Thr mutation is also toxic in ALS, and it is a nonpolar amino acid with an aliphatic R group changed to a polar amino acid with an
uncharged R group. This mutation is also located on the beta-strand structure. The presence of amino acid residues with a hydroxyl group increases the hydrogen bonding capacity. The Glu 116
to Gly is another ALS linked mutation, which is a negatively charged R group changed to a nonpolar amino acid with the smallest R group. This change would influence the interaction of the
neighboring residues and impact the three dimensional (3-D) structural integrity. The Gly 117 to Val is considered toxic in ALS as it is proven in in vitro and in vivo models. Gly is a
nonpolar aliphatic residue that is substituted to Val, and although the Val has the same nonpolar aliphatic R group, this point mutation reduces the conformational flexibility of the loop
region between the alpha-helix and beta-strand (Fig. 3). The location of each mutation in a fully folded PFN-1 structure (some on the surface and some deep in the structure) with annotation
of the properties of each amino acid residue is listed in Fig. 4. RMSD MEASURES FOR THE COMPARISON OF TWO MOLECULAR ENSEMBLES The RMSD diagrams represent the spatial displacement rate and
moving parts of the protein model during the simulation, and it serves as a measure of protein stability due to a mutation. The smaller the value of RMSD during the simulation translates to
the lower protein stability. During each simulation period, compared to the initial (crystal) state of the simulation, and only with respect to the major atoms of the molecule, amino acid
units were calculated for each structure with and without a mutation29. The RMSD data generated from a triplicate runs for 200 ns each and compared to the state of the changes with the
original structure of the crystal as shown in (Fig. 5). In a short movie, we show the timing and area of the molecules with the largest deviations, wherein C70G movie at 90–110 ns, M113T at
5–25 ns, E116G at 0–20 ns, and for G117V at 10–25 ns are in the display (Supplementary Movie). RADIUS OF GYRATION (RG) CALCULATIONS AND INTERATOMIC DISTANCES DURING MD SIMULATIONS To find
deviation in the parameters relating to the structure of proteins in terms of compression and density as a measure of the stability of the protein after the MD simulation were analyzed. The
Rg data globular proteins are based on the principle that the more compact the protein is, the more stable it will be30 making Rg is suitable to investigate the characteristics for
simulating protein structure that we ran for 50–200 ns MD simulation and measured the radius of gyration. The rate of change for M113T is 0.04 nm and the rate of change of RG for C70G is
0.01 nm. The E116G and G117G have Rg changes close to 0.02 nm (Fig. 6). ROOT MEAN SQUARE FLUCTUATION (RMSF) Investigation of the dynamic behavior of Cα atoms in the structure of proteins
contains sufficient information to investigate the impact of each motion that reflects in the general motions of the structure. To find the changes in the motion and structural fluctuation
we used Cα oscillations (backbone) in an RMSF test. We found the rate of change in fluctuation for M113T to be 0.5 nm maximum in the backbone, 0.6 nm in sidechain in the area of 90–100 amino
acid sequences. The rate of change fluctuation for C70G was 0.35 nm in the backbone and 0.55 nm in sidechain in the area of 35–45 residues. The rate of change in the fluctuation for E116G
is 0.35 in the backbone and 0.45 nm maximum in the sidechain and the area of 90–100 residues. In G117G we found the rate change of fluctuation to be 0.35 nm in the backbone and 0.45 nm in
the side chain with two peaks in the area of 90–100 and 115–125 residues (Fig. 7). SUPERIMPOSITION OF PFN-1 STRUCTURE In order to compare the PFN-1 structure affected by mutation after MD
simulation to the PFN-1WT, it was needed to set up, train, and observe their differences by overlapping the initial structure of the crystal over the final structure obtained after the
simulation. We generated data and analyzed it with the goal in mind to determine and measure the structural changes in the protein structure using 200 ns simulations. Data show an overlap in
the state of the primary crystal structure and each of the mutated structures. Visual analysis, before and after MD simulations, allowed us to test our hypotheses, examine protein function
and enabled us to corroborate our data with previously published laboratory and clinical data. In the visual analysis of PFN-1 in its crystallized form, the protein binding segments to other
structures were precisely pinpointed and the amino acids that play a role in binding to actin are identified31. We have identified Hydrogen bonding and hydrophobic interaction amino acid
residues in the actin-binding domain. The amino acid residues, Arg 74, Gly 120, Lys 125 and Thr 89, Val 60, Ser 71, Lys 90, and Lys 69, are found to participate in hydrogen bonding. The
counter binding residues that form actin molecules are His 371, Glu 361, Tyr 169, Tyr 166, Ile 287, Asp 286, and 288. The amino acid residues participating in hydrophobic interaction are
identified and shown in (Fig. 8)32. The wild-type structures are coded white in Fig. 9 and refer to the crystal structure, and the predefined colors for the mutant structures as given in the
figure legends (Fig. 9). This figure is created using data from RMSD, residue-residue distance maps (RRDisMap). The RMSD values (in nm) for C70G, M113T, E116G and G117V are 0.159, 0.237,
0.272, and 0.231, respectively. Structural superimposition and difference in RRDisMap were calculated for selected conformers by their evaluation of the RRDistMap. The α-helices, β-strands,
and turns, whose interface form the core of the domain are expected to differ for the wild-type and mutant PFN-1 structures. The differences in wild-type and mutant PFN-1’s core structure
was further evaluated. This was done by calculating the overlapped area of residues in the secondary structures, such as helices. The UCSF chimera program was used and the calculated
parameters of distance, and the standard deviation denoted in the analysis as shown in (Fig. 10)33. Superimposition of wild-type and mutant 3-D structures was created based on 1FIK with
original crystal structure from protein data-bank. Those data points were used as a base and the sequence data entered to generate 3-D structures for the mutants with 200 ns MD simulations.
The changes in the RMSD values related to the wild-type structure of PFN-1 before and after the simulation were determined. The RMSD value for wild-type was found to be 1.957 nm and the
value for M113T was 2.078 nm which is greater than the wild-type. The C70G and G117V mutations’ RMSD values were 1.560 and 1.598 nm respectively. The RMSD value of E116G is very close to the
wild-type value (Fig. 9) and in acceptable cut-off range, which is consistent with the clinical observation that this mutation was reported to be benign, in contrast to other mutants that
were found to cause ALS4. THE IMPACT OF THE MUTATION ON THE FUNCTION OF PFN-1 To investigate the effect of the mutation on the PFN-1 function, we used the "molecular docking to
ensembles of protein structures approach followed by MD simulations" as described in the “Methods and materials” section. The docking energy of riluzole and edaravone with PFN-1 to
determine whether they interact, the degree of interaction, and at which location was first characterized using PFN-1WT and mutant forms34. The docking data for each molecule listed in three
rows (Supplementary Table S3). The first row (from left to right) refers to the lowest binding energy state (most stable), shown as a negative value, and the second number refers to the
repetitions of distinct clusters at the lowest binding energy. The second row belongs to the docking binding energy with the highest number in the cluster (abundance in cluster) followed by
the number in clusters per mutation. The third row refers to the values that are indicating less favorable and unlikely conditions/conformation to occur as the negative binding energies tend
to be higher in value. Therefore, the binding energy value and the number in clusters are shown in (Supplementary Table S3). CAPTURING, MODELING, AND OPTIMIZATION OF STRUCTURES We describe
our findings in capturing and preparing small molecule structures (riluzole and edaravone) to perform molecular docking simulations based on the previously discussed topics as shown in (Fig.
2). The results of molecular docking simulations of PFN-1WT and mutant PFN-1 structures over the small molecular structures were extracted with details (Supplementary Table S3) and (Fig.
11). We have presented analytical images of the molecular docking simulation (Fig. 12a–c). The steps and parameters of simulating molecular docking are described above. CONFORMATIONAL AND
STRUCTURAL CHANGES The in silico evidence drives our speculation that structural changes due to a given mutation (e.g. E116G) disturb the protein structure in the actin-binding region.
Therefore, the actin-binding region is critically important and highly vulnerable to mis-conformation and explains the loss of binding affinity to actin molecules. The Rg changes indicate
the value of protein structure compression (Fig. 6). The RRDistMaps variations indicate the largest difference in the E116G mutation as shown in (Fig. 10). Interestingly the RMSD data
consistently show the largest change in E116G mutation. Furthermore, RMSD data analysis revealed that the M113T mutation is located deep in the central region of PFN-1 compared to the
wild-type crystal structure listed in (Supplementary Table S1). This mutation is not juxtaposed to the actin-binding domain; however, it may alter the 3-D structural integrity and stability
of the PFN-1. It is of great interest that RMSF analysis of MD simulations demonstrated altered dynamic properties for these mutant PFN-1 that we studied. The largest alteration in the
protein dynamics was found to be caused by G117V mutations. FUNCTIONAL CHANGES IN PFN-1 AND INTERACTION OF RILUZOLE AND EDARAVONE The effects of point mutations on "protein
function" according to parameters set in silico and before and after scenarios, which are as shown in (Fig. 12a–c), unravel atomic details of the alteration that could be the basis for
“gain-of-function” or “loss-of-function” of PFN-1 in ALS. We have carefully utilized the computer-generated images and checked the binding energy (Supplementary Table S3). Since ALS patients
with the PFN-1 mutation may receive treatment with the current FDA-approved drugs, we sought to examine the interaction of riluzole and edaravone with mutant PFN-1. Our objective was to
examine whether the patients with PFN-1 mutations could potentially be able to expect some clinical effect from the available and FDA approved therapies for ALS. These two drugs appear to
have an affinity to bind the mutant PFN-1, which, if confirmed, would be of great value for the impact on patients. For instance, we observed the highest affinity for the PFN-1C71G structure
of FDA-approved drugs under two conditions, before and after the mutations. Interestingly, we found that G117V and C70G mutations have the least affinity for these drugs (Fig. 11). Although
it is beyond the scope of this study to investigate these differences in the affinity to interact with riluzole or edaravone, a future study may reveal important knowledge on how each
mutant PFN-1 or other ALS-causing mutated proteins (e.g. _SOD1_ gene, TDP-43 neurofilament or gene encoding FUS) interact with drugs that patients carrying any of these mutations are
receiving (Fig. 11). The images of the mutants and wild-type PFN-1 interacting with these two ALS drugs are fully analyzed and depicted in the Fig. 12a–c. The in silico tools we employed for
our investigation in this study were instrumental in the generation of these invaluable data and enabled us to learn how each of the four mutations contributed to the structural deviations.
Our data appear to be consistent with the in vitro and in vivo observations reported by us and others. In contrast, others employed algorithmic programs such as PhD-SNP, PMUT, PolyPhen- 2,
SIFT, SNAP, SNPS&GO, SAAP, nsSNPAnalyzer, SNPeffect4.0, and I-Mutant2.0 to predict the functional and stability of mutant PFN-1 and their conclusion was to re-confirm what was already
known. Therefore, the outcome of these studies was useful although impacted by a shorter simulation of 100 ns as the study by Pereira et al.35. The approach that we have used is popular and
frequently utilized in protein structural analysis adding to our confidence for the choice of tools that allowed us a study with a greater depth that revealed nuances that weren’t known.
CONCLUSION Biophysical and MD simulation analysis series of wild-type and mutant PFN-1 proteins with the variation revealed critical information that we have generated on the conformational
and functional changes in mutant PFN-1. This may elucidate the underlying mechanism of neurotoxicity observed in ALS. Further investigation and confirmation of these in silico data can shed
light on the toxicity of mutant PFN-1 and may turn highly informative in pre-clinical studies for the development of a novel therapy for ALS patients with mutant PFN-1, as well as possible
treatments to prevent in sALS cases. Once confirmed, this approach may be used on other ALS-causing mutants and suspected proteins to be involved in the neurotoxicity of both sALS and fALS.
The in silico method has great power and real potential to facilitate the investigation of the changes in the mutant protein outside the wet laboratory7. It also enables researchers to
develop and test variety of hypotheses using computer programs otherwise wouldn’t be possible in the wet laboratory. This study on the four ALS-causing point mutations in the actin-binding
domain of PFN-1 protein reveals the structural deviations in the structure and the possible adverse impact on the PFN-1 interaction and its function. Altogether, our analysis using modeling
and simulation tools detected important changes in the PFN-1 which revealed atomic-level mechanisms of PFN-1 protein structure. The structural deviations in the overall structure measured by
RMSD, RMSF, the Radius of Gyration, RRDisMap, as a new series of parametric indexes into the stability and 3-D formation are interesting and reveal nuances for the mutant PFN-1. This data
also shed light on the quaternary structure of the protein that may impact the function(s) of PFN-1 and could partly explain the toxic gain/loss-of-function as the mechanisms in the
neurodegeneration in ALS. Since there are two FDA-approved drugs for ALS, we investigated whether these drugs would interact with mutant PFN-1. We found both new therapeutic medications,
riluzole, and edaravone interact with mutant PFN-1, where restoring the stability of the PFN-1 structure due to the interaction of these drugs may take place. Therefore, ALS patients
carrying PFN-1 mutations may inquire about getting these drugs prescribed which can attenuate disease progression. REFERENCES * Longo, D.L. Robert H. Brown, D. Phil., MD, and Ammar
AlChalabi, Ph. D., FRCP, Dip. _Stat. N. Engl. J. Med._ 377, 162–172 (2017). * Dervishi, I. & Ozdinler, P. H. Incorporating upper motor neuron health in ALS drug discovery. _Drug Discov.
Today_ 23(3), 696–703 (2018). Article Google Scholar * Nguyen, H. P., Van Broeckhoven, C. & van der Zee, J. ALS genes in the genomic era and their implications for FTD. _Trends Genet._
34(6), 404–423 (2018). Article CAS Google Scholar * Wu, C.-H. _et al._ Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. _Nature_ 488(7412), 499–503 (2012).
Article ADS CAS Google Scholar * Alkam, D., Feldman, E. Z., Singh, A. & Kiaei, M. Profilin1 biology and its mutation, actin (g) in disease. _Cell. Mol. Life Sci._ 74(6), 967–981
(2017). Article CAS Google Scholar * Gau, D., Veon, W., Shroff, S. G. & Roy, P. The VASP–profilin1 (Pfn1) interaction is critical for efficient cell migration and is regulated by
cell–substrate adhesion in a PKA-dependent manner. _J. Biol. Chem._ 294(17), 6972–6985 (2019). Article CAS Google Scholar * Potapov, V., Cohen, M. & Schreiber, G. Assessing
computational methods for predicting protein stability upon mutation: good on average but not in the details. _Protein Eng. Des. Sel._ 22(9), 553–560 (2009). Article CAS Google Scholar *
Hodos, R. A., Kidd, B. A., Shameer, K., Readhead, B. P. & Dudley, J. T. In silico methods for drug repurposing and pharmacology. _Wiley Interdiscip. Rev. Syst. Biol. Med._ 8(3), 186–210
(2016). Article Google Scholar * Ideker, T., Galitski, T. & Hood, L. A new approach to decoding life: Systems biology. _Annu. Rev. Genom. Hum. Genet._ 2(1), 343–372 (2001). Article
CAS Google Scholar * Kirschner, M. W. The meaning of systems biology. _Cell_ 121(4), 503–504 (2005). Article CAS Google Scholar * Noble, D. Claude Bernard, the first systems biologist,
and the future of physiology. _Exp. Physiol._ 93(1), 16–26 (2008). Article Google Scholar * Pettersen, E. F. _et al._ UCSF Chimera—A visualization system for exploratory research and
analysis. _J. Comput. Chem._ 25(13), 1605–1612 (2004). Article CAS Google Scholar * Srivastava, A., Nagai, T., Srivastava, A., Miyashita, O. & Tama, F. Role of computational methods
in going beyond X-ray crystallography to explore protein structure and dynamics. _Int. J. Mol. Sci._ 19(11), 3401 (2018). Article Google Scholar * Abraham, M., van der Spoel, D., Lindahl,
E., Hess, B. GROMACS user manual version 2018. www.gromacs.org (2018). * Van Der Spoel, D. _et al._ GROMACS: Fast, flexible, and free. _J. Comput. Chem._ 26(16), 1701–1718 (2005). Article
Google Scholar * Lemkul, J. From proteins to perturbed Hamiltonians: A suite of tutorials for the GROMACS-2018 molecular simulation package [article v1.0]. _Living J. Comput. Mol. Sci._
1(1), 5068 (2018). Google Scholar * Turner, P., Stambulchik, E., Unix-Like, A. Grace (plotting tool). (2012). * Kim, S. _et al._ PubChem 2019 update: Improved access to chemical data.
_Nucleic Acids Res._ 47(D1), D1102–D1109 (2019). Article Google Scholar * GaussView V. 4.1, Roy Dennington II, Todd Keith and John Millam, Semichem. Inc, Shawnee Mission, KS. (2007). *
Frisch, M. Gaussian 03 Rev. E. 01. http://www.gaussian.com/. (2004). * Morris, G. M. _et al._ AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. _J. Comput.
Chem._ 30(16), 2785–2791 (2009). Article CAS Google Scholar * Wallace, A. C., Laskowski, R. A. & Thornton, J. M. LIGPLOT: A program to generate schematic diagrams of protein-ligand
interactions. _Protein Eng. Des. Sel._ 8(2), 127–134 (1995). Article CAS Google Scholar * Nekouei, M. _et al._ Changes in biophysical characteristics of PFN1 due to mutation causing
amyotrophic lateral sclerosis. _Metab. Brain Dis._ 33(6), 1975–1984 (2018). Article CAS Google Scholar * Petrov, D., Mansfield, C., Moussy, A. & Hermine, O. ALS clinical trials
review: 20 years of failure. Are we any closer to registering a new treatment?. _Front. Aging Neurosci._ 9, 68 (2017). Article Google Scholar * Jubb, H. C. _et al._ Mutations at
protein–protein interfaces: Small changes over big surfaces have large impacts on human health. _Prog. Biophys. Mol. Biol._ 128, 3–13 (2017). Article CAS Google Scholar * Maiorov, V. N.
& Crippen, G. M. Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. _J. Mol. Biol._ 235, 625–634 (1994). Article CAS Google
Scholar * Cazals, F. & Tetley, R. Characterizing molecular flexibility by combining least root mean square deviation measures. _Proteins Struct. Funct. Bioinform._ 87(5), 380–389
(2019). Article CAS Google Scholar * Low, B.-C. Root mean square fluctuation of a weak magnetic field in an infinite medium of homogeneous stationary turbulence. _Astrophys. J._ 173, 549
(1972). Article ADS Google Scholar * Kufareva, I. & Abagyan, R. _Methods of Protein Structure Comparison_ 231–257 (Springer, 2011). Google Scholar * Lobanov, M. Y., Bogatyreva, N.
& Galzitskaya, O. Radius of gyration as an indicator of protein structure compactness. _Mol. Biol._ 42(4), 623–628 (2008). Article CAS Google Scholar * Berjanskii, M. & Wishart,
D. S. NMR: Prediction of protein flexibility. _Nat. Protoc._ 1(2), 683 (2006). Article CAS Google Scholar * Meng, E. C. _et al._ Tools for integrated sequence-structure analysis with UCSF
Chimera. _BMC Bioinform._ 7(1), 1–10 (2006). Article Google Scholar * Chen, J. E., Huang, C. C. & Ferrin, T. E. RRDistMaps: A UCSF Chimera tool for viewing and comparing protein
distance maps. _Bioinformatics_ 31(9), 1484–1486 (2015). Article CAS Google Scholar * Ngan, C. H. _et al._ FTMAP: Extended protein mapping with user-selected probe molecules. _Nucleic
Acids Res._ 40(W1), W271–W275 (2012). Article CAS Google Scholar * Pereira, G. R. C., Tellini, G. H. A. S. & De Mesquita, J. F. In silico analysis of PFN1 related to amyotrophic
lateral sclerosis. _PLoS ONE_ 14(6), e0215723 (2019). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS This work was partially supported by UAMS startup fund to M.K. We
acknowledge and thank Lily Kiaei, a high school intern at RockGen Therapeutics, LLC., for her contributions in editing the manuscript, critical discussion, and feedback. Authors express
their appreciation and gratitude to Dr. Mohsen Sharifi for his valuable discussion on the methods used to generate data and his edits on this manuscript. Authors acknowledge Dr. Dara
Rahmati, director of IT at the High Performance Computing (HPC) Center, Institute for Research in Fundamental Sciences (IPM), for providing access to computer servers. Authors also
acknowledge Dr. Amirhossein Jahangir at the Computer Engineering Department at Sharif University providing continues support during the use of computer servers. AUTHOR INFORMATION Author
notes * These authors jointly supervised this work: Changiz Eslahchi and Mahmoud Kiaei. AUTHORS AND AFFILIATIONS * Department of Computer and Data Sciences, Faculty of Mathematical Sciences,
Shahid Beheshti University, Tehran, Iran Ahmad Shahir Sadr & Changiz Eslahchi * School of Biological Sciences, Institute for Research in Fundamental Sciences (IPM), 193955746, Tehran,
Iran Changiz Eslahchi * Department of Phytochemistry, Medicinal Plants and Drugs Research Institute, Shahid Beheshti University, Tehran, Iran Alireza Ghassempour * Department of Pharmacology
and Toxicology, College of Medicine, University of Arkansas for Medical Sciences, Little Rock, AR, 72205, USA Mahmoud Kiaei * Department of Neurology, College of Medicine, University of
Arkansas for Medical Sciences, Little Rock, AR, 72205, USA Mahmoud Kiaei * Department of Geriatrics, College of Medicine, University of Arkansas for Medical Sciences, Little Rock, AR, 72205,
USA Mahmoud Kiaei * RockGen Therapeutics, LLC., c/o Bioventures, LLC, 4301 W. Markham St., #831, Little Rock, AR, 72205, USA Mahmoud Kiaei Authors * Ahmad Shahir Sadr View author
publications You can also search for this author inPubMed Google Scholar * Changiz Eslahchi View author publications You can also search for this author inPubMed Google Scholar * Alireza
Ghassempour View author publications You can also search for this author inPubMed Google Scholar * Mahmoud Kiaei View author publications You can also search for this author inPubMed Google
Scholar CONTRIBUTIONS A.S.S., C.E., A.G., and M.K. contributed to the design and implementation of the research, to the analysis of the results, and to the writing of the manuscript.
CORRESPONDING AUTHORS Correspondence to Changiz Eslahchi or Mahmoud Kiaei. ETHICS DECLARATIONS COMPETING INTERESTS M.K. is the founder, president, and CEO of RockGen Therapeutics, LLC.
ADDITIONAL INFORMATION PUBLISHER'S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION
SUPPLEMENTARY INFORMATION. Supplementary Video 1. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits
use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the
Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless
indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory
regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Sadr, A.S., Eslahchi, C., Ghassempour, A. _et al._ In silico studies reveal
structural deviations of mutant profilin-1 and interaction with riluzole and edaravone in amyotrophic lateral sclerosis. _Sci Rep_ 11, 6849 (2021). https://doi.org/10.1038/s41598-021-86211-4
Download citation * Received: 15 October 2020 * Accepted: 11 March 2021 * Published: 25 March 2021 * DOI: https://doi.org/10.1038/s41598-021-86211-4 SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative