Play all audios:
ABSTRACT Coding transcript-derived siRNAs (ct-siRNAs) produced from specific endogenous loci can suppress the translation of their source genes to balance plant growth and stress response.
In this study, we generated _Arabidopsis_ mutants with deficiencies in RNA decay and/or post-transcriptional gene silencing (PTGS) pathways and performed comparative sRNA-seq analysis,
revealing that multiple RNA decay and PTGS factors impede the ct-siRNA selective production. Genes that produce ct-siRNAs often show increased or unchanged expression and typically have
higher GC content in sequence composition. The growth and development of plants can perturb the dynamic accumulation of ct-siRNAs from different gene loci. Two nitrate reductase genes,
_NIA1_ and _NIA2_, produce massive amounts of 22-nt ct-siRNAs and are highly expressed in a subtype of mesophyll cells where _DCL2_ exhibits higher expression relative to _DCL4_, suggesting
a potential role of cell-specific expression of ct-siRNAs. Overall, our findings unveil the multifaceted factors and features involved in the selective production and regulation of ct-siRNAs
and enrich our understanding of gene silencing process in plants. SIMILAR CONTENT BEING VIEWED BY OTHERS ARABIDOPSIS SGS3 IS RECRUITED TO CHROMATIN BY CHR11 TO SELECT RNA THAT INITIATE
SIRNA PRODUCTION Article Open access 26 March 2025 INTERPLAY BETWEEN CODING AND NON-CODING REGULATION DRIVES THE ARABIDOPSIS SEED-TO-SEEDLING TRANSITION Article Open access 26 February 2024
IN-FRAME EDITING OF TRANSCRIPTION FACTOR GENE _RDD1_ TO SUPPRESS MIR166 RECOGNITION INFLUENCES NUTRIENT UPTAKE, PHOTOSYNTHESIS, AND GRAIN QUALITY IN RICE Article Open access 24 June 2022
INTRODUCTION In various eukaryotes, small non-coding RNAs (sRNAs) play crucial roles in silencing genes at both post-transcriptional (PTGS) and transcriptional (TGS) levels1,2. Plant sRNA
population is mainly classified into two groups: microRNAs (miRNAs) and small interfering RNAs (siRNAs). Among these, miRNAs generate from precursors transcribed from _MIRNA_ genes by RNA
polymerase II (Pol II) and cleaved by DCL1. In contrast, siRNAs are primarily derived from double-stranded RNAs (dsRNAs) that are catalyzed by RNA-dependent RNA Polymerase (RDR) proteins
from the transcripts of transposons, transgenes, and viral RNAs3. Generally, DCL4 is responsible for cleaving most RDR6-dependent dsRNAs into 21-nt secondary siRNAs. Subsequently, the mature
miRNAs and siRNAs are loaded into Argonaute (AGO) proteins to form RNA-induced silencing complexes (RISC), which can mediate the cleavage or translational repression of target genes4. A
novel class of siRNAs known as coding transcript-derived siRNAs (ct-siRNAs) has been discovered in _Arabidopsis_ plants deficient in various RNA decay factors5. RNA decay is a complex
process that involves the 5’-3’ exonuclease XRN (EXORIBONUCLEASE) and the 3’-5’ multi-subunit exonuclease protein complex (exosome)6,7. The 5’-3’ or 3’-5’ RNA decay can occur in both the
nucleus and cytoplasm, with different cofactors involved. In _Arabidopsis_, the cytoplasmic XRN4/EIN5 mainly degrades uncapped mRNAs and RNA fragments cleaved by miRISC/siRISC8, while XRN2
and XRN3 in the nucleus remove abnormal RNAs during transcription9,10,11. Loss of EIN5, SKI2, and XRN3 proteins can lead to the production of massive ct-siRNAs11,12,13. As exosome cofactors,
members of Superkiller complex (SKI complex), such as SKI2, SKI3, SKI7, and SKI8, are required for 3’-5’ cytoplasmic RNA decay7. The homologous nuclear-localized MTR4 assists in removing
rRNA precursors and maturation by-products, while the nucleocytoplasmic-localized HEN2 is involved in the degradation of polyadenylated nuclear exosome substrates14,15,16. A recent study
detected distinct accumulation patterns of ct-siRNAs at miRNA targets in plants deficient in the HEN2 and SKI2 activity17. Other exosome cofactors with unknown functions have also been
identified in _Arabidopsis_, such as RST1 and RIPR, and deficiency of these factors can lead to the production of ct-siRNAs from over one hundred endogenous coding transcripts18,19. RNA
decay is a multi-step process that involves removing the poly(A) tail, mediated by the CCR4-NOT complex and PAN2-PAN3, followed by the binding of the shortened poly(A) by
LSM1-7(SM-like)/PAT1 and the recruitment of the mRNA decapping complex. After the removal of 5’ cap, mRNAs can be directionally degraded either from the 5’-3’ by EIN5 or 3’-5’ by
exosome20,21. The poly(A) truncation process is often accompanied by modifications to the poly(A) tail. In plants, specific mRNAs undergoing poly(A) truncation are initially uridylated by
Uridylyltransferase 1 (URT1) before being degraded in the directional 5’-3’ manner, thereby inhibits 3’-5’ RNA decay22. The decapping complex, which is responsible for removing the canonical
5’ m7G cap of mRNAs, consists of Nudix family members and Decapping 2 (DCP2) cofactors, such as DCP1 and DHH123. DCP1, DCP2, DCP5, and VARICOSE (VCS) have been found to play important roles
in post-embryonic plants24,25. Additionally, mRNAs carrying non-canonical 5’ NAD+ caps can be degraded by the non-Nudix family hydrolase Decapping and exoribonuclease protein 1
(DXO1)26,27,28. Plants deficient in URT1, DCP2, VCS, and DXO1 enhanced the accumulation of ct-siRNAs27,29,30. FRY1 (FIERY1) is another 5’-3’ RNA decay factor that promotes the abundance and
function of miRNAs by inhibiting the biogenesis of ribosomal RNA-derived siRNAs (risiRNAs)12. Therefore, RNA decay factors intricately coordinate to regulate the fate of mRNAs, ensuring
normal gene expression by preventing aberrant mRNAs from being captured by the PTGS pathway. Our previous study found that in _ein5 ski2_ plants, a minimum of 441 protein-coding genes can
produce ct-siRNAs, mainly 21-nt in length. The biogenesis of these ct-siRNAs relied on DCL4/DCL2, RDR6, and SGS3, with partial dependence on AGO113. In contrast, in _ein5 dcl4_ and _ski2
dcl4_ plants, a massive amount of RDR6- and DCL2-dependent 22-nt ct-siRNAs accumulated, leading to more severe growth and developmental defects31. Approximately 50% of the total 22-nt
ct-siRNAs originated from _NIA1_ and _NIA2_. These ct-siRNAs are predominantly loaded into AGO1, leading to the inhibition of NIA1 and NIA2 protein levels by stimulating secondary siRNA
amplification and inducing strong gene silencing effects31. Highly abundant 22-nt ct-siRNAs play a crucial role in regulating plant responses to nitrogen deficiency, ABA signaling, and salt
stress31. Although the source genes of ct-siRNAs represent only a small portion of the genome-wide expressed genes, the distinct accumulation of 22-nt ct-siRNAs at different loci in plants
deficient in RNA decay and PTGS factors or under various stresses suggests that ct-siRNA production is regulated by unknown selective and regulatory mechanisms. A previous study reported
that the 5’-3’ RNA decay factor EIN5 selectively degrades _cis_-acting elements containing the CTCCGT motif, thereby more effectively preventing them as substrates for ct-siRNA production32.
Additionally, transgenes characterized by a high GC content in their sequence composition have been observed to enhance protein translation rates and slowdown RNA degradation in plants by
modulating the codon-tRNA matching efficiency32. Therefore, studying the characteristics of source genes is essential for understanding the determining mechanisms of ct-siRNA selective
production from distinct endogenous coding genes in plants. In this study, our aim was to elucidate the selective production and regulatory mechanism of ct-siRNAs. To achieve this, we
constructed a series of mutants with deficiencies in RNA decay and PTGS factors, followed by performing sRNA-seq, RNA-seq, and single-nucleus RNA-seq (snRNA-seq). Comparative analysis
revealed that multiple RNA decay and PTGS factors strongly inhibit the ct-siRNA selective production. Genes with high GC content in their sequence composition contribute to the accumulation
of highly abundant ct-siRNAs. Transgenic experiments involving truncated _NIA1_ and _NIA2_ fragments suggested that ct-siRNA-induced off-target silencing may lead to the transitive silencing
of _NIA1_ and _NIA2_. Additionally, we unveiled the importance of the spatiotemporal expression of ct-siRNA source genes at both the developmental stage and single-cell level in the
selective production of ct-siRNAs. Overall, our study advances our understanding of RNA silencing and provides new insights into the role of ct-siRNAs in regulating plant development and
responses to stress. RESULTS RNA DECAY AND PTGS FACTORS REGULATE 21-NT AND 22-NT CT-SIRNA SELECTIVE PRODUCTION Our previous study demonstrated that loss of the cytoplasmic RNA decay and/or
DCL4 activity in _Arabidopsis_ induces the production of abundant DCL2-dependent 22-nt ct-siRNAs from specific endogenous loci, leading to the silencing of their source genes and the defects
of plant growth and development31. To investigate the selective production and regulatory mechanism of ct-siRNAs, we generated a series of mutants with deficiencies in RNA decay and/or PTGS
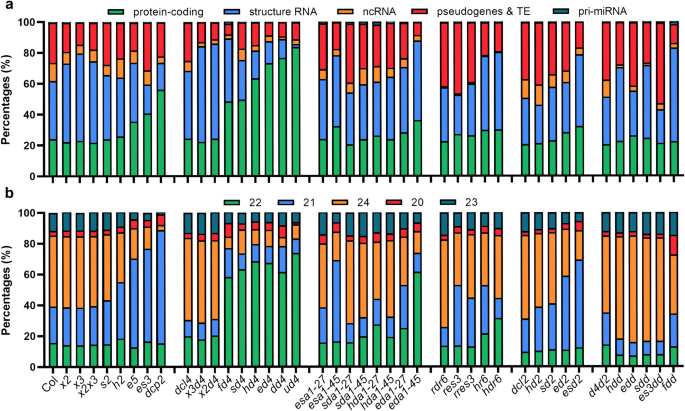
factors and performed sRNA sequencing. By analyzing the abundance of siRNAs accumulated in these mutants, we found that RNA decay and PTGS factors can markedly suppress the production of
siRNAs from protein-coding loci (Fig. 1a, Supplementary Fig. 1a). Mutation of _FRY1_ could induce the biogenesis of risiRNAs from ribosomal RNAs, which belong to a class of structural
RNAs12. Notably, there is a reciprocal relationship between the expression levels of siRNAs produced from structural RNAs and coding genes in these mutants. Minor changes in the accumulation
of siRNAs from other genomic regions suggest that endogenous mRNAs might preferentially entering the PTGS pathway as substitute substrates for structural RNAs (Fig. 1a, Supplementary Fig.
1a). Given that sRNAs generated from the sense strand of their source genes can represent either functional siRNAs or RNA degradation fragments, while those originating from the antisense
strand are typically considered as genuine siRNAs processed by RDR6 and DCLs, we particularly focused on antisense strand-derived ct-siRNAs. We observed that mutants with defects in RNA
decay and/or PTGS pathways produced varying levels of ct-siRNAs, mainly 21-nt and 22-nt in length (Fig. 1b, Supplementary Fig. 1b). These ct-siRNAs significantly accumulated in eight
mutants, including _ein5-1 ski2-3_, _dcp2_, _fry1-6 dcl4-2_, _ski2-2 dcl4-2_, _hen2-1 dcl4-2_, _ein5-1 dcl4-2_, _dxo1-1 dcl4-2_, and _urt1-1 dcl4-2_. Among these mutants, _dcp2_ exhibited
the most pronounced accumulation of 21-nt ct-siRNAs, followed by _ein5-1 ski2-3_, while the other six mutants with DCL4 defects were mainly enriched in 22-nt ct-siRNAs (Fig. 1b,
Supplementary Fig. 1b). We also found that 21-nt ct-siRNA biogenesis relied on DCL4, AGO1, and RDR6, and could shift to 22-nt or 24-nt when DCL4 was deficient, or when both DCL2 and DCL4
functions were simultaneously lost (Fig. 1b, Supplementary Fig. 1b). These results suggested that DCL proteins, including DCL4 and DCL2, competitively inhibit ct-siRNA production in plants
deficient in RNA decay. Indeed, DCL2 is typically considered less competitive than DCL4 in processing RDR6-dependent dsRNAs into siRNAs. However, compared to the _ein5-1 ski2-3_ mutant, the
abundance of 21-nt ct-siRNAs declined in the _ein5-1 ski2-3 dcl2-1_ plants although it remained at higher levels than in the wild-type Col-0 and _ein5-1 ski2-3 dcl4-2 dcl2-1_ mutant
(Supplementary Fig. 1b). This suggests that DCL2 could still contribute to the production of 21-nt ct-siRNAs even when DCL4 is functional. Therefore, our findings demonstrate that RNA decay
and PTGS factors impede the selective biogenesis of 21-nt and 22-nt ct-siRNAs to varying levels and exhibit cumulative effects. DISPERSION OF CT-SIRNAS IN MUTANTS DEFICIENT IN RNA DECAY AND
PTGS PATHWAYS Our recent study revealed that 22-nt ct-siRNAs could strongly inhibit the translation of their source genes instead of cleaving the transcripts31. In this study, we tried to
identify genome-wide hotspot genes that repeatedly producing high levels of 22-nt ct-siRNAs among the eight mutants mentioned above. In double mutants deficient in RNA decay and DCL4
activity, including _ein5 dcl4_, _dxo1 dcl4_, _hen2 dcl4_, _ski2 dcl4_, and _urt1 dcl4_, _NIA1_ and _NIA2_ produced almost half of the 22-nt ct-siRNAs among the top 20 ct-siRNA source genes
(Fig. 2a). Other loci, such as _DIACYLGLYCEROL ACYLTRANSFERASE 3_ (_DGAT3_), _GLOBAL TRANSCRIPTION FACTOR GROUP E 2/7_ (_GTE2/7_), and _SMAX1-LIKE 4/5_ (_SMXL4/5_), consistently contributed
a high percentage of 22-nt ct-siRNAs (Fig. 2a). Considering the amounts of 22-nt ct-siRNAs derived from the top 20 source genes account for more than 80% of the total 22-nt ct-siRNAs (Fig.
2a), all the top 20 source genes in the eight mutants were regarded as hotspot genes producing 22-nt ct-siRNAs, resulting in a union set of 52 genes (Fig. 2b). Analyzing the expression
patterns, functions, and sequence features of these hotspot genes will assist in elucidating the potential mechanism of ct-siRNA selective production. Consistent with our previous findings,
mRNA levels of these hotspot genes remained unchanged or were even up-regulated in _ein5-1 dcl4-2_ and _ski2-2 dcl4-2_ plants (Fig. 2c), leading us to further estimate the dynamic production
of ct-siRNAs during plant growth and development, as well as the expression patterns of source genes in different cell types later in this work. While there was considerable overlap in the
genes producing 22-nt ct-siRNAs among the eight mutants, we still found several loci specifically generating this class of siRNAs in less than three mutants (Fig. 2d). For example, 21-nt and
22-nt ct-siRNAs originating from _EBF1_ were only detected in _dcp2_ and _ein5-1 ski2-3_, while 22-nt ct-siRNAs generating from _HSP70-1_ were specifically detected in _ein5-1 dcl4-2_ and
_fry1-6 dcl4-2_ (Fig. 2d). The abundance of 21-nt ct-siRNAs were also estimated for the selected genes for comparisons with 22-nt ct-siRNAs (Fig. 2d). The distinct accumulations of 21-nt and
22-nt ct-siRNAs at several loci in different mutants indicated diverse inhibitory effects of specific RNA decay and PTGS factors on ct-siRNA production. To validate this hypothesis, we
assessed the genome-wide production of 22-nt ct-siRNAs in all the eight mutants. Compared with Col-0, the profiles of 22-nt ct-siRNAs displayed substantial variations among the eight
mutants, with those deficient in 5’-3’ and 3’-5’ mRNA decay being clustered into two distinct clusters (Fig. 2e). Despite the shared biological functions among the ct-siRNA source genes, the
genes in mutants deficient in 5’-3’ mRNA decay were mainly enriched in photosynthesis, metabolic processes, and defense-related functions, while genes in mutants affecting 3’-5’ mRNA decay
were specifically enriched in signaling regulation, cell communication and organ development-related functions (Fig. 2f). These findings suggest that RNA decay and PTGS factors can
specifically or synergistically inhibit the selective production of ct-siRNAs. The dispersed accumulation of ct-siRNAs at specific genes suggests that RNA decay and PTGS factors regulate
ct-siRNA selective production at both quantity and functional levels. SOURCE GENE CHARACTERISTICS CONTRIBUTING TO CT-SIRNA SELECTIVE PRODUCTION To investigate the potential features of
ct-siRNA source genes, we focused on their biological functions, expression levels, and sequence characteristics in the eight mutants mentioned above. In double mutants deficient in RNA
decay and/or DCL4 activity, the production of ct-siRNAs was increased to varying levels among source genes. We analyzed the biological functions of genes with similar fold-change ranges in
ct-siRNA accumulation (Supplementary Fig. 2). Interestingly, the infrequent overlap among Gene Ontology (GO) terms annotated by genes exhibiting different fold-change scales indicates that
the selective production of ct-siRNAs is related to the functions of their source genes (Supplementary Fig. 2). Notably, genes with a thousand-fold increase in ct-siRNA production (log2FC
range 11-14.5 in Supplementary Fig. 2) were involved in processes such as nitric oxide biosynthesis, nitrate assimilation, or stress response to light or hormone stimuli, whereas genes with
slightly increased ct-siRNA accumulations (log2FC range 2-5) tended to regulate cell death, photosynthesis, auxin and hormone transport, and development (Supplementary Fig. 2). In contrast,
when plants deficient in RNA decay and DCL4 activity, no significant difference was found in the expression levels of genes producing 22-nt ct-siRNAs compared to those not producing them
(Supplementary Fig. 3a). These findings suggest that the accumulation of ct-siRNAs correlates with the biological functions of their source genes rather than expression levels. Regarding the
ct-siRNA generation in relation to the sequence composition of the source gene, we found that compared to genes unable to produce 22-nt ct-siRNAs, genes producing them tended to have longer
sequences and extended 5’ UTRs (Fig. 3a, b), while no significant difference was observed in the 3’ UTR length and intron number (Supplementary Fig. 3b, c). Higher GC content in
bacteria-originated transgenes can enhance expression and protein accumulation through mechanisms such as decreased mRNA degradation, improved translation efficiency, and optimized
epigenetic modifications33,34. However, it is still unclear whether GC content plays a role in ct-siRNA production from plant endogenous coding genes. The GC content of genome-wide coding
regions follows a normal distribution, approximately ranging from 30% to 55%35. Interestingly, we found that ct-siRNA producing genes exhibited a higher GC content in all the mutants we
tested (Supplementary Fig. 3d, e). The GC content in the flanking 1Kb regions upstream or downstream of source genes also showed positive correlations with the generation of 22-nt ct-siRNAs
(Fig. 3c). In human cells, the GC content plays a central role in mRNA fate, with the translation efficiencies and stability of GC-rich mRNAs were significantly higher than AU-rich mRNAs35.
Therefore, GC-rich mRNAs with GC ≥ 50% may become substrates for PTGS pathway and contribute to more abundant ct-siRNA production when both RNA decay and DCL4 are deficient. Conversely,
regions with low GC content (GC ≤ 30%) generated lower abundances of ct-siRNAs, possibly due to limited translational efficiency (Fig. 3c). In conclusion, our results demonstrate that the
selective production of ct-siRNAs is closely associated with the function, sequence length, and GC content of their source genes. This indicates that an array of gene characteristics can
collectively contribute to the selective production of ct-siRNAs. TRUNCATED _NIA1_ AND _NIA2_ FRAGMENTS WITH HIGH GC CONTENT INDUCE CT-SIRNA PRODUCTION In _Arabidopsis_, _NIA1_ and _NIA2_
encoding nitrate reductases, are essential for nitrate assimilation and can generate highly abundant 22-nt ct-siRNAs when nitrogen nutrition is scarce31. These ct-siRNAs efficiently inhibit
NIA1 and NIA2 protein levels, thereby reducing energy consumption and ensuring plant survival31. In plants deficient in several RNA decay factors and DCL4 activity, such as _ein5-1 dcl4-2_,
_fry1-6 dcl4-2_, _dxo1-1 dcl4-2_, _hen2-1 dcl4-2_, _ski2-2 dcl4-2_, and _urt1-1 dcl4-2_ mutants, we observed a massive amount of 22-nt ct-siRNAs accumulated at the _NIA1_ and _NIA2_ loci
(Fig. 2a, b, d). The question then arises: why do _NIA1_ and _NIA2_ genes frequently produce large quantities of ct-siRNAs to trigger endogenous gene silencing when both RNA decay and PTGS
factors are deficient? To address this question above, we truncated the CDS sequences of _NIA1_ and _NIA2_ into consecutive 600-nt fragments and generated transgenic plants by expressing
each fragment fused with the 35S promoter and the green fluorescent protein (GFP) sequence (Fig. 4a). Our observation revealed that only transgenic plants expressing specific fragments, like
_NIA1-5_, _NIA1-6_, _NIA2-1_, _NIA2-5_, and _NIA2-6_, exhibited a strong ability to induce transgenic silencing of GFP, while other transgenic plants exhibited weak or no silencing effects
(Fig. 4b). By further analyzing the sRNA-seq data from transgenic plants expressing _NIA1-3_, _NIA1-6_, _NIA2-1_, _NIA2-3_, and _NIA2-5_, we observed an obvious correlation between siRNA
production from _NIA1_ or _NIA2_ and GFP gene silencing, but not GFP intensity in transgenic plants (Fig. 4b–d). This phenomenon may be attributed to the insufficiency of GFP fluorescence as
a quantitative measure of gene silencing or to the possibility that the abundance of siRNAs precedes or lags behind the immediate state of gene silencing (Fig. 4c, d). Intriguingly, the
sRNA-seq data analysis further revealed that transgenes of the aforementioned truncated _NIA1_ and _NIA2_ fragments could all stimulate siRNA production from both genes to varying levels.
This indicates that ct-siRNAs originating from _NIA1_ or _NIA2_ can enforce transitive silencing of the source gene and its homologous gene (Fig. 4d). This observation was further confirmed
by Northern blot analysis of two _NIA1-6_ transgenic lines, which triggered siRNA production from both _NIA1_ and _NIA2_ (Fig. 4e). Consistent results were observed in the case of the
_NIA2-1_, _NIA2-5_, and _NIA2-6_ transgenic lines. However, a detailed examination of siRNA accumulation peaks at _NIA1_ and _NIA2_ in different transgenic lines revealed substantial
differences (Fig. 4f), suggesting that multiple factors influence siRNA production. Upon investigating sequence similarity using Circoletto with default settings
(http://tools.bat.infspire.org/circoletto/), we found that _NIA2-5_ is the only fragment partially homologous to _NIA1-5_ (Fig. 4g), and its transgenic plant exhibited transitive siRNA
production on _NIA1_ (Fig. 4f). Additionally, we noticed that the GC content of full-length _NIA1_ and _NIA2_, as well as all truncated fragments, exceeded the average GC content of whole
transcriptome CDS sequences (44.37%) (Fig. 4h). Notably, among these fragments, _NIA2-1_ with the highest GC content at 55.5% exhibited a concentrated siRNA peak within the fragment boundary
(Fig. 4f, h), implying that GC content is a crucial factor influencing siRNA generation. Our genetic findings confirmed a strong correlation among GC content, siRNA production, and gene
silencing. Therefore, it is crucial to calculate GC content to avoid high GC sequences and achieve efficient transgenesis, especially those sequences with a GC content exceeding 55%, which
can frequently trigger gene silencing. CT-SIRNA DYNAMICALLY ACCUMULATED AT DIFFERENT PLANT GROWTH AND DEVELOPMENT STAGES To investigate whether the accumulation of ct-siRNAs is dynamically
regulated during plant growth and development, we conducted time-series sRNA-seq on _ein5-1 dcl4-2_ and _ski2-2 dcl4-2_ plants. Commencing from the point at which the homozygous mutants
first displayed identifiable phenotypes. We observed that 21-nt and 22-nt ct-siRNAs were rarely detected in Col-0, _ski2-2 dcl2-1 dcl4-2_, and _ein5-1 dcl2-1 dcl4-2_ plants but showed a
dynamic accumulation pattern when both RNA decay and DCL4 activity were deficient (Fig. 5a, b). In _ein5-1 dcl4-2_ and _ski2-2 dcl4-2_ plants, the expression of 22-nt ct-siRNAs gradually
increased and reached its peak at 15-day-old and 20-day-old, respectively. In contrast, 21-nt ct-siRNA accumulation peaked at 14-day-old and 12-day-old, respectively (Fig. 5a, b). We ranked
the hotspot genes with high levels of ct-siRNA production to identify the source genes contributing to the dynamic accumulation of ct-siRNAs during plant growth and development (Fig. 5c).
Among these genes, _NIA1_ and _NIA2_ consistently produced the highest proportion of 22-nt ct-siRNAs in both _ein5-1 dcl4-2_ and _ski2-2 dcl4-2_ plants (Fig. 5c). Interestingly, we found
that 5’-3’ and 3’-5’ RNA decay factors had different effects on the production of 22-nt ct-siRNAs from various substrates (Fig. 5c). When classifying the source genes based on their
functions, we found that the hotspot genes producing 22-nt ct-siRNAs in both _ein5-1 dcl4-2_ and _ski2-2 dcl4-2_ plants were primarily encoded transcription factors, heat-shock proteins,
multiple enzymes, hormone responsive proteins, and other functional genes (Fig. 5c). The predominant production of 22-nt ct-siRNAs from genes that encoding transcription factors GTE2 and
GTE7, as well as genes encoding heat-shock proteins, was only detected in _ein5-1 dcl4-2_, while genes encoding HD-ZIP transcription factors (PHB, PHV, HB-8, and REV) and several enzymes
were exclusively identified in _ski2-2 dcl4-2_ (Fig. 5c). To identify distinct groups of genes that co-accumulated ct-siRNAs during different stages of plant growth and development, we
performed clustering analysis of source genes based on the abundance of accumulated ct-siRNAs. Specifically, we focused on coding genes with Transcripts Per Million (TPM) levels of
accumulated 22-nt ct-siRNAs greater than 10 in at least two stages, as well as that differentially accumulated with a |log2FC|> 1 when cross-comparing any two stages. Our analysis
identified 18 clusters comprising 583 and 423 genes in _ein5-1 dcl4-2_ and _ski2-2 dcl4-2_ plants, respectively (Fig. 5d). In _ein5-1 dcl4-2_ plants, we observed a continual increase in
22-nt ct-siRNA production from 145 genes (clusters 6, 9, 10, 13, and 18), which are functionally involved in regulating RNA metabolism and the cellular response to hypoxia and oxygen.
Simultaneously, we found a gradual decrease in the accumulation of 22-nt ct-siRNAs from 115 genes (clusters 4, 8, and 15) that are functionally involved in photosynthesis, light harvesting,
and translational elongation (Fig. 5d). In _ski2-2 dcl4-2_ plants, a continuous increase expression of 22-nt ct-siRNAs was observed in 20 genes (clusters 13, 15, and 16) that played critical
roles in anther, stamen, and floral development, while the abundance of 22-nt ct-siRNAs gradually decreased in 28 genes (clusters 4, 6, 14, and 17) that were not enriched in any specific
biological processes (Fig. 5d). These findings suggest that the accumulation of ct-siRNAs from discrete gene loci exhibits a fluctuating pattern of changes during various stages of plant
growth and development. This alternation in abundance over stages could also account for the apparent differences in ct-siRNA selective production observed in sRNA-seq snapshots. CT-SIRNA
SOURCE GENES ARE EXPRESSED IN SPECIFIC CELL TYPES In our previous study, we observed that specific endogenous coding genes, like _NIA1_ and _NIA2_, accumulated large amounts of ct-siRNAs in
_ein5-1 dcl4-2_ and _ski2-2 dcl4-2_ plants31. We also found that their mRNA levels remained either unchanged or upregulated in both mutants compared to Col-0 in the bulk RNA-seq31. However,
it remains unknown whether these ct-siRNA source genes are expressed in specific cell types and whether their expression patterns contribute to the production of ct-siRNAs. In recent years,
snRNA-seq has emerged as a powerful tool for studying cell-specific gene expression. Thus, we employed snRNA-seq to investigate the expression of ct-siRNA source genes at the single-cell
level. We utilized the 10X Genomics snRNA-seq platform to amplify and profile the transcriptome of cells from 21-day-old _Arabidopsis_ seedlings without roots, including Col-0, _ein5-1
dcl4-2_, _ski2-2 dcl4-2_, and _hen2-1 dcl4-2_ plants. After quality control at both cell and gene levels, a pool of 8323 cells with 59,950 genes were obtained from Col-0 (1875 cells),
_ein5-1 dcl4-2_ (3475 cells), _ski2-2 dcl4-2_ (776 cells), and _hen2-1 dcl4-2_ (2197 cells) plants (Fig. 6a, b). To identify distinct cell populations based on gene expression profiles, we
employed graph-based clustering approach by Seurat package to identify clusters36. We then selected cell type-specific marker genes from the PCMDB database37,38 and the studies to define
cell types to these clusters39,40,41,42,43,44,45. Ultimately, we manually annotated 21 clusters into 9 functional cell types (Fig. 6a, Supplementary Fig. 4). Among all cell types, mesophyll
cells accounted for the largest proportion, where _NIA1_ and _NIA2_ showed high expression levels (Fig. 6a–c). As mesophyll cells can be further divided into subtypes such as palisade tissue
and spongy tissue, we continued to define the subtypes of mesophyll cells. According to previously identified single-cell sequencing markers expressed in mesophyll46,47,48, we distinguished
the mesophyll cells mainly into eight subtypes (Supplementary Fig. 5). Analyzing the expression of ct-siRNA producing hotspot genes at the single-cell level, we observed that _NIA1_ and
_NIA2_ were robustly expressed in the MC2 subtype of mesophyll cells (Fig. 6d, Supplementary Fig. 6), which closely resembled the palisade tissue. Notably, _dcl4-2_ plants still express a
_DCL4_ chimeric with T-DNA sequence31. We also observed a notable increase in the relative expression of _DCL2_ versus _DCL4_ in this mesophyll subtype in _ein5-1 dcl4-2_, _ski2-2 dcl4-2_,
and _hen2-1 dcl4-2_ mutants compared to Col-0 plants, which might contribute to the higher abundance of 22-nt ct-siRNAs produced from _NIA1_ and _NIA2_ (Fig. 6d, Supplementary Figs. 6, 7).
These results suggest that 22-nt ct-siRNA production may be cell type-specific. In line with this, we found that the expressions of _NIA1_ and _NIA2_ were downregulated in the MC2 subtype of
mesophyll cells in _ein5-1 dcl4-2_, _ski2-2 dcl4-2_, and _hen2-1 dcl4-2_ mutants relative to Col-0 plants (Fig. 6d). Compared to the upregulated expression levels of _NIA1_ and _NIA2_
observed in bulk RNA-seq (Fig. 2c), our findings suggest that gene silencing can occur at the single-cell level and may be specific to certain cell types. The expression of ct-siRNA source
genes and PTGS pathway genes at single-cell level can also contribute to ct-siRNA selective generation. Thus, when the target gene fused with the 35S promoter induces gene silencing, early
consideration of tissue-specific promoters should be given to achieve efficient transgenesis and molecular breeding. DISCUSSION Here we reported the production of 21-nt and 22-nt ct-siRNAs
from endogenous mRNAs and uncovered particularly the synergistic inhibitory effects of mRNA decay and PTGS factors (Fig. 7). Among the RNA decay factors, HEN2, EIN5, DCP2, and the
combination of EIN5 and SKI2 emerged as key players influencing/hindering the biogenesis of 21-nt ct-siRNAs. Meanwhile, other factors, including FRY1, SKI2, HEN2, DXO1, EIN5, and URT1, in
conjunction with DCL4, specifically suppress the production of 22-nt ct-siRNAs. However, the PTGS factors DCL2 and DCL4 exhibited functional redundancy in ct-siRNA production, highlighting
the complexity of the regulatory network involved in ct-siRNA biogenesis. The production of ct-siRNAs was influenced by the characteristics of their source genes, including gene length, 5’
UTR length and GC contents. Furthermore, snRNA-seq data analysis revealed that _NIA1_ and _NIA2_ exhibited a substantial accumulation of 22-nt ct-siRNAs in plants deficient in both EIN5/SKI2
and DCL4 and displayed an increased expression in a subtype of mesophyll cells, where a higher expression of _DCL2_ relative to _DCL4_ was observed. RNA decay factors affect the selective
generation of ct-siRNAs, and these effects generally reflect the disparities in decay at the 5’ and 3’ ends. This manifests in the clustering of ct-siRNA expression profiles, biological
functions of source genes, and dynamic expression patterns throughout plant growth and development. This selective regulation mechanism may be influenced by the functional redundancy among
the RNA decay factors. Our earlier studies found that plants deficient in RNA decay factors, in either the 5’-3’ or 3’-5’ direction, had no impact on ct-siRNA generation. However,
simultaneous mutations of non-homologous RNA decay factors, EIN5 and SKI2, led to an overproduction of ct-siRNAs in the mutants, accompanied by severe growth defect phenotypes13. In this
study, we employed EIN5, XRN2, and XRN3 in the 5’-3’ RNA decay direction and SKI2 and HEN2 in the 3’-5’ RNA decay direction. These factors, whether with sequence homology or functional
redundancy, are speculated potential contributors to the selective generation of ct-siRNAs. Additionally, the selective production of ct-siRNAs may be influenced by the PTGS factors and
their subcellular localization. Several previous studies have reported that DCL4 and DCL2 can form a dicing body in the nucleus. Notably, our recent research on phase separation has revealed
that RDR6 and SGS3 can form liquid-liquid phase separation bodies in the cytoplasm49, thereby promoting endogenous gene silencing. While this aspect falls beyond the scope of this study, it
merits attention in future investigations. Our previous study has demonstrated that the nitrate reductase genes _NIA1_ and _NIA2_ produce large amounts of 22-nt ct-siRNAs to efficiently
inhibit their protein levels, potentially promoting plant survival under stress conditions by conserving energy31. The efficient RNAi could be mediated by stronger transitivity or a
substantial number of 22-nt siRNAs, capable of amplifying the silencing effect on their primary target or homologous gene, either in a cis or trans manner50,51. Previous study has indicated
that a 500-nt overlap between homologous genes is sufficient to establish efficient and frequent transitive silencing, whereas homologies of 250-nt and 98-nt resulted in reduced and minimal
co-suppression effect, respectively52. In this study, we found that 22-nt ct-siRNAs were frequently produced from _NIA1_ and _NIA2_ in plants particularly deficient in both RNA decay factors
and DCL4 activity. This raises the question of whether ct-siRNAs induce transitive silencing between _NIA1_ and _NIA2_. Gene silencing signal tends to expand towards the 3’ region of the
transcript52,53. Our observation confirmed that the transgenic plants expressing the 3’ fragments of _NIA1_ (_NIA1-5_ and _NIA1-6_) and _NIA2_ (_NIA2-5_ and _NIA2-6_) efficiently induced the
silencing of both genes, with ct-siRNAs enriched in the 3’ region and less spread to the 5’ region (Fig. 4f). It is widely accepted that at least 21-nt homology between genes can induce the
co-suppression of homologous genes. Even though the CDS sequences of _NIA1_ and _NIA2_ share no more than successive 20-nt of identical sequence, it remains unclear how ct-siRNAs induce the
transitivity silencing of homologous genes and which part of ct-siRNAs serve as efficient inducers. Previous studies have suggested that off-target silencing could be induced by
approximately 70-nt fragments containing at least three mismatches within any 21-nt sequence shared between homologous genes54. Consequently, the transitivity and frequent silencing of
_NIA1_ and _NIA2_ may be caused by ct-siRNA induced off-target silencing. When RNA decay and/or PTGS factors are deficient, ct-siRNAs can produce from either aberrant or normal mRNA
transcripts. Despite analyzing RNA-seq data from different mutants, we did not detect a notable downregulation in the expression of ct-siRNA source genes. This presented a contradiction to
the co-suppression effect as the expression levels of ct-siRNA producing genes were unchanged or even upregulated. According to an important previous discovery34, one possible explanation is
that the increased production of 21-nt and 22-nt ct-siRNAs corresponds to a decreased level of 24-nt siRNAs and reduced DNA methylation, leading to upregulated gene expression. On the other
hand, the recent development of single-cell transcriptome sequencing technology allows us to measure gene expression at the single-cell level within samples encompassing multiple tissues
and cell types. This technology has enabled us to find that hotspot genes with high-frequency accumulation of ct-siRNAs, like _NIA1_ and _NIA2_, were predominantly expressed in mesophyll
cells. Interestingly, we have also found that _DCL2_ showed a higher expression level than other subtypes of mesophyll cells, which may provide an explanation for the selective production of
22-nt ct-siRNAs from _NIA1_ and _NIA2_. More importantly, we observed the downregulated expression of _NIA1_ and _NIA2_ in the same subtype of mesophyll cells, aligning with our initial
expectation that the production of abundant ct-siRNAs would decrease the expression of their source transcripts. Furthermore, we observed that downregulated expression of ct-siRNA source
genes in specific cells might be compensated by upregulated expression of these transcripts in a variety of other cell types (Supplementary Fig. 7), resulting in their unchanged or increased
expression in bulk RNA-seq data. Our results suggest that gene silencing induced by ct-siRNAs potentially occur in specific cells, and whether this triggers compensatory upregulation of
genes in neighbouring cells is an interesting biological question. Given the lack of spatial information in single-cell transcriptomics, there is an urgent need for further research to
leverage the maturation of spatial transcriptomics and spatial small RNA detection technologies to address this limitation. As a fundamental surveillance mechanism, RNA decay eliminates
aberrant mRNAs, preventing them from being captured by the PTGS pathway and ultimately processed into rogue ct-siRNAs. The fate of aberrant mRNAs, whether they undergo decay or are silenced
by ct-siRNAs, may be determined by various factors involved in RNA decay and PTGS pathways, sequence composition, biological function, and cell-specific expression of ct-siRNA source genes.
It is unclear how a single factor may affect ct-siRNA selective production, while multiple factors should be considered in both qualitatively and quantitatively. METHODS PLANT MATERIALS AND
GROWTH CONDITIONS The _Arabidopsis_ plants of the Columbia (Col-0) accession were exclusively used. Commercially available Murashige and Skoog (MS) medium, along with nitrogen-depleted MS
salt obtained from Phyto Technology Laboratories (Catalog: M524, M531), to prepare the full-nutrition MS medium and nitrogen-depleted medium (pH 5.7–5.8, 1% sucrose, 10 g/L agar),
respectively. Seeds were surface-sterilized and plated on the medium55. Seeds pretreated with stratification for 3 days at 4 °C were kept in the greenhouse for another 6–7 days (22 °C, 16
h/8 h photoperiod) before transferring the seedlings to the soil or phenotyping. GENETIC ANALYSIS AND GENOTYPING The mutants and transgenic materials employed in this study were either
maintained in our laboratory or purchased from SALK. The _ein5-1_ alleles were derived from an x-ray mutagenized population (ecotype Col-0)56. The T-DNA insertional mutant _ski2-3_ was
acquired from SALK and subsequently validated by PCR amplification57. Point mutations including _rdr6-11_58, _ago1-47_, _ago1-45_59 and _hen1-8_60 were genotyped. The homozygous double and
triple mutants (_dcl2-1_ _dcl4-2_61, _ein5-1 dcl4-2_, _ski2-2 dcl4-2_, _ein5-1 ski2-3, ein5-1 dcl4-2 dcl2-1_, _ski2-2 dcl4-2 dcl2-1_, _ein5-1 dcl4-2 ago1-45_, _ein5-1 dcl4-2 ago1-27_31) were
generated through genetic crosses and identified from the F2 or F3 populations. Each mutation was confirmed by PCR-based genotyping and phenotypic analysis, or through the use of
antibiotic-resistant markers. To generate the _ein5-1 ski2-3 dcl4-2 dcl2-1_ quadruple mutant, we genotyped the F2 and F3 plants propagated from the cross between _ein5-1 ski2-3_ hemizygote
and _dcl4-2 dcl2-1_. While no _ein5-1 ski2-3 dcl4-2_ plant was verified from the segregating population derived from the _ein5-1 ski2-3_ hemizygote and _dcl4-2 dcl2-1_ cross. In these
experiments, the genotyping of _ski2-3, dcl4-2_, and _dcl2-1_ loci were conducted via PCR, and the _ein5-1_ mutation (1-bp deletion, frameshift) was confirmed through ethylene-related
phenotyping62 and validated through Sanger sequencing. RNA-SEQ AND SRNA-SEQ ANALYSIS RNA-seq and sRNA-seq data analysis was performed as described in our previous study31. GENE ENRICHMENT
ANALYSIS Gene enrichment analysis was performed using the BiNGO plugin63 of Cytoscape software64 with default parameters. GC CONTENT ANALYSIS We split the coding sequences of the
_Arabidopsis_ reference genome (TAIR10, https://www.arabidopsis.org/) into 100-bp bins and calculated the GC contents, which were used to visualize the distribution of the whole genome GC
content. In this study, GC content ≤ 30%, 30% < GC content < 50%, and GC content ≥ 50%, were defined as low, medium, and high GC regions, respectively. SINGLE NUCLEUS DATA
PREPROCESSING AND ANALYSIS The leaf tissue of 21-day-old Col-0, _ein5-1 dcl4-2_, _ski2-2 dcl4-2_ and _hen2-1 dcl4-2_ plants were harvested. We used the 10X Genomics snRNA-seq platform
(http://10xgenomics.com/) to profile over 15,000 nuclei. The FASTQ files were generated from Illumina BCL files using the _mkfastq_ function of Cell Ranger (version 6.1.2)
(http://10xgenomics.com/) and processed to count matrix by _count_ pipeline. The R package Seurat (version 3.1.5)65 was used to conduct single-cell data analysis. After filtering out
low-quality genes in each nucleus, the retained 8757 nuclei with the percentage of mitochondrial genes (percent.mt < 5) and chloroplast genes (percent.ct < 10) were used to carry out
the downstream analysis. The Seurat package was used to identify distinct cell populations based on gene expression profiles36. Cell populations were manually annotated to the functional
cell-type clusters combining the cell markers from the PlantscRNAdb and PCMDB database37,38. The “FindSubCluster” function with resolution = 0.6 was used to identify subclusters in mesophyll
cell publications. TRANSGENIC MATERIALS We truncated _NIA1_ and _NIA2_ CDS sequences to the consecutive 600-nt fragments and fused each truncated fragment with 35S promoter and green
fluorescent protein (GFP) sequence to construct transgenic materials. The sequences of truncated _NIA1_ and _NIA2_ CDS fragments of Fig. 4a are described in Supplementary Data 1. STATISTICS
AND REPRODUCIBILITY Genes that differentially accumulated 21-nt or 22-nt ct-siRNAs were identified by comparing the mutants deficient in RNA decay factors and/or DCL4 activity, against the
Col-0 using the R package – Deseq2 (version 1.38.3)66, with a cutoff of _padj_ < 0.05 and absolute log2FC (Fold Change) > 1. At least three biological replicates were used for these
analyses. Statistical analyses were conducted on source gene sequence length, 5’ UTR length, 3’ UTR length, intron number, and GC ratio using R (version 4.2.2). The source data for gene
sequence length and 5’ UTR length are provided in Supplementary Data 1. Statistical significance was determined through a two-tailed Student’s _t_-test (***_p_ < 0.001, ****_p_ <
0.0001) by comparing genes enriched in 21-nt or 22-nt ct-siRNAs with non-22-nt siRNA-producing genes. Statistical analysis of fluorescence intensity in transgenic plant expressing each
truncated _NIA1_ and _NIA2_ fragment was performed using GraphPad Prism 9. At least three technical replicates were detected for each sample, except for the transgenic plant expressing
_NIA2-2_, which had one replicate. Error bars were represented using the standard deviation (SD). Additionally, the abundance of sRNAs produced from transgenic plant expressing each
truncated _NIA1_ and _NIA2_ fragment was detected with two biological replicates. Genes were chosen for clustering analysis based on the accumulation of 22-nt ct-siRNAs, with TPM > 10 in
at least two stages, and an absolute log2FC > 1 when comparing any two stages. Each stage for the _ein5-1 dcl4-2_ and _ski2-2 dcl4-2_ mutant plants had one replicate. A total of 18
clusters were identified among the stages of the _ein5-1 dcl4-2_ and _ski2-2 dcl4-2_ plants using R package – pheatmap (version 1.0.12), respectively. REPORTING SUMMARY Further information
on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY The part of raw sRNA-seq data and all RNA-seq data used in this study have
been published by our previous work31, which were deposited on the NCBI Gene Expression Omnibus67 under the accession GSE136164. The raw sRNA-seq and snRNA-seq data generated by this study
can be accessed on the National Genomics Data Center under the BioProject PRJCA024518. The numerical source values underlying Fig. 1a, b, Fig. 2a–c, e, f, Fig. 3a–c, Fig. 4a, c, d, h, Fig.
5a–d, and Fig. 6a, d can be found in Supplementary Data 1. All other data related to this study can also be available upon reasonable request to the corresponding or 1st author. Uncropped
and unedited gel images are added in Supplementary Fig. 8. CHANGE HISTORY * _ 06 MAY 2024 A Correction to this paper has been published: https://doi.org/10.1038/s42003-024-06241-2 _
REFERENCES * Elkind, Y. et al. Abnormal plant development and down-regulation of phenylpropanoid biosynthesis in transgenic tobacco containing a heterologous phenylalanine ammonia-lyase
gene. _Proc. Natl. Acad. Sci. USA_ 87, 9057–9061 (1990). Article CAS PubMed PubMed Central Google Scholar * Borges, F. & Martienssen, R. A. The expanding world of small RNAs in
plants. _Nat. Rev. Mol. Cell Biol._ 16, 727–741 (2015). Article CAS PubMed PubMed Central Google Scholar * Ferrer-Orta, C., Agudo, R., Domingo, E. & Verdaguer, N. Structural
insights into replication initiation and elongation processes by the FMDV RNA-dependent RNA polymerase. _Curr. Opin. Struct. Biol._ 19, 752–758 (2009). Article CAS PubMed Google Scholar
* Bologna, N. G. & Voinnet, O. The diversity, biogenesis, and activities of endogenous silencing small RNAs in _Arabidopsis_. _Annu Rev. Plant Biol._ 65, 473–503 (2014). Article CAS
PubMed Google Scholar * Liu, L. & Chen, X. RNA quality control as a key to suppressing RNA silencing of endogenous genes in plants. _Mol. Plant_ 9, 826–836 (2016). Article CAS PubMed
Google Scholar * Houseley, J. & Tollervey, D. The many pathways of RNA degradation. _Cell_ 136, 763–776 (2009). Article CAS PubMed Google Scholar * Houseley, J., LaCava, J. &
Tollervey, D. RNA-quality control by the exosome. _Nat. Rev. Mol. Cell Biol._ 7, 529–539 (2006). Article CAS PubMed Google Scholar * Souret, F. F., Kastenmayer, J. P. & Green, P. J.
AtXRN4 degrades mRNA in _Arabidopsis_ and its substrates include selected miRNA targets. _Mol. Cell_ 15, 173–183 (2004). Article CAS PubMed Google Scholar * Zakrzewska-Placzek, M.,
Souret, F. F., Sobczyk, G. J., Green, P. J. & Kufel, J. _Arabidopsis_ _thaliana_ XRN2 is required for primary cleavage in the pre-ribosomal RNA. _Nucleic Acids Res._ 38, 4487–4502
(2010). Article CAS PubMed PubMed Central Google Scholar * Fang, X. et al. Chloroplast-to-nucleus signaling regulates MicroRNA biogenesis in _Arabidopsis_. _Dev. Cell_ 48, 371–382.e4
(2019). Article CAS PubMed Google Scholar * Krzyszton, M. et al. Defective XRN3-mediated transcription termination in _Arabidopsis_ affects the expression of protein-coding genes. _Plant
J._ 93, 1017–1031 (2018). Article CAS PubMed Google Scholar * You, C. et al. FIERY1 promotes microRNA accumulation by suppressing rRNA-derived small interfering RNAs in _Arabidopsis_.
_Nat. Commun._ 10, 4424 (2019). Article PubMed PubMed Central Google Scholar * Zhang, X. et al. Suppression of endogenous gene silencing by bidirectional cytoplasmic RNA decay in
_Arabidopsis_. _Science_ 348, 120–123 (2015). Article CAS PubMed Google Scholar * Western, T. L., Cheng, Y., Liu, J. & Chen, X. HUA ENHANCER2, a putative DExH-box RNA helicase,
maintains homeotic B and C gene expression in _Arabidopsis_. _Development_ 129, 1569–1581 (2002). Article CAS PubMed Google Scholar * Lange, H., Sement, F. M. & Gagliardi, D. MTR4, a
putative RNA helicase and exosome co-factor, is required for proper rRNA biogenesis and development in _Arabidopsis thaliana_. _Plant J._ 68, 51–63 (2011). Article CAS PubMed Google
Scholar * Lange, H. et al. The RNA Helicases AtMTR4 and HEN2 target specific subsets of nuclear transcripts for degradation by the nuclear exosome in _Arabidopsis thaliana_. _PLoS Genet._
10, e1004564 (2014). Article PubMed PubMed Central Google Scholar * Vigh, M. L., Bressendorff, S., Thieffry, A., Arribas-Hernández, L. & Brodersen, P. Nuclear and cytoplasmic RNA
exosomes and PELOTA1 prevent miRNA-induced secondary siRNA production in _Arabidopsis_. _Nucleic Acids Res._ 50, 1396–1415 (2022). Article CAS PubMed PubMed Central Google Scholar *
Lange, H. et al. RST1 and RIPR connect the cytosolic RNA exosome to the Ski complex in _Arabidopsis_. _Nat. Commun._ 10, 3871 (2019). Article PubMed PubMed Central Google Scholar * Auth,
M., Nyikó, T., Auber, A. & Silhavy, D. The role of RST1 and RIPR proteins in plant RNA quality control systems. _Plant Mol. Biol._ 106, 271–284 (2021). Article CAS PubMed PubMed
Central Google Scholar * Tharun, S. Lsm1-7-Pat1 complex: A link between 3′ and 5′-ends in mRNA decay? _RNA Biol._ 6, 228–232 (2009). Article CAS PubMed Google Scholar * Łabno, A.,
Tomecki, R. & Dziembowski, A. Cytoplasmic RNA decay pathways - Enzymes and mechanisms. _Biochim Biophys. Acta Mol. Cell Res._ 1863, 3125–3147 (2016). Article Google Scholar * Sement,
F. M. et al. Uridylation prevents 3′ trimming of oligoadenylated mRNAs. _Nucleic Acids Res_. 41, 7115–7127 (2013). Article CAS PubMed PubMed Central Google Scholar * Siwaszek, A.,
Ukleja, M. & Dziembowski, A. Proteins involved in the degradation of cytoplasmic mRNA in the major eukaryotic model systems. _RNA Biol._ 11, 1122–1136 (2014). Article PubMed PubMed
Central Google Scholar * Xu, J., Yang, J. Y., Niu, Q. W. & Chua, N. H. _Arabidopsis_ DCP2, DCP1, and VARICOSE form a decapping complex required for postembryonic development. _Plant
Cell_ 18, 3386–3398 (2006). Article CAS PubMed PubMed Central Google Scholar * Xu, J. & Chua, N. H. _Arabidopsis_ decapping 5 is required for mRNA decapping, P-body formation, and
translational repression during postembryonic development. _Plant Cell_ 21, 3270–3279 (2009). Article CAS PubMed PubMed Central Google Scholar * Kwasnik, A. et al. Erratum:
_Arabidopsis_ DXO1 links RNA turnover and chloroplast function independently of its enzymatic activity. _Nucleic Acids Res._ 47, 4910 (2019). Article PubMed PubMed Central Google Scholar
* Pan, S. et al. _Arabidopsis_ DXO1 possesses deNADding and exonuclease activities and its mutation affects defense-related and photosynthetic gene expression. _J. Integr. Plant Biol._ 62,
967–983 (2020). Article CAS PubMed Google Scholar * Jiao, X. et al. 5′ end nicotinamide adenine dinucleotide cap in human cells promotes RNA decay through DXO-mediated deNADding. _Cell_
168, 1015–1027.e10 (2017). Article CAS PubMed PubMed Central Google Scholar * De Alba, A. E. M. et al. In plants, decapping prevents RDR6-dependent production of small interfering RNAs
from endogenous mRNAs. _Nucleic Acids Res._ 43, 2902–2913 (2015). Article Google Scholar * Scheer, H. et al. The TUTase URT1 connects decapping activators and prevents the accumulation of
excessively deadenylated mRNAs to avoid siRNA biogenesis. _Nat. Commun._ 12, 1298 (2021). Article CAS PubMed PubMed Central Google Scholar * Wu, H. et al. Plant 22-nt siRNAs mediate
translational repression and stress adaptation. _Nature_ 581, 89–93 (2020). Article CAS PubMed Google Scholar * Rymarquis, L. A., Souret, F. F. & Green, P. J. Evidence that XRN4, an
_Arabidopsis_ homolog of exoribonuclease XRN1, preferentially impacts transcripts with certain sequences or in particular functional categories. _RNA_ 17, 501–511 (2011). Article CAS
PubMed PubMed Central Google Scholar * Jay De Rocher, E., Vargo-Gogola, T. C., Diehn, S. H. & Green, P. J. Direct evidence for rapid degradation of bacillus thuringiensis toxin mRNA
as a cause of poor expression in plants. _Plant Physiol._ 117, 1445–1461 (1998). Article PubMed Central Google Scholar * Sidorenko, L. V. et al. GC-rich coding sequences reduce
transposon-like, small RNA-mediated transgene silencing. _Nat. Plants_ 3, 875–884 (2017). Article CAS PubMed Google Scholar * Courel, M. et al. Gc content shapes mRNA storage and decay
in human cells. _Elife_ 8, e49708 (2019). Article PubMed PubMed Central Google Scholar * Hao, Y. et al. Integrated analysis of multimodal single-cell data. _Cell_ 184, 3573–3587 (2021).
Article CAS PubMed PubMed Central Google Scholar * Jin, J. et al. PCMDB: A curated and comprehensive resource of plant cell markers. _Nucleic Acids Res._ 50, D1448–D1455 (2022). Article
CAS PubMed Google Scholar * Chen, H. et al. PlantscRNAdb: A database for plant single-cell RNA analysis. _Mol. Plant_ 14, 855–857 (2021). Article CAS PubMed Google Scholar * Kim, J.
Y. et al. Distinct identities of leaf phloem cells revealed by single cell transcriptomics. _Plant Cell_ 33, 511–530 (2021). Article PubMed PubMed Central Google Scholar * Fu, Y., Gu,
Y., Zheng, Z., Wasteneys, G. & Yang, Z. _Arabidopsis_ interdigitating cell growth requires two antagonistic pathways with opposing action on cell morphogenesis. _Cell_ 120, 687–700
(2005). Article CAS PubMed Google Scholar * Perrin, R. M., Wang, Y., Yuen, C. Y. L., Will, J. & Masson, P. H. WVD2 is a novel microtubule‐associated protein in _Arabidopsis
thaliana_. _Plant J._ 49, 961–971 (2007). Article CAS PubMed Google Scholar * Peaucelle, A. et al. _Arabidopsis_ phyllotaxis is controlled by the methyl-esterification status of
cell-wall pectins. _Curr. Biol._ 18, 1943–1948 (2008). Article CAS PubMed Google Scholar * Yang, W., Wightman, R. & Meyerowitz, E. M. Cell cycle control by nuclear sequestration of
_CDC20_ and _CDH1_ mRNA in plant stem cells. _Mol. Cell_ 68, 1108–1119 (2017). Article CAS PubMed PubMed Central Google Scholar * Procko, C. et al. Leaf cell-specific and single-cell
transcriptional profiling reveals a role for the palisade layer in UV light protection. _Plant Cell_ 34, 3261–3279 (2022). Article PubMed PubMed Central Google Scholar * Takahashi, H. et
al. The roles of three functional sulphate transporters involved in uptake and translocation of sulphate in _Arabidopsis thaliana_. _Plant J._ 23, 171–182 (2000). Article CAS PubMed
Google Scholar * Guo, X. et al. Single‐cell transcriptome reveals differentiation between adaxial and abaxial mesophyll cells in _Brassica_ _rapa_. _Plant Biotechnol. J._ 20, 2233 (2022).
Article CAS PubMed PubMed Central Google Scholar * Tian, C. et al. A gene expression map of shoot domains reveals regulatory mechanisms. _Nat. Commun._ 10, 141 (2019). Article PubMed
PubMed Central Google Scholar * Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. _Nucleic Acids Res._ 28, 27–30 (2000). Article CAS PubMed PubMed Central
Google Scholar * Tan, H. et al. Phase separation of SGS3 drives siRNA body formation and promotes endogenous gene silencing. _Cell Rep._ 42, 111985 (2023). Article CAS PubMed Google
Scholar * Axtell, M. J. Classification and comparison of small RNAs from plants. _Annu Rev. Plant Biol._ 64, 137–159 (2013). Article CAS PubMed Google Scholar * McHale, M., Eamens, A.
L., Finnegan, E. J. & Waterhouse, P. M. A 22-nt artificial microRNA mediates widespread RNA silencing in _Arabidopsis_. _Plant J._ 76, 519–529 (2013). Article CAS PubMed PubMed
Central Google Scholar * Bleys, A., Vermeersch, L., Van Houdt, H. & Depicker, A. The frequency and efficiency of endogene suppression by transitive silencing signals is influenced by
the length of sequence homology. _Plant Physiol._ 142, 788–796 (2006). Article CAS PubMed PubMed Central Google Scholar * Braunstein, T. H., Moury, B., Johannessen, M. &
Albrechtsen, M. Specific degradation of 3′ regions of GUS mRNA in posttranscriptionally silenced tobacco lines may be related to 5′-3′ spreading of silencing. _RNA_ 8, 1034–1044 (2002).
Article CAS PubMed PubMed Central Google Scholar * Zhou, B. & Zeng, L. Elucidating the role of highly homologous _Nicotiana_ _benthamiana_ ubiquitin E2 gene family members in plant
immunity through an improved virus-induced gene silencing approach. _Plant Methods_ 13, 1–17 (2017). Article CAS Google Scholar * Vogel, J. P., Woeste, K. E., Theologis, A. & Kieber,
J. J. Recessive and dominant mutations in the ethylene biosynthetic gene _ACS5_ of _Arabidopsis_ confer cytokinin insensitivity and ethylene overproduction, respectively. _Proc. Natl. Acad.
Sci. USA_ 95, 4766–4771 (1998). * Kieber, J. J., Rothenberg, M., Roman, G., Feldmann, K. A. & Ecker, J. R. _CTR1_, a negative regulator of the ethylene response pathway in _Arabidopsis_,
encodes a member of the Raf family of protein kinases. _Cell_ 72, 427–441 (1993). Article CAS PubMed Google Scholar * Alonso, J. M. et al. Genome-wide insertional mutagenesis of
_Arabidopsis thaliana_. _Science_ 301, 653–657 (2003). Article PubMed Google Scholar * Peragine, A., Yoshikawa, M., Wu, G., Albrecht, H. L. & Poethig, R. S. _SGS3_ and
_SGS2_/_SDE1_/_RDR6_ are required for juvenile development and the production of trans-acting siRNAs in _Arabidopsis_. _Genes Dev._ 18, 2368–2379 (2004). Article CAS PubMed PubMed Central
Google Scholar * Smith, M. R. et al. Cyclophilin 40 is required for microRNA activity in _Arabidopsis_. _Proc. Natl Acad. Sci._ 106, 5424–5429 (2009). Article CAS PubMed PubMed Central
Google Scholar * Yu, B. et al. siRNAs compete with miRNAs for methylation by HEN1 in _Arabidopsis_. _Nucleic Acids Res._ 38, 5844–5850 (2010). Article CAS PubMed PubMed Central Google
Scholar * Henderson, I. R. et al. Dissecting _Arabidopsis thaliana_ DICER function in small RNA processing, gene silencing and DNA methylation patterning. _Nat. Genet._ 38, 721–725 (2006).
Article CAS PubMed Google Scholar * Guzmán, P. & Ecker, J. R. Exploiting the triple response of _Arabidopsis_ to identify ethylene-related mutants. _Plant Cell_ 2, 513–523 (1990). *
Maere, S., Heymans, K. & Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. _Bioinformatics_ 21, 3448–3449 (2005).
Article CAS PubMed Google Scholar * Shannon, P. et al. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. _Genome Res._ 13, 2498–2504 (2003).
Article CAS PubMed PubMed Central Google Scholar * Stuart, T. et al. Comprehensive integration of single-cell data. _Cell_ 177, 1888–1902.e21 (2019). Article CAS PubMed PubMed
Central Google Scholar * Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. _Genome Biol._ 15, 1–21 (2014). Article
Google Scholar * Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. _Nucleic Acids Res._ 30, 207–210 (2002).
Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by the National Natural Science Foundation of China (Grant no. 32170574),
the National Key Research and Development Program of China (Grant no. 2018YFA0507101), the National Natural Science Foundation of China (Grant no. 32261160572), the Shenzhen Science and
Technology Innovation Program (Grant no. 20200925161843002), the Shenzhen Science and Technology Program (Grant no. JCYJ20190809163019421), the Key R&D Program of Shandong Province
(Grant no. ZR202211070163), the Shandong Provincial Natural Science Foundation (Grant no. ZR2023QC026), the Young Taishan Scholars Program and Yuandu Scholars Program. We thank Dr. Kaiwen Lv
(Peking University) for assistance in small RNA analysis. We are grateful to Dr. Jixian Zhai (SUSTech) for critical comments on the subject. We thank Dr. Zhiyuan Sun (SUSTech) for the help
in single cell RNA-seq. AUTHOR INFORMATION Author notes * These authors contributed equally: Li Feng, Wei Yan, Xianli Tang. AUTHORS AND AFFILIATIONS * Peking University Institute of Advanced
Agricultural Sciences, Shandong Laboratory of Advanced Agriculture Sciences in Weifang, Weifang, Shandong, 261325, China Li Feng, Dongdong Lu, Yongqi Liu, Xiehai Song, Muhammad Ali &
Bosheng Li * Institute of Plant and Food Science, Department of Biology, Southern University of Science and Technology, Shenzhen, Guangdong, 518055, China Wei Yan, Xianli Tang, Huihui Wu,
Yajie Pan, Qianyan Ling-hu, Yuelin Liu, Liang Fang & Hongwei Guo Authors * Li Feng View author publications You can also search for this author inPubMed Google Scholar * Wei Yan View
author publications You can also search for this author inPubMed Google Scholar * Xianli Tang View author publications You can also search for this author inPubMed Google Scholar * Huihui Wu
View author publications You can also search for this author inPubMed Google Scholar * Yajie Pan View author publications You can also search for this author inPubMed Google Scholar *
Dongdong Lu View author publications You can also search for this author inPubMed Google Scholar * Qianyan Ling-hu View author publications You can also search for this author inPubMed
Google Scholar * Yuelin Liu View author publications You can also search for this author inPubMed Google Scholar * Yongqi Liu View author publications You can also search for this author
inPubMed Google Scholar * Xiehai Song View author publications You can also search for this author inPubMed Google Scholar * Muhammad Ali View author publications You can also search for
this author inPubMed Google Scholar * Liang Fang View author publications You can also search for this author inPubMed Google Scholar * Hongwei Guo View author publications You can also
search for this author inPubMed Google Scholar * Bosheng Li View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS B.S.Li and H.W.Guo conceived of
the project and designed the experiments; X.L.Tang, Q.Y.Ling-hu, Y.J.Pan, Y.L.Liu, and H.H.Wu prepared the genetic materials; small RNA sequencing experiment with contributions from
X.L.Tang, Y.J.Pan, Y.L.Liu, and H.H.Wu; L.Feng, W.Yan, Y.J.Pan, Y.Q.Liu, M.Ali, and B.S.Li performed the bioinformatics analysis; B.S.Li, X.H.Song, Y.Q.Liu, D.D.Lu, and L.Fang performed
single cell RNA sequencing; L.Feng, B.S.Li, W.Yan, and H.W.Guo wrote the manuscript with input from all co-authors. CORRESPONDING AUTHORS Correspondence to Hongwei Guo or Bosheng Li. ETHICS
DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Communications Biology_ thanks Niankui Li, Jungnam Cho and the other,
anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: David Favero. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral
with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGS. DESCRIPTION OF SUPPLEMENTARY MATERIALS SUPPLEMENTARY
DATA 1 REPORTING SUMMARY RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation,
distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and
indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to
the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will
need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE
CITE THIS ARTICLE Feng, L., Yan, W., Tang, X. _et al._ Multiple factors and features dictate the selective production of ct-siRNA in _Arabidopsis_. _Commun Biol_ 7, 474 (2024).
https://doi.org/10.1038/s42003-024-06142-4 Download citation * Received: 10 November 2023 * Accepted: 03 April 2024 * Published: 18 April 2024 * DOI:
https://doi.org/10.1038/s42003-024-06142-4 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative