Play all audios:
ABSTRACT Breast cancer is a heterogenous disease that can be classified into multiple subtypes including the most aggressive basal-like and triple-negative subtypes. Understanding the
heterogeneity within the normal mammary basal epithelial cells holds the key to inform us about basal-like cancer cell differentiation dynamics as well as potential cells of origin. Although
it is known that the mammary basal compartment contains small pools of stem cells that fuel normal tissue morphogenesis and regeneration, a comprehensive yet focused analysis of the
transcriptional makeup of the basal cells is lacking. We used single-cell RNA-sequencing and multiplexed RNA _in-situ_ hybridization to characterize mammary basal cell heterogeneity. We used
bioinformatic and computational pipelines to characterize the molecular features as well as predict differentiation dynamics and cell–cell communications of the newly identified basal cell
states. We used genetic cell labeling to map the in vivo fates of cells in one of these states. We identified four major distinct transcriptional states within the mammary basal cells that
exhibit gene expression signatures suggestive of different functional activity and metabolic preference. Our in vivo labeling and ex vivo organoid culture data suggest that one of these
states, marked by _Egr2_ expression, represents a dynamic transcriptional state that all basal cells transit through during pubertal mammary morphogenesis. Our study provides a systematic
approach to understanding the molecular heterogeneity of mammary basal cells and identifies previously unknown dynamics of basal cell transcriptional states. SIMILAR CONTENT BEING VIEWED BY
OTHERS INTEGRATING SINGLE-CELL RNA-SEQUENCING AND FUNCTIONAL ASSAYS TO DECIPHER MAMMARY CELL STATES AND LINEAGE HIERARCHIES Article Open access 29 July 2020 DUCTAL KERATIN 15+ LUMINAL
PROGENITORS IN NORMAL BREAST EXHIBIT A BASAL-LIKE BREAST CANCER TRANSCRIPTOMIC SIGNATURE Article Open access 12 July 2022 MAMMARY CELL GENE EXPRESSION ATLAS LINKS EPITHELIAL CELL REMODELING
EVENTS TO BREAST CARCINOGENESIS Article Open access 02 June 2021 INTRODUCTION The mammary gland contains an epithelial bilayer of basal and luminal cells that function in the production and
secretion of milk from mother to offspring1,2. Multiple stem and progenitor cell populations have been identified in the basal and luminal layers, and the basal layer harbors multipotent
stem cells that are capable of generating both basal and luminal progenies and reconstituting a functional mammary gland upon transplantation3,4,5. Distinct, small pools of stem cells in the
basal layer have been identified by the expression of different markers such as _Procr_6, _Bcl11b_7_, Lgr5/Tspan8_8, _Cdh5_9, as well as by lineage-tracing _Axin2-_expressing cells10.
However, basal cell heterogeneity in the normal mammary gland has been understudied despite knowledge of the existence of such stem cells. In particular, it is not clear how non-stem basal
cells are transcriptionally organized or what their specific function and differentiation status might be. Recent studies have utilized single-cell RNA-sequencing (scRNA-seq) to examine the
normal mammary gland at single-cell resolution in both human11 and mouse9,12,13. These studies have provided foundational insights into the transcriptional landscape of the mammary
epithelium. Other studies have also coupled transcriptional and epigenetic modalities at single-cell resolution to help uncover regulators of cellular identity within the mammary
epithelium13,14. While these studies have advanced the understanding of the mammary epithelium, there still lacks a comprehensive and in-depth characterization of cellular states and
differentiation landscape within the basal layer. In this work, we aimed to characterize the transcriptional heterogeneity of mammary basal cells using scRNA-seq. Using differential gene
expression analysis, we find four major basal cell transcriptional states. We provide a bioinformatic characterization of the molecular features of the cell states that suggest differential
functional and metabolic activities. Using RNAScope, we validate the heterogeneous expression and differential enrichment of several scRNA-seq-identified basal transcriptional state markers,
_Acta2_, _Tspan8_, and _Egr2_, in the intact mammary gland tissue. Lastly, we provide genetic cell labeling data to suggest that _Egr2_-expressing transcriptional state represents a dynamic
one that all basal cells transit through during pubertal mammary morphogenesis. These findings regarding basal cell heterogeneity in the normal tissue lay the foundation for future work to
probe the heterogeneity of their malignant counterparts in basal-like and triple negative breast cancer subtypes. METHODS MICE Wild-type C57BL/6 mice (Stock #000,664) and _ROSA26__mTmG_ mice
(Stock #007,576) were purchased from the Jackson Laboratory. _Egr2-Cre_ mice were reported in a previous study15, and the following primers were used for genotyping: Forward (_Egr2-Cre_):
CGC TTC CTC GTG CTT TAC GGT AT (480-bp product); Forward (WT): TCA TCA GTC GGG TTA GAG CTG (312-bp product); Reverse: GGG CTG AGG AAG ACG ACT TTA. _Egr2-Cre_;_ROSA26__mTmG_ mice used for
experimental analysis were generated by crossing _Egr2-Cre_ males with _ROSA26__mTmG__ females_. Crossing _Egr2-Cre;ROSA26__mTmG_ males with WT females resulted in 100% GFP expression in the
mammary epithelium of female _Egr2-Cre;ROSA26__mTmG_ offspring (data not shown), suggesting germline expression of _Egr2_. Mice were maintained in a pathogen-free facility, following
procedures that conform and have been approved by the University of California Irvine Institutional Animal Care and Use Committee. FLOW CYTOMETRY Flow cytometry was performed as
described16,17. Briefly, mammary glands were isolated, minced with a razor blade, and incubated with 300 U/mL collagenase (Sigma, C9891) and 100 U/mL hyaluronidase (Sigma, H3506) for 90 min
at 37 °C. The cells were treated with red blood cell lysis buffer (Sigma, R7757) for 5 min at room temperature before treating with 0.25% Trypsin (Gibco, 25,200) for 2 min at 37 °C and
dispase II (2 mg/mL) (Stem Cell Technologies, 07,913) with 0.1 mg/mL DNase I (Sigma, DN25) for 2 min at 37 °C. Cells were stained for 30 min in the dark at room temperature using APC-CD45
(BD Biosciences, 559,864; 1:250), APC-CD31 (BD Biosciences, 551,262; 1:250), APC-Ter119 (BD Biosciences, 557,909; 1:250), PE/C7-Epcam (Bio Legend, 118,215; 1:250), and PerCP-Cy5.5-Cd49f (Bio
Legend, 555,735; 1:250). Cells were washed and stained with Sytox blue (Invitrogen, S34857; 1:1000) before flow cytometry. SCRNA-SEQ ANALYSIS scRNA-seq experiments were performed in two
separate runs using the 10X Genomics platform on FACS-sorted mammary epithelial cells from 8–9-week-old virgin females. Experimental details were as described in another study18. The
sequencing data were analyzed using R version 3.6.1 and Seurat 3.1.0. Low quality cells were filtered based on mitochondrial DNA content, total number of transcripts, and total number of
genes detected. Basal cells (_Krt14_+ cluster) were computationally isolated for further analysis. STRING version 11.0 was used to examine the protein–protein interactions using the marker
genes identified by differential expression test using Seurat. Disconnected nodes were removed from the graphs, and the default interaction score (medium confidence; 0.4) was used to
identify interactions. RNA Velocity analyses were performed using the R package velocyto.R (linear model) and nlvelo (non-linear model). The R package CellChat was used for analyzing the
ligand-receptor communications of cells. RNASCOPE RNAScope was performed as previously described19 using ACD Bio’s reagents. Briefly, #4 mammary glands from mice were frozen in OCT and
cryosectioned at 10 µm. Sectioned tissues were fixed for 1 h at room temperature with 4% PFA, and the RNAScope Multiplex Fluorescent v2 assay was run per manufacturer’s recommendations using
the following probes: _Acta2_ (319,531-C3), _Tspan8_ (842,941-C1), and _Egr2_ (497,871-C2). Images were obtained using a Zeiss LSM700 confocal microscope and quantified using FIJI software.
WHOLE-MOUNT IMMUNOFLUORESCENCE IMAGING Mammary glands (#4) of 3-week-old _Egr2-Cre;ROSA26__mTmG_ and _ROSA26__mTmG_ mice were surgically isolated from adjacent tissues and spread on glass
slides. Images were acquired using a Keyence BZ-X710 microscope (Keyence Corporation, Itasca, Illinois, USA). ETHICS AND APPROVAL AND CONSENT TO PARTICIPATE All mouse experiments have been
approved by and conform to the regulatory guidelines of the Institutional Animal Care and Use Committee of the University of California, Irvine. The study is reported in accordance with
ARRIVE guidelines. RESULTS IDENTIFICATION OF FOUR BASAL CELL TRANSCRIPTIONAL STATES IN ADULT VIRGIN MOUSE MAMMARY GLAND Previously, we performed scRNA-seq analysis on fluorescence-activated
cell sorting (FACS)-isolated mammary epithelial cells (including both basal and luminal populations) from 8–9-week-old virgin females to characterize the heterogeneous expression of
epithelial-to-mesenchymal transition (EMT)-associated genes18. However, a systematic characterization of the transcriptional diversity of basal cells has not been done in that study. To
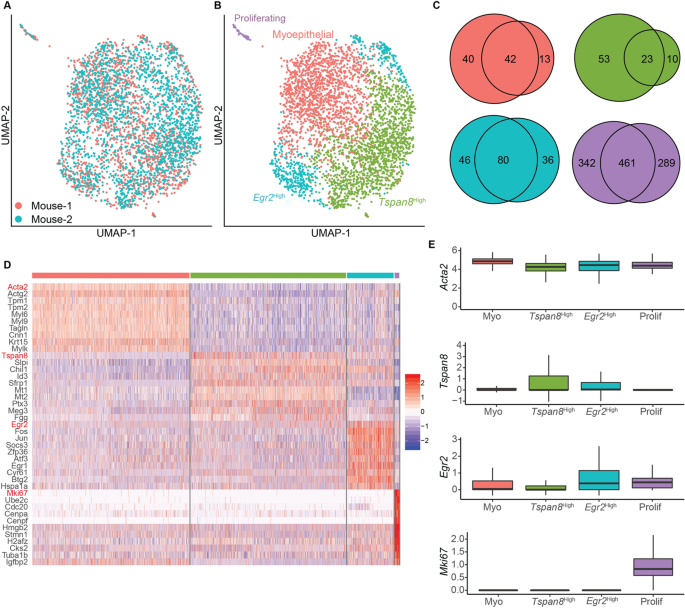
achieve this, we computationally subset the 3,651 cells that express basal cell marker _Krt14_ from the dataset for further analysis. Visualizing these basal cells in a UMAP projection
suggests that there are no obvious batch effects (Fig. 1A). Clustering and differential gene expression analyses revealed the presence of four clusters: (1) a cluster enriched for classical
myoepithelial genes (_Acta2, Actg2)_ and thus termed “myoepithelial”; (2) a _Tspan8_High cluster enriched for genes the high expression of which has been previously shown to identify stem
cells in the mammary basal compartment (_e.g. Tspan8_8, _Epcam_20); (3) an _Egr2_High cluster enriched for early response- and stress-related genes (e.g. _Egr2_, _Fos_, _Jun_)21; and (4) a
small cluster marked by proliferation-associated genes (e.g., _Mki67_, _Pcna_) (Fig. 1B–D; Supplemental Table 1). We also observed the same four cell states when each mouse was analyzed
individually (Fig. S1A–D), and the marker genes used to identify each cell state in the mice showed a high degree of overlap (Fig. 1C). Importantly, the marker genes that discriminated the
different transcriptional states were differentially expressed but not mutually exclusive (Fig. 1E). To examine the robustness of these transcriptional states, we also sequenced FACS-sorted
mammary epithelial cells from 8–9-week-old virgin mice deficient in EMT-inducing transcription factor _Zeb1_18 and computationally subset basal cells for further analysis (Fig. S1E, F). In
addition to a proliferating basal cell population, three other basal transcriptional states were observed in each mutant mouse analyzed (Fig. S1G–J). The top markers of each of these
transcriptional states were largely similar, albeit not identical, to those in the wild-type mice. Therefore, even in _Zeb1_-deficient mammary glands where basal stem cell function is
compromised18, similar molecular heterogeneity of bulk basal cells still exists, featuring four major transcriptional states. We also interrogated the published scRNA-seq data on
_KRT14_-expressing cells of the human breast from three individuals11. Generally, top marker genes for the three “non-proliferating” mouse basal cell states were detected at much lower
frequencies in these human samples (Supplemental Table 2). For example, _Acta2_, which has been previously reported to be expressed in > 96% of mouse mammary basal cells20, was detected
in 99.6% of the mouse basal cells in our scRNA-seq analysis but _ACTA2_ was only detected in 38.6% of the human _KRT14_+ cells. Moreover, _Egr2_ was expressed in 29.8% of mouse basal cells,
but _EGR2_ was only detected in 5.6% of human _KRT14_+ cells. Nevertheless, we were still able to observe transcriptional states in the human _KRT14_+ population that resembled the
“myoepithelial” and proliferating basal cell states in mouse (Fig. S1K–M). These data reveal both disparities and similarities in the transcriptional states of mammary basal cells between
mouse and human. MOLECULAR FEATURES OF THE BASAL CELL STATES Next, we sought to characterize the molecular features of the basal cell states in mice. Intrigued by their upregulated
expression of known stemness-associated genes _Tspan8_ and _Epcam_, we wondered if _Tspan8_High cells also display enriched expression of other genes reported to mark mammary basal/stem
cells (_e.g. Procr_6, _Bcl11b_7, _Cdh5_9, _Lgr5_8). Using a gene scoring approach to calculate the average expression of a “stemness gene” signature (Supplementary Table 3), we found that
the _Egr2_High basal cells had the highest average score whereas the proliferating basal cells the lowest (Fig. 2A). Overall, the discriminating power of this signature is non-remarkable.
Differential metabolic preference of mammary basal and luminal cells has been suggested, such that basal cells may prefer a glycolytic metabolism while luminal cells display increased
oxidative phosphorylation22,23. This metabolic paradigm is of interest, given that cancerous cells have increased glycolytic activity24. Using a set of hallmark gene sets defined by the
Molecular Signatures Database to score for glycolysis and oxidative phosphorylation (OxPhos) (Supplementary Table 3), we found that the proliferating basal cells have the highest glycolytic
and OxPhos signatures (Fig. 2B,C), suggesting increased requirement for energy to support cellular growth and division. Of the “non-proliferating” basal cell states, the _Egr2_High cells
scored the highest for the glycolytic signature, and the “myoepithelial” cells scored the highest for OxPhos. Interestingly, the _Tspan8_High cells scored the lowest for both signatures.
These data reveal previously unrecognized metabolic heterogeneity within the mammary basal layer. To gain further insights into the potential activity of each basal transcriptional state, we
examined the protein–protein interaction (PPI) networks of all of the marker genes that define each state. We utilized STRING25 to derive direct (physical) and indirect (functional)
associations between the marker genes. Interestingly, the number of PPIs were vastly different across the cell states, with proliferating basal cells exhibiting the highest number of PPIs
and the highest number of marker genes (Fig. 2D,E; Fig. S2A), which is consistent with their highly specialized cellular activity. Although the difference in the number of marker genes for
each “non-proliferating” cell state was small, the difference in the number of PPIs was more pronounced (Fig. 2E; Fig. S2A). For example, the numbers of PPIs in the _Tspan8_High and
“myoepithelial” cells were 15 (lowest of all four states) and 113 (second highest), respectively, whereas their numbers of unique marker genes were 31 (lowest) and 39 (second lowest). This
data suggest that the marker genes used to define the “myoepithelial” cells have a higher degree of physical and functional associations and may point to a functional consequence.
Consistently, a gene ontology (GO) term analysis using Enrichr26,27 to probe the GO Biological Processes 2018 library revealed terms related to muscle contraction for the “myoepithelial”
marker genes (Fig. S2B). Next, we performed RNA Velocity28, a computational method that calculates the relative abundances of spliced and unspliced RNA to infer the future states of single
cells. Based on the directions of the vectors arrows, which are known to associate with possible state transitions, we did not observe a clear differentiation trajectory across the different
basal cell states regardless of whether we used a linear28 (Fig. 2F) or non-linear model19 (Fig. 2G) of RNA Velocity. However, we found consistent differences in the length of the vector
arrows that suggest differential RNA dynamics among the different states. The “myoepithelial” and _Tspan8_High cells showed small RNA velocities (short or no arrows), known to associate with
either quiescent or terminally differentiated cells29,30. The _Egr2_High cells exhibited large RNA velocities in both linear and non-linear models (Fig. 2F,G), suggesting that these cells
may be in a more active and transitional cellular state compared to the others. The proliferating cells exhibited large RNA velocities in the linear but not in the perhaps more realistic
non-linear model19, and in both cases, the arrows pointed away from but not back to the “non-proliferating” cells (Fig. 2F,G), raising the possibility that they may not be able to readily
switch back to a “non-proliferating” state19. Lastly, we used CellChat31 to explore ligand-receptor pairs and infer potential signaling cross-talks within the basal cell layer. CellChat
identified ten signaling pathways that were significantly enriched within basal cells, and outgoing and incoming signals were largely heterogeneous across the cell states (Fig. 2H,I). It
appeared that the _Tspan8_High cells send the most outgoing signals, and the “myoepithelial” cells receive the most incoming signals. Interestingly, non-canonical WNT (ncWNT) signaling
surfaced as the most prominent in “myoepithelial” cells, which appeared to be the primary cells responding to WNT signals from the _Tspan8_High cells (i.e., paracrine) and, to a lesser
extent, from the “myoepithelial” cells themselves (i.e., possibly autocrine) (Fig. 2J). Collectively, our findings suggest that the _Tspan8_High transcriptional state associates with low
number of marker genes, low PPIs, low glycolysis, slow RNA dynamics, but can potentially serve as a signaling niche32, whereas the _Egr2_High transcriptional state is the most dynamic of all
states and the “myoepithelial” transcriptional state represents the most specialized state associated with a “mature” myoepithelial fate. VALIDATING BASAL TRANSCRIPTIONAL HETEROGENEITY AND
DETECTING DYNAMIC _EGR2_ EXPRESSION IN THE INTACT MAMMARY TISSUE We next sought to examine the molecular heterogeneity in bulk basal cells in the intact mammary tissue by using RNAScope to
validate the differential expression of a top marker gene from each “non-proliferating” basal transcriptional state. Specifically, we probed for the expression of _Acta2_, _Tspan8_, and
_Egr2_ mRNAs simultaneously in mammary gland of adult virgin mice. Indeed, expression of each transcriptional state marker gene was detected in only a subset of basal cells (Fig. 3A–E; Fig.
S3A–H). There did not appear to be any notable spatial patterning of these transcriptional state markers, except that a small number of _Tspan8_+ cells that are positive for K14 (indicating
a basal cell fate2,33) seem to occupy positions between basal and luminal layers (Fig. 3A-D; Fig. S3A–H). Consistent with scRNA-seq data, _Acta2_ showed the highest coverage in basal cells
(~ 39%) (Fig. 3E). This is less than the 99% detection rate in our scRNA-seq data, likely reflecting differential sensitivity of different detection methods. _Tspan8_ transcripts were
detected in ~ 23% of the basal cells (Fig. 3E), and its most abundant expression was actually seen in the luminal layer, a finding confirmed by our scRNA-seq data (Fig. S3I). _Egr2_ mRNAs
were detected in ~ 27% of the basal cells (Fig. 3E) and expression was barely detectable in luminal cells, and this basal-restricted pattern was also seen in our scRNA-seq data (Fig. S3J).
Hierarchically clustering basal cells by their expression (number of RNAScope signal foci/per nucleus) of the marker genes revealed groups of single basal cells that expressed a single
marker gene (Fig. 3E). Overall, only a small fraction of basal cells (~ 3%) expressed all three markers, and a sizable fraction (~ 35%) of cells did not exhibit detectable expression of any
of the makers examined (Fig. 3A′–D′,A″–D″,E). Together, these data uncover the scRNA-seq-predicted transcriptional heterogeneity in the basal compartment of the intact mammary tissue.
Expression of early-response genes such as _Egr2_ has been reported as an artifact of stresses induced by the tissue dissociation/sequencing protocol21, but our detection of
_Egr2_-expressing basal cells in the mammary tissue suggests the physiological existence of an _Egr2_High basal transcriptional state. To probe this further, we performed RNAScope on mammary
gland of different reproductive stages using the _Egr2_ probe alone. In adult virgin gland, a significant enrichment of _Egr2_ foci was again observed in the basal layer relative to the
luminal layer, with _Egr2_ mRNA being detected in ~ 28% of basal cells but in only ~ 2% of luminal cells (Fig. 4A–C). Basal-enriched _Egr2_ expression was also abundantly detected in the
mammary ducts of pregnant mice at day 3 of pregnancy (Fig. 4D). By day 15 of pregnancy, _Egr2_-expressing cells became less frequent in the ductal basal compartment, and were barely
detectable in the alveolar basal compartment (Fig. 4E,F). We also probed the expression of _Egr2_ using previously published microarray data on whole mammary gland across different stages of
the reproductive cycle34, which revealed significantly upregulated _Egr2_ expression during puberty (5–6 weeks of age) and early pregnancy (day 3) but very low expression during late
pregnancy (day 17–19), lactation, and involution (Fig. 4G,H). Taken together, these data demonstrate physiologic and dynamic expression of _Egr2_ in the mammary gland that coincides with
periods of active epithelial morphogenesis and ductal expansion. GENETIC EVIDENCE FOR BASAL-BIASED EXPANSION OF _EGR2_-EXPRESSING CELLS DURING PUBERTAL MAMMARY GLAND DEVELOPMENT _Egr2_ is of
interest because its expression marks a population of actively expanding hair follicle progenitor cells15 and it encodes a transcription factor that regulates the expression of _Notch1_, a
critical gene involved in mammary basal-luminal binary differentiation35. To track _Egr2_-expressing cells in vivo, we crossed _Egr2-Cre_ (_Krox20-Cre_) mice15 with _ROSA26__mTmG_ reporter
mice to generate _Egr2-Cre_; _ROSA26__mTmG_ females, where _Egr2_-expressing cells and their progenies are fluorescently marked by GFP expression (Fig. 5A). To visualize the spatial location
of _Egr2_-expressing cells and progenies, we performed whole-mount imaging analysis of GFP and tdTomato fluorescence in mammary gland of 3-week-old _Egr2-Cre_; _ROSA26__mTmG_ mice. While no
GFP+ cells were detected in the _ROSA26__mTmG_ control mice as expected, such cells were found throughout the rudimentary ducts as well as in terminal end buds of _Egr2-Cre_; _ROSA26__mTmG_
mice (Fig. 5B). Flow cytometry analysis using GFP in conjunction with cell lineage surface markers (e.g., Cd49f, Epcam) revealed that 15–27% of the basal cells in mammary gland from
3-week-old (pre-puberty) _Egr2-Cre_; _ROSA26__mTmG_ females were GFP+ (Fig. 5C,D). By mid-puberty (6.5 weeks of age) and in adulthood (10 weeks of age), the number of GFP+ basal cells
dramatically increased to near 100% of all basal cells (Fig. 5C,D). The surface expression levels of Cd49f and Epcam in GFP+ and GFP- basal cells were similar (Fig. S4A). At all ages
examined, less than 10% of the luminal cells showed GFP expression (Fig. 5C,D). Taken together with the scRNA-seq and RNAScope findings above that fewer than a third of the basal cells in
adult virgin gland showed detectable _Egr2_ mRNA expression, these flow data suggest that virtually all basal cells in the adult gland had transited through an _Egr2_-expressing state at
some point during pubertal mammary gland development and/or are derived from _Egr2_-expressing basal cells. Further illustrating the dynamic nature of _Egr2_ expression in basal cells,
FACS-sorted GFP- basal cells from mammary glands of 3-week-old _Egr2-Cre_; _ROSA26__mTmG_ females were able to activate GFP expression de novo under organoid culture conditions, whereas GFP+
basal cells remained positive over multiple passages (Fig. S4B). DISCUSSION To date, several scRNA-seq studies have been conducted on human and mouse mammary glands, each presenting a
unique perspective on epithelial cellular composition, differentiation dynamics, and stem cell prediction. Previous studies have also delved into basal cell heterogeneity and the presence of
rare stem cells in the basal layer6,7,9,10,36,37,38, but the gene expression program that underlies bulk basal cell dynamics and differentiation has not been clearly elucidated. Our study
adds to the list of existing datasets and provides a deeper and comprehensive analysis of the transcriptional heterogeneity within mammary basal cells. Our analysis shows that bulk basal
cells exist in at least four distinct transcriptional states that can be identified by their unique enrichment for the expression of specific gene signatures associated with potential
stemness (e.g., _Tspan8_high state), differentiation status (e.g., mature myoepithelial state), and/or rapid cellular dynamics and responses (e.g., _Egr2_High and proliferating states).
Intriguingly, our data does not point to a unidirectional, hierarchical differentiation trajectory originating from one state and ending in another. Instead, they suggest that mammary basal
cells adopt dynamic non-hierarchical transcriptional states, with the exception that the proliferating state may not readily revert back to any of the “non-proliferating” state. This
dynamics implies the inherent plasticity of any given basal cell, a notion that is consistent with the demonstrated functional plasticity of adult basal cells especially upon
transplantation, and that mature myoepithelial cells possess regenerative stem cell activity that can manifest under appropriate conditions4,5,10,20. While it may be technically challenging
to sort and purify the basal cell subsets in different transcriptional states due to overlapping expression of cell surface markers, future experiments to generate and analyze Cre-expressing
alleles driven by temporally controlled promoters of genes encoding select basal transcriptional state markers will enable lineage tracing of the fates and activities of basal cells in each
state during mammary development, regeneration, and tumorigenesis. We were able to confirm the expression of several basal transcriptional state markers in the intact mammary gland using in
situ mRNA detection and found them to be largely non-overlapping albeit not mutually exclusive. It has been reported that nearly all basal cells in the mammary gland, including stem cells,
express _Acta2_ at the mRNA and protein levels20. Our scRNA-seq data support this notion by revealing that > 99% of basal cells show detectable expression of _Acta2._ This said, we found
that a fraction of them show heightened expression of _Acta2_ and other myoepithelial-related genes; these are likely the same cells in which _Acta2_ expression was over the detection
threshold of RNAScope analysis and they may represent mature myoepithelial cells. The _Egr2_High transcriptional state is of particular interest because of its faster cellular dynamics
relative to the other states. Our RNAScope experiments detected the mRNA expression of _Egr2_ in a subset of basal cells in the intact tissue. Moreover, in scRNA-seq, single-probe, and
multi-probe RNAScope experiments, we observed remarkable consistency in the precise frequency (27–30%) of _Egr2_-positive cells, indicating robustness of the observation. In the hair
follicle, another leading model of adult stem cell biology, _Egr2_ expression marks the matrix cells, which are highly proliferative but transit-amplifying progenitor cells that terminally
differentiate to produce the hair shaft15. In the mammary gland, such transient amplifying progenitor cells remain elusive. Our proof-of principle data based on _Egr2-Cre_-mediated GFP
fluorescence is consistent with the possibility that _Egr2-_expressing mammary basal cells are such progenitor cells and serve as the workhorse during pubertal mammary gland development to
generate nearly the entire basal cell compartment of mature gland. However, an alternative, and perhaps more likely based on our RNA Velocity and organoid culture data, possibility is that
_Egr2_High is simply a transcriptional state that all basal cells transit through at some point during pubertal mammary development. It will be worthwhile to generate _Egr2-Cre_ER mice in
the future to seek definitive evidence for the function and fate of the _Egr2_+ basal cell subset during mammary gland development, regeneration, and tumorigenesis. Overall, our study
provides a systematic analysis of mammary basal cell heterogeneity and a useful reference for future investigation into how basal cell gene expression changes during breast cancer initiation
and progression. A thorough understanding of the transcriptional heterogeneity of normal mammary basal cells and their malignant counterparts might be particularly relevant in the
development of differentiation therapies for basal-like and triple negative breast cancers. CONCLUSIONS Our results have identified four major transcriptional states within the mammary basal
cells that exhibit gene expression signatures suggestive of different functional activity and metabolic preference. Our in vivo data suggest that one of these transcriptional states, marked
by _Egr2_ mRNA expression, represents an actively expanding and/or obligatory transitional state during pubertal mammary morphogenesis. These findings regarding basal cell heterogeneity in
the normal tissue lay the foundation for future work to probe the heterogeneity of the malignant counterparts in basal-like and triple negative breast cancer subtypes. DATA AVAILABILITY The
single-cell datasets used and analyzed during the current study are available under Accession #GSE155636 (https://www.ncbi.nlm.nih.gov/gds). Code will be provided upon request. All other
data generated or analyzed during this study are included in this published article and its supplementary information files. All relevant information about materials used in the study is
provided in the Methods section of the text. ABBREVIATIONS * FACS: Fluorescence activated cell sorting * GO: Gene ontology * Myo: Myoepithelial * PPI: Protein–protein interaction *
scRNA-seq: Single-cell RNA-sequencing * UMAP: Uniform manifold approximation projection REFERENCES * Macias, H. & Hinck, L. Mammary gland development. _Rev. Dev. Biol._
https://doi.org/10.1002/wdev.35 (2012). Article Google Scholar * Inman, J. L., Robertson, C., Mott, J. D. & Bissell, M. J. Mammary gland development: Cell fate specification, stem
cells and the microenvironment. _Development (Cambridge)_ https://doi.org/10.1242/dev.087643 (2015). Article Google Scholar * Faulkin, L. J. & Deome, K. B. Regulation of growth and
spacing of gland elements in the mammary fat pad of the C3H mouse. _J. Natl. Cancer Inst._ 24, 953–969 (1960). PubMed Google Scholar * Shackleton, M. _et al._ Generation of a functional
mammary gland from a single stem cell. _Nature_ https://doi.org/10.1038/nature04372 (2006). Article PubMed Google Scholar * Stingl, J. _et al._ Purification and unique properties of
mammary epithelial stem cells. _Nature_ https://doi.org/10.1038/nature04496 (2006). Article PubMed Google Scholar * Wang, D. _et al._ Identification of multipotent mammary stemcells by
protein C receptor expression. _Nature_ https://doi.org/10.1038/nature13851 (2015). Article PubMed PubMed Central Google Scholar * Cai, S. _et al._ A quiescent Bcl11b high stem cell
population is required for maintenance of the mammary gland. _Cell Stem Cell_ https://doi.org/10.1016/j.stem.2016.11.007 (2017). Article PubMed Google Scholar * Fu, N. Y. _et al._
Identification of quiescent and spatially restricted mammary stem cells that are hormone responsive. _Nat. Cell Biol._ https://doi.org/10.1038/ncb3471 (2017). Article PubMed Google Scholar
* Sun, H. _et al._ Single-cell RNA-Seq reveals cell heterogeneity and hierarchy within mouse mammary epithelia. _J. Biol. Chem._ https://doi.org/10.1074/jbc.RA118.002297 (2018). Article
PubMed PubMed Central Google Scholar * Van Amerongen, R., Bowman, A. N. & Nusse, R. Developmental stage and time dictate the fate of Wnt/β-catenin- responsive stem cells in the
mammary gland. _Cell Stem Cell_ https://doi.org/10.1016/j.stem.2012.05.023 (2012). Article PubMed Google Scholar * Nguyen, Q. H. _et al._ Profiling human breast epithelial cells using
single cell RNA sequencing identifies cell diversity. _Nat. Commun._ https://doi.org/10.1038/s41467-018-04334-1 (2018). Article PubMed PubMed Central Google Scholar * Pal, B. _et al._
Construction of developmental lineage relationships in the mouse mammary gland by single-cell RNA profiling. _Nat. Commun._ https://doi.org/10.1038/s41467-017-01560-x (2017). Article PubMed
PubMed Central Google Scholar * Giraddi, R. R. _et al._ Single-cell transcriptomes distinguish stem cell state changes and lineage specification programs in early mammary gland
development. _Cell Rep._ 24, 1653-1666.e7 (2018). Article CAS Google Scholar * Pervolarakis, N. _et al._ Integrated single-cell transcriptomics and chromatin accessibility analysis
reveals regulators of mammary epithelial cell identity. _Cell Rep._ 33, 10058 (2020). Article Google Scholar * Liao, C. P., Booker, R. C., Morrison, S. J. & Le, L. Q. Identification of
hair shaft progenitors that create a niche for hair pigmentation. _Genes Dev._ https://doi.org/10.1101/gad.298703.117 (2017). Article PubMed PubMed Central Google Scholar * Gu, B.,
Watanabe, K., Sun, P., Fallahi, M. & Dai, X. Chromatin effector Pygo2 mediates wnt-notch crosstalk to suppress luminal/alveolar potential of mammary stem and basal cells. _Cell Stem
Cell_ 13, 48–61 (2013). Article CAS Google Scholar * Watanabe, K. _et al._ Mammary morphogenesis and regeneration require the inhibition of EMT at terminal end buds by ovol2
transcriptional repressor. _Dev. Cell_ 29, 59–74 (2014). Article CAS Google Scholar * Han, Y. _et al._ Coordinate control of basal epithelial cell fate and stem cell maintenance by core
EMT transcription factor Zeb1. _Cell Rep._ 38, 110240 (2022). Article CAS Google Scholar * Haensel, D., Jin, S., Gratton, E., Nie, Q. & Correspondence, X. Defining epidermal basal
cell states during skin homeostasis and wound healing using single-cell transcriptomics. _Cell Rep._ 30, 3932-3947.e6 (2020). Article CAS Google Scholar * Prater, M. D. _et al._ Mammary
stem cells have myoepithelial cell properties. _Nat. Cell Biol._ https://doi.org/10.1038/ncb3025 (2014). Article PubMed PubMed Central Google Scholar * Van Den Brink, S. C. _et al._
Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. _Nat. Methods_ 14, 935–936 (2017). Article Google Scholar * Mahendralingam, M. _et al._
Mammary epithelial cells have lineage-restricted metabolic identities. _biorxiv_ 79, 8173 (2019). Google Scholar * Casey, A. E. _et al._ Mammary molecular portraits reveal lineage-specific
features and progenitor cell vulnerabilities. _J. Cell Biol._ 217, 2951–2974 (2018). Article CAS Google Scholar * Heiden, M. G. V., Cantley, L. C. & Thompson, C. B. Understanding the
warburg effect: The metabolic requirements of cell proliferation. _Science_ 324, 1029–1033 (2009). Article ADS Google Scholar * Szklarczyk, D. _et al._ STRING v11: Protein-protein
association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. _Nucleic Acids Res._ 47, D607–D613 (2019). Article CAS Google Scholar *
Chen, E. Y. _et al._ Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. _BMC Bioinform._ 14, 1005 (2013). Google Scholar * Kuleshov, M. V. _et al._ Enrichr: a
comprehensive gene set enrichment analysis web server 2016 update. _Nucleic Acids Res._ 44, W90–W97 (2016). Article CAS Google Scholar * La Manno, G. _et al._ RNA velocity of single
cells. _Nature_ 560, 494–498 (2018). Article ADS Google Scholar * Svensson, V. & Pachter, L. RNA velocity: Molecular kinetics from single-cell RNA-Seq. _Mol. Cell_ 72, 7–9 (2018).
Article CAS Google Scholar * Zywitza, V., Misios, A., Bunatyan, L., Willnow, T. E. & Rajewsky, N. Single-cell transcriptomics characterizes cell types in the subventricular zone and
uncovers molecular defects impairing adult neurogenesis. _Cell Rep._ 25, 2457-2469.e8 (2018). Article CAS Google Scholar * Suoqin, J. _et al._ Inference and analysis of cell-cell
communication using Cell Chat. _bioRxiv._ 2, 1008 (2011). Google Scholar * Hsu, Y. C., Pasolli, H. A. & Fuchs, E. Dynamics between stem cells, niche, and progeny in the hair follicle.
_Cell_ https://doi.org/10.1016/j.cell.2010.11.049 (2011). Article PubMed PubMed Central Google Scholar * Rios, A. C., Fu, N. Y., Lindeman, G. J. & Visvader, J. E. In situ
identification of bipotent stem cells in the mammary gland. _Nature_ https://doi.org/10.1038/nature12948 (2014). Article PubMed Google Scholar * Williams, M. M. _et al._ ErbB3 drives
mammary epithelial survival and differentiation during pregnancy and lactation. _Breast Cancer Res._ https://doi.org/10.1186/s13058-017-0893-7 (2017). Article PubMed PubMed Central Google
Scholar * Bouras, T. _et al._ Notch signaling regulates mammary stem cell function and luminal cell-fate commitment. _Cell Stem Cell_ https://doi.org/10.1016/j.stem.2008.08.001 (2008).
Article PubMed Google Scholar * Liu, W., Wu, T., Dong, X. & Zeng, Y. A. Neuropilin-1 is upregulated by Wnt/β-catenin signaling and is important for mammary stem cells. _Sci. Rep._
https://doi.org/10.1038/s41598-017-11287-w (2017). Article PubMed PubMed Central Google Scholar * Soady, K. J. _et al._ Mouse mammary stem cells express prognostic markers for
triple-negative breast cancer. _Breast Cancer Res._ https://doi.org/10.1186/s13058-015-0539-6 (2015). Article PubMed PubMed Central Google Scholar * Regan, J. L. & Smalley, M. J.
Integrating single-cell RNA-sequencing and functional assays to decipher mammary cell states and lineage hierarchies. _NPJ Breast Cancer._ https://doi.org/10.1038/s41523-020-00175-8 (2020).
Article PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS The authors would like to thank Lu Le’s lab for the _Egr2-Cre_ mice, the Genomics High Throughput
Facility, the Institute for Immunology FACS Core Facility, and the Optical Biology Core at UC Irvine for expert service. FUNDING This work was supported by NIH Grant R01-GM123731 and
R01-AR068074 (X.D.). G.G. was supported by a diversity supplement (R01GM123731-02S2) and was a NSF-Simons Center for Multiscale Cell Fate Research predoctoral fellow through NSF Grants
DMS1763272 and Simons Foundation Grant 594598 (Q.N.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. AUTHOR
INFORMATION Author notes * These authors contributed equally: Guadalupe Gutierrez and Peng Sun. AUTHORS AND AFFILIATIONS * Department of Biological Chemistry, School of Medicine, University
of California, D250 Med Sci I, Irvine, CA, 92697-1700, USA Guadalupe Gutierrez, Peng Sun, Yingying Han & Xing Dai * The NSF-Simons Center for Multiscale Cell Fate Research, University of
California, Irvine, CA, 92697, USA Guadalupe Gutierrez & Xing Dai Authors * Guadalupe Gutierrez View author publications You can also search for this author inPubMed Google Scholar *
Peng Sun View author publications You can also search for this author inPubMed Google Scholar * Yingying Han View author publications You can also search for this author inPubMed Google
Scholar * Xing Dai View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS G.G. and X.D. conceived the study and designed the analysis. G.G.
carried out the computational/bioinformatics analysis. G.G., P.S., and Y.H. performed the experiments. G.G. and X.D. wrote the manuscript, and P.S. and Y.H. read, edited, and approved the
final manuscript. CORRESPONDING AUTHOR Correspondence to Xing Dai. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER'S
NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURE S1. SUPPLEMENTARY
FIGURE S2. SUPPLEMENTARY FIGURE S3. SUPPLEMENTARY FIGURE S4. SUPPLEMENTARY TABLE 1. SUPPLEMENTARY TABLE 2. SUPPLEMENTARY TABLE 3. SUPPLEMENTARY LEGENDS. RIGHTS AND PERMISSIONS OPEN ACCESS
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as
long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third
party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the
article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the
copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Gutierrez, G., Sun, P.,
Han, Y. _et al._ Defining mammary basal cell transcriptional states using single-cell RNA-sequencing. _Sci Rep_ 12, 4893 (2022). https://doi.org/10.1038/s41598-022-08870-1 Download citation
* Received: 17 November 2021 * Accepted: 08 March 2022 * Published: 22 March 2022 * DOI: https://doi.org/10.1038/s41598-022-08870-1 SHARE THIS ARTICLE Anyone you share the following link
with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative