Play all audios:
ABSTRACT The accelerating growth of make-on-demand chemical libraries provides unprecedented opportunities to identify starting points for drug discovery with virtual screening. However,
these multi-billion-scale libraries are challenging to screen, even for the fastest structure-based docking methods. Here we explore a strategy that combines machine learning and molecular
docking to enable rapid virtual screening of databases containing billions of compounds. In our workflow, a classification algorithm is trained to identify top-scoring compounds based on
molecular docking of 1 million compounds to the target protein. The conformal prediction framework is then used to make selections from the multi-billion-scale library, reducing the number
of compounds to be scored by docking. The CatBoost classifier showed an optimal balance between speed and accuracy and was used to adapt the workflow for screens of ultralarge libraries.
Application to a library of 3.5 billion compounds demonstrated that our protocol can reduce the computational cost of structure-based virtual screening by more than 1,000-fold. Experimental
testing of predictions identified ligands of G protein-coupled receptors and demonstrated that our approach enables discovery of compounds with multi-target activity tailored for therapeutic
effect. SIMILAR CONTENT BEING VIEWED BY OTHERS ARTIFICIAL INTELLIGENCE–ENABLED VIRTUAL SCREENING OF ULTRA-LARGE CHEMICAL LIBRARIES WITH DEEP DOCKING Article 04 February 2022 AN ARTIFICIAL
INTELLIGENCE ACCELERATED VIRTUAL SCREENING PLATFORM FOR DRUG DISCOVERY Article Open access 05 September 2024 THE IMPACT OF LIBRARY SIZE AND SCALE OF TESTING ON VIRTUAL SCREENING Article 03
January 2025 MAIN The number of possible drug-like molecules has been estimated to be more than 1060, which exceeds the size of chemical libraries evaluated in early drug discovery by many
orders of magnitude1. In fact, only ~13 million compounds are currently available in-stock from chemical suppliers, which clearly illustrates the limited coverage of chemical space2.
Advances in synthetic organic chemistry have provided access to increasingly larger compound collections and make-on-demand libraries currently contain >70 billion readily available
molecules3,4. The diverse scaffolds available in these libraries represent a major opportunity for drug discovery, but identifying the compounds relevant for a specific target in this
enormous chemical space remains a major challenge. Recently, structure-based virtual screens of ultralarge libraries have identified ligands of important therapeutic targets, demonstrating
that expanding the coverage of chemical space can accelerate early drug discovery5,6,7. The most recently published docking screens have reached billions of compounds, but evaluation of
these massive libraries is demanding due to the substantial computational resources required8,9. The make-on-demand databases will also continue to grow and probably reach trillions of
compounds in the near future, which will be unfeasible to screen even with the fastest structure-based docking algorithms. Therefore, there is an urgent need for more efficient virtual
screening approaches able to evaluate these vast chemical libraries10. Recent breakthroughs in artificial intelligence have revived interest in using quantitative structure–activity
relationship (QSAR) models in drug discovery. QSAR has been widely used by the pharmaceutical industry to predict both on- and off-target activities, as well as physicochemical and
pharmacokinetic properties11. By representing compounds using molecular descriptors, machine learning methods can rapidly evaluate large compound databases. Traditionally, QSAR models have
been trained on experimental data12, but there is an increasing interest in predicting which compounds in make-on-demand libraries are likely to receive favorable scores from computationally
expensive virtual screening methods13,14,15. This combination of machine learning and molecular docking screening has the potential to enable virtual screens of multi-billion-scale compound
libraries at a modest computational cost. In this work, we developed an ultrafast workflow based on conformal prediction (CP) for screening of vast chemical libraries. The CP framework can
be applied to any machine learning classifier and allows the user to control the error rate of the predictions16,17. Mondrian conformal predictors provide class-specific confidence levels
that ensure validity for both the majority and minority class. This approach is therefore well suited for handling inherently imbalanced datasets such as virtual screening applications,
which focus on identifying a small number of top-scoring compounds in a chemical library18. The framework has been utilized in QSAR applications to predict pharmacokinetic properties and
bioactivity19,20. Strategies to improve the virtual screening efficiency using the CP framework have been explored, but these workflows did not achieve the efficiency required to evaluate
multi-billion-scale libraries21. Applications of more recently developed techniques such as gradient boosting, deep neural networks and transformers to early-phase drug discovery have been
successful22,23,24. Here we combined the CP framework with several state-of-the-art classification algorithms to develop a workflow for accelerated structure-based virtual screening. Our
most efficient protocol identifies the top-scoring compounds in ultralarge compound libraries and reduces the number of molecules to be explicitly docked by three orders of magnitude. We
show that application of machine learning to guide docking screens of multi-billion-scale compound databases enables efficient discovery of ligands targeting G protein-coupled receptors,
which is one of the most important families of drug targets25. In particular, our workflow can screen billions of compounds against several targets to identify ligands with activity at
multiple targets relevant for the same disease. RESULTS The protocol combining CP and molecular docking to navigate ultralarge compound libraries is described in ‘Machine
learning-accelerated virtual screening pipeline’ in Methods (Fig. 1, Supplementary Section 1 and Supplementary Fig. 1). In the development of this approach, we first conducted benchmarking
docking screens against eight protein targets. The resulting datasets were used to select suitable algorithms and molecular descriptors. In the second step, the method was further optimized
to enable virtual screens of multi-billion-scale libraries and applied to predict ligands of the A2A adenosine (A2AR) and D2 dopamine (D2R) receptors. BENCHMARKING OF CONFORMAL PREDICTORS
Molecular docking screens against eight therapeutically relevant proteins were carried out to initiate performance evaluation of the CP workflow. A detailed description of the protein
targets and preparation of the molecular docking calculations is provided in Supplementary Table 1, Supplementary Fig. 2 and ‘Preparation of proteins for docking’ in Methods. Eleven million
randomly sampled rule-of-four (Ro4, molecular weight <400 Da and cLog_P_ < 4) molecules from the Enamine REAL space were prepared for molecular docking and screened against each
target. In total, more than 493 trillion protein–ligand complexes were predicted, resulting in a final benchmarking set of 88 million unique protein–ligand complexes and their corresponding
scores. For each target, chemical structures of the compounds and their corresponding docking scores were used to create training (106 compounds) and test (107 compounds) sets for evaluating
the CP framework. The energy threshold for the active (minority) class was determined based on the top-scoring 1% of each screen. For each protein target, we assessed the performance of
three different machine learning algorithms: CatBoost26, deep neural networks27 and Robustly Optimized Bidirectional Encoder Representations from Transformers Approach (RoBERTa)28. To
explore diverse representations of small molecules, we trained our algorithms on three different types of features: (1) Morgan2 fingerprints, the RDKit implementation of the
substructure-based ECFP4 descriptor29, which have consistently performed among the best features in previous virtual screening benchmarks30; (2) recently developed continuous data-driven
descriptors (CDDD)31, which provide dense latent representations of molecules; and (3) transformer-based descriptors derived from a pretrained RoBERTa encoder, which serve as the input for
fine-tuning the RoBERTa models28. Detailed descriptions of the hyperparameters used in the training of each classifier are provided in Supplementary Section 2 (Supplementary Table 2 and
Supplementary Figs. 3–5). Five independent classifiers were trained on 1 million labeled features, of which 80% was used for proper training and the remaining 20% for calibration. The
features of the compounds in the test set (10 million compounds) were then assigned 10 normalized _P_ values (five _P_1 and five _P_0 values) by using each individual classification model
and its corresponding calibration sets. The two resulting sets of _P_1 and _P_0 values were aggregated into a single pair of _P_1 and _P_0 values by taking the respective medians
(Supplementary Fig. 1). On the basis of the aggregated _P_ values and a selected significance level (_ε_), the Mondrian CP framework was used to divide the compounds into virtual active,
virtual inactive, both (meaning, either virtual active or inactive) and null (no assignment) sets (Fig. 2a). The performance on the benchmarking set was assessed using the significance level
at which the predictor resulted in the maximal number of useful (single label) predictions, _ε_opt (Fig. 2b). The metrics to assess the performance of the models (sensitivity, precision,
efficiency and prediction error rate) are defined in ‘Training and evaluation of machine learning classifiers’ in Methods. Following the CP framework, exchangeability between the training
and test sets led to strong agreement between the prediction error rate and the selected significance level16 (Fig. 2c). To minimize the number of compounds requiring explicit docking while
maximizing predictive power, we aimed to determine the optimal size of the training set, exploring a range between 25,000 and 1 million compounds. Improved sensitivity, precision, and
significance values were obtained for all targets when increasing the training set size (Fig. 2d–f and Supplementary Tables 3–8). As the performance of the models stabilized at a training
size of 1 million molecules, this size was established as the standard for the training of new models. Conformal predictors composed of CatBoost classifiers trained on Morgan2 fingerprints
achieved the best average precision and had comparable or slightly better significance and sensitivity values compared with other combinations. In addition, this configuration required the
least computational resources, both in the training of the classifier, predictions for the test set and storage of molecular descriptors (Supplementary Table 9). Hyperparameters (class
imbalance and number of aggregated models), robustness (influence of noise and scrambling of the training data), target dependency and the exchangeability criterion were also investigated.
These results are provided in detail in Supplementary Figs. 6–12. OPTIMIZED WORKFLOW FOR ULTRALARGE CHEMICAL LIBRARIES To optimize the performance of the workflow for ultralarge databases,
we conducted further analyses of datasets containing docking scores for 235 million compounds from the ZINC15 library32, focusing on two benchmarking proteins (A2AR and D2R). For each
target, a conformal predictor composed of five independent CatBoost classifiers was trained on 1 million compounds (Morgan2 representation), followed by predictions for the remainder of the
library. As the docking scores were available for all the compounds in the library, efficient strategies to identify the top-scoring molecules could be established. In the CP framework, the
selected significance level determines the size of the predicted virtual active set, which is the library that will be docked to the target. The significance level was first set to achieve
the maximal efficiency (A2AR _ε_opt = 0.12 and D2R _ε_opt = 0.08), and close to all compounds received a single label (>98% for both targets). CP reduced the ultralarge library from 234
million to 25 million and 19 million compounds for A2AR and D2R, respectively, with high sensitivity values (0.87 and 0.88, respectively; Fig. 3a). The workflow would hence be able to
identify close to 90% of the virtual actives by docking only ~10% of the ultralarge library and the CP framework guaranteed that the percentage of incorrectly classified compounds did not
exceed 12% and 8% respectively. For libraries of this size, molecular docking screens of the entire predicted virtual active set would be viable. However, further reduction of the database
would be required to apply our workflow to multi-billion-scale libraries. In these cases, docking calculations for even a small percentage of the library would be computationally demanding.
In theory, decreasing the significance level should lead to a reduction of the virtual active set and enrich predictions in which the conformal predictor has the highest confidence. This
approach was evaluated by gradually reducing the significance level and assessing how the distribution of docking scores in the virtual active set was influenced. As anticipated, lowering
the significance level did reduce the virtual active set size (Fig. 3a) and led to substantial shifts of the docking score distribution toward better energies for both protein targets (Fig.
3b). At the lowest evaluated significance level (0.01), the database was reduced to 3.0 million and 2.6 million molecules for A2AR and D2R, respectively, and the largest shifts in docking
score distributions were obtained. For example, the most populated bin in the docking score distribution for the training set was −23.8 kcal mol−1 for D2R, which was improved to −47.7 kcal
mol−1 and −50.9 kcal mol−1 for significance levels of 0.08 (D2R _ε_opt) and 0.01, respectively. At the strictest significance level (0.01), 80% and 64% of the 10,000 top-scoring A2AR and D2R
molecules (corresponding to 0.004% of the chemical library) could still be identified. These results showed that the significance level can be tuned to achieve substantial database
reduction and retain most of the very top-scoring candidates for the subsequent docking step. An alternative approach to reduce the size of the set to evaluate by molecular docking is to
sort the compounds based on the difference between the _P_1 and _P_0 values (the quality of information, _P_1 − _P_0). This metric can be used to prioritize subsets in which the predictor
has the highest confidence (Fig. 3c) and correlated with docking ranks (Supplementary Fig. 13). The enrichment of the top-scoring 10,000 molecules from the A2AR and D2R screens was assessed
based on prioritizing the compounds using the quality of information. Remarkably, the workflow identified more than 90% of the very top-scoring molecules after only 3% (A2AR) and 5% (D2R) of
the 234 million remaining compounds had been evaluated (Fig. 3c). Notably, reproducible recall values were obtained by independently generated conformal predictors, demonstrating that a
random selection of training set will lead to similar selection of molecules. Data-dimensionality reduction (Uniform Manifold Approximation and Projection, UMAP) of the Morgan2 fingerprints
indicated that these prioritized molecules bear structural similarity to the actives present in the training set (Fig. 3d). This observation was also supported by analysis of Tanimoto
similarity. The molecules in which the predictor had higher confidence generally showed greater structural similarity to actives from the training set (Fig. 3e). Using the quality of
information to reduce the docked set of compounds hence had a similar effect to decreasing the significance level, and these two techniques can be combined in prospective screens of
multi-billion-scale libraries. To assess whether the use of conformal predictors leads to a reduction in structural diversity among prioritized molecules compared with large-scale docking
screens, we analyzed the 1% top-ranked molecules from both approaches for D2R. Although a smaller fraction of the 1% top-ranked compounds prioritized by the conformal predictor had unique
Bemis–Murcko scaffolds (13% compared with 23% from docking), a pairwise Tanimoto coefficient analysis demonstrated that the decorated versions of these scaffolds were not significantly less
diverse than those identified by molecular docking alone (Supplementary Fig. 14). Collectively, these results demonstrated that top-scoring compounds could be identified using the conformal
predictor. To assess its ability to find experimentally confirmed actives, known A2AR and D2R ligands from the ChEMBL database33 were evaluated. Models trained only on docking data correctly
classified 92% and 86% of these ligands as virtual actives. This highlights the importance of benchmarking against known actives to validate workflows before conducting prospective virtual
screens (Fig. 3f). PROSPECTIVE VIRTUAL SCREEN OF A MULTI-BILLION-SCALE LIBRARY A primary goal in the development of the workflow was that the machine learning step must be able to reduce a
multi-billion-scale database to a few million promising compounds, which was evaluated for A2AR and D2R. Docking of the training set, training of the conformal predictor and predictions for
3.5 billion compounds for one target could be performed in approximately 2,500 core-hours. The significance level was set to 0.005, resulting in 25 million and 24 million predicted virtual
actives for A2AR and D2R, respectively. Of these, 5 million compounds per target were prioritized for docking calculations based on the quality of information (corresponding to a 700-fold
reduction of the library), which required 10,344 core-hours per target. Compared with explicit docking of the 3.5 billion compounds, the workflow hence achieved a 568-fold reduction of
compute cost. For both targets, the docking score distribution of the 5 million prioritized compounds was substantially shifted toward more favorable energies. For example, the most
populated bin in the docking score distribution of the training set was −25.1 kcal mol−1 for D2R, which was shifted to −51.6 kcal mol−1 for the predicted virtual actives (Fig. 4a). A large
fraction of the predicted compounds (49%) had a docking score better than the energy threshold (−49.7 kcal mol−1) used for labeling of the training set, corresponding to a 49-fold enrichment
of virtual actives. Similar docking energy distributions were obtained when only 1 million predicted virtual actives were selected for molecular docking, demonstrating that users can
control the extent of database reduction and even achieve up to a 3,500-fold decrease in library size. To assess whether ligands could be discovered using the workflow, we selected 31
top-ranked compounds from the D2R screen of 3.5 billion compounds and evaluated these in a radioligand binding assay at a concentration of 10 μM (Supplementary Table 10 and Supplementary
Data). Of these, compounds 1 and 2 showed significant radioligand displacement and affinity values (_K_i) values were determined for these D2R ligands (_K_i = 3.0 μM and _K_i = 3.8 μM,
respectively; Fig. 4b, Supplementary Table 11 and Supplementary Fig. 15). To further characterize compounds 1 and 2, we performed a functional assay quantifying D2R-mediated changes in
intracellular cAMP. Compounds 1 and 2 were full agonists of the D2R with potency values (EC50) values of 10 μM and 14 μM, respectively (maximal effect, _E_max = 99% and _E_max = 100%,
relative to the maximal effect of dopamine; Supplementary Fig. 16). These results demonstrate that our protocol enables identification of starting points for drug discovery by docking only a
small subset of compounds from a multi-billion-scale library. MACHINE LEARNING-GUIDED DESIGN OF POLYPHARMACOLOGY One of the potential advantages of screening multi-billion-scale compound
databases is that improved coverage of chemical space could enable discovery of ligands with complex properties, which may be difficult to find in smaller libraries containing only a few
million molecules. One potential application is to design compounds with activity at multiple targets that are relevant for the same disease, which could lead to synergistic therapeutic
effects. For example, the treatment of many central nervous system disorders requires the modulation of multiple targets (polypharmacology)34. Parkinson’s disease is a neurological disorder
that can be treated by modulating the activity of D2R and A2AR. However, the identification of dual-target ligands of A2AR and D2R is challenging because of the lack of similarity between
the binding sites of the receptors35. To assess whether a machine learning approach could facilitate the search for dual-target ligands, A2AR and D2R were prepared for docking calculations.
As the treatment of Parkinson’s disease necessitates activation of D2R through agonist binding, we generated a model of the active receptor state using homology modeling based on an
agonist-bound cryogenic electron microscopy (cryo-EM) structure of the D3 subtype (Protein Data Bank (PDB) accession code: 7CMV), which is described in detail in ‘Homology modeling of
active-state D2 dopamine receptor’ in Methods (Fig. 5a). To identify compounds blocking A2AR signaling, we prepared an antagonist-bound crystal structure (PDB accession code: 8GNE) for
molecular docking. In this structure, the salt bridge formed by the residues His264 and Glu169 has been disrupted and could potentially accommodate the ammonium cation characteristic of
dopaminergic ligands. After optimizing the receptor models and docking parameters, strong enrichment of known ligands was obtained for both targets (Supplementary Fig. 17). A new training
set of 1 million random molecules from a lead-like library (see ‘Docking library preparation’ in Methods) containing more than 3 billion compounds was then docked to both receptors. As
anticipated, the resulting docking score distributions illustrate that compounds that bind to both targets would be difficult to identify in small libraries. The overlap between the
top-ranked compounds (top 1%) was less than 0.02% (Fig. 5b). For both targets, conformal predictors were trained and used to predict the remaining billions of compounds in the lead-like
library. To enhance the likelihood of obtaining favorable docking scores across both targets, the predicted molecules were ranked according to the sum of their quality of information
(_P_A2AR,1 – _P_A2AR,0 + _P_D2R,1 – _P_D2R,0: in which _P__φ_,1 and _P__φ_,0 correspond to the confidences that a specific molecule belongs to the active and inactive class, respectively,
for target _φ_) and the 5 million top-ranked compounds were then prioritized for explicit docking. The docking score distributions of the machine learning-prioritized compounds were
substantially shifted toward better energies for both targets (17- and 34-fold for A2AR and D2R, respectively; Fig. 5c), leading to an enrichment of dual virtual actives. More than 3.8% of
the 5 million docked molecules had energies better than both score thresholds (top 1%), corresponding to a 191-fold enrichment of dual virtual actives compared with docking of a random
library. The molecules were subsequently sorted according to the sum of their individual docking ranks (rankA2AR + rankD2R), and the top-ranked molecules were then visually inspected for
their complementarity with the respective binding sites. Encouragingly, the molecules formed hydrogen bonds with residues known to be important for ligand binding to the A2AR (Asn2536.55)
and D2R (Asp1143.32) (Ballesteros-Weinstein residue numbering scheme36 denoted as superscripts). A set of 45 compounds was prioritized for make-on-demand synthesis and successfully obtained
within 4–5 weeks. The compounds were first tested in an A2AR radioligand binding assay at a concentration of 20 μM (Supplementary Table 12 and Supplementary Data). Of these, the binding
affinities of four compounds (4–6) that showed significant radioligand displacement were determined, leading to _K_i values ranging from 1.3 μM to 20 μM (Supplementary Table 13). Compounds
4–6 were subsequently tested at D2R at a concentration of 20 μM, which showed that compound 5 also binds to this target (_K_i,D2 = 14 μM, _K_i,A2A ≈ 20 μM; Fig. 5d, Supplementary Table 14
and Supplementary Fig. 18). The dual-target compound 5 was predicted to form hydrogen bonds with orthosteric binding site residues Asn2536.55 and Asp1143.32 for A2AR and D2R, respectively
(Fig. 5e,f). These observations indicate that our virtual screening workflow can be applied to identify starting points for the development of drugs with multi-target profiles tailored for
treatment of complex diseases. DISCUSSION The rapid expansion of commercial chemical libraries has sparked the development of diverse structure-based virtual screening methods aiming to
reduce the computational cost of exploring chemical space. Several of these approaches incorporated machine learning techniques to efficiently evaluate libraries ranging from millions to
billions of compounds13,14,15,37. Compared with previously developed methods tackling ultralarge libraries, our approach is based on CP, a robust framework that enables control over the
error rate of the predictions16. We extend the application of conformal predictors to multi-billion-scale libraries by leveraging class-specific confidence levels to identify top-scoring
molecules. Our approach achieves equivalent or better recall values and database reductions as other workflows, without the need for resource-intensive active learning. Our comparison of
substructure-based fingerprints with more recently developed data-driven descriptors demonstrates that traditional fingerprints suffice for this type of application, in agreement with a
recently published study37. Integrating these key advantages led to a workflow capable of traversing vast chemical libraries but requiring only modest computational cost associated with the
training and prediction steps. Notably, our results demonstrate that the effectiveness of machine learning as a proxy for molecular docking is target dependent, underscoring the need for
large and diverse benchmarking sets to achieve generalizable methods. To catalyze further development of methods in this field, we share both our virtual screening workflow, which is
compatible with any docking software, and extensive benchmarking sets for eight diverse protein targets. Other methods to explore large chemical libraries via molecular docking use a
hierarchical approach38,39, which is fundamentally different from the machine learning techniques. For example, the V-SYNTHES method is based on the same principles as fragment-based ligand
discovery. First, a small set of fragment-sized compounds called synthons, which represent substructures of larger compounds in the multi-billion-scale library, is docked to the binding
site. In a second step, larger molecules that embed top-scoring synthons are docked to identify compounds with improved docking scores. Currently, hierarchical approaches do not require the
use of machine learning because the number of synthons and their corresponding elaborations are small enough to allow for explicit molecular docking. Furthermore, as the synthon library is
not exchangeable with the database of fully enumerated molecules, a conformal predictor trained on synthon docking results is unlikely to accurately predict top-scoring compounds. As the
libraries continue to grow, a comparison of the effectiveness of the hierarchical and machine learning approaches could reveal how these complementary techniques could be combined to further
accelerate virtual screens. A unique aspect of our work is the combination of a powerful machine-learning method with experimental evaluation of predictions, which reveals the potential and
limitations of this approach. Our first screen showed that agonists of D2R, an important drug target for neuropsychiatric and neurodegenerative diseases, could be identified by the
workflow. Interestingly, the hit rate from the docking screen was comparable to previous screens using smaller libraries40. Although screens of large libraries has yielded exceptionally
potent compounds, these results suggest that further progress may be partially limited by the accuracy of molecular docking. As recent studies have shown, flaws in docking scoring functions
may lead to an accumulation of false positives among the top-scoring compounds in large libraries10,41. By reducing the number of compounds requiring explicit docking, our approach enables
resources to be reallocated toward accurate re-scoring of top-ranked compounds using more advanced methods. Despite these challenges, our second prospective screen for multi-target ligands
also illustrates the tremendous opportunities provided by access to larger libraries, which will soon reach the trillion scale. Encouragingly, prospective screens against A2AR and D2R
identified a dual-target ligand, which represents a promising starting point for the development of drugs for the treatment of Parkinson’s disease34. Hence, expanding the sampling across
broader regions of chemical space can enable the discovery of ligands with complex properties that may not be found in smaller libraries. A further extension of our virtual screening
approach could involve the multi-objective design of ligands with specific selectivity, physiochemical and pharmacokinetic properties by integrating conformal predictors trained for
different tasks. Collectively, our results demonstrate how docking screens guided by conformal predictors can accelerate the development of small-molecule therapeutics. METHODS DOCKING
LIBRARY PREPARATION The Enamine REAL Database (November 2019 version, 12.3 billion compounds) was reduced to a rule-of-four chemical subspace by excluding compounds with a molecular weight
over 400 Da and cLog_P_ over 4 using RDKit42. The rule-of-four subspace contained 3,541,746,925 compounds. A random sample containing 15 million compounds (0.4%) from this library was
obtained after shuffling the Simplified Molecular Input Line Entry System (SMILES) with Terashuf43. Molecules were prepared for docking using DOCK3.7 standard protocols44. CXCalc (ChemAxon’s
Marvin package Marvin 18.10.0) was used to calculate predominant protomers at relevant pH levels (6.9, 7.4 and 7.9). Conformational ensembles were generated with OMEGA (OpenEye, version
2020.2) and were capped at 400 conformations and an inter-conformer root mean squared deviation (r.m.s.d.) diversity threshold of 0.25 Å. In-house preparation of these rule-of-four molecules
approximately took 18 CPU-seconds per compound. Preparation of 1 million molecules present in our training set for docking was completed in approximately 5,000 CPU-hours. In the prospective
screens for dual-target compounds, the Enamine REAL Database (November 2022 version, 33.5 billion compounds) was reduced to a subspace of 3,137,276,984 lead-like molecules (ZINC Database
definition: 20 ≤ heavy atom count ≤ 25 and −5 ≤ cLog_P_ ≤ 3.5) using a similar procedure to that described above. The conformational ensembles of lead-like molecules were capped at 200
conformations and an inter-conformer r.m.s.d. diversity threshold of 0.5 Å. A random sample of 1 million rule-of-four WuXi GalaXi molecules (March 2024 version) was prepared using the same
protocol as described above. The WuXi GalaXi rule-of-four space contained 1,371,598,090 compounds. MOLECULAR DESCRIPTORS Canonical SMILES were used to generate three different molecular
descriptors as input data for the machine learning classifiers. Morgan2 descriptors were generated using RDKit29,42. Continuous data-driven descriptors were generated using the CDDD Python
library31. The RoBERTa model generated descriptors directly from the SMILES. We used a pretrained RoBERTa model45 to generate the internal encoded representation of each molecule using the
simpletransformers Python library46. PREPARATION OF PROTEINS FOR DOCKING Experimental structures of the selected protein targets were extracted from the PDB. Details regarding preparation of
crystal structures for molecular docking are provided in Supplementary Table 1. Unless stated otherwise, water molecules and other solutes were removed from the experimental structures. The
N- and C-termini were capped with acetyl and methyl groups, respectively, using PyMOL47. The atoms of the bound ligands were used to generate matching spheres in the binding site. DOCK3.7
uses a flexible ligand algorithm that superimposes rigid segments of a molecule’s pre-calculated conformational ensemble on top of the matching spheres44. Histidine protonation states were
assigned manually after visual inspection of the hydrogen-bonding network. The remainder of the protein structure was protonated by REDUCE48 and assigned AMBER49 united atom charges. The
dipole moments of key residues involved in recognition of the bound ligands were increased to favor interactions with these (Supplementary Fig. 2). This technique is common practice for
users of DOCK3.7 to improve docking performance and has been used in previous virtual screens50. The atoms of the co-crystallized ligands were used to create two sets of sphere layers on the
protein surface (referred to as thin spheres). One set of thin spheres described the low dielectric region of the binding site. A second set of thin spheres was used to calibrate ligand
desolvation penalties. Scoring grids were pre-calculated using QNIFFT51 for Poisson–Boltzmann electrostatic potentials, SOLVMAP52 for ligand desolvation, and CHEMGRID53 for AMBER van der
Waals potentials. For each protein target, known ligands were retrieved from the ChEMBL database or previous studies, followed by generation of property-matched decoys according to the
procedure described in ref. 54. Actives and decoys control sets were docked to the protein structures to evaluate the influence of different docking grid parameters (the radii of
electrostatic and desolvation thin spheres). Finally, ligand enrichment and predicted binding poses were used to select the optimal grid parameters50. HOMOLOGY MODELING OF ACTIVE-STATE D2
DOPAMINE RECEPTOR Alignment of the human D2 and D3 dopamine receptor sequences was performed using the GPCRdb (https://gpcrdb.org/)55. One hundred homology models of the activated D2R bound
to agonist ligand (PD128907) were constructed using MODELLER56 version 10.2 based on a cryo-EM structure of the D3 dopamine receptor complexed with the Gi protein57 (PDB accession code:
7CMV) as a template. Residues 5 to 11 were restrained to form an alpha helix and the residue pairs Cys77–Cys152 and Cys235–Cys237 were set to form disulfide bridges. Molecular docking grids
of homology models were constructed using the same protocols as for the antagonist-bound D2 dopamine receptor crystal structure (PDB accession code 6CM4; Supplementary Table 1). The
resulting docking grids were then evaluated for their ability to reproduce the modeled binding mode of PD128907 in the corresponding homology models and enrich 25 known D2R agonists among a
set of in-house-generated decoys54. A final receptor model was selected based on agonist enrichment calculations and the presence of the conserved salt bridge interaction with Asp1143.32 in
docking screens. MOLECULAR DOCKING CALCULATIONS The orientational sampling parameter was set to 5,000 matches for both the rule-of-four benchmarking set and molecules selected based on
machine learning predictions. Molecules in the ultralarge docking screens (235 million lead-like molecules from the ZINC15 database32) were docked using 1,000 matches. During the generation
of the benchmarking dataset, for each docked compound, 18,652 orientations were calculated on average, and for each orientation, an average of 1,654 conformations were sampled. The
best-scoring pose of each ligand was optimized using a simplex rigid-body minimizer. COMPOUND SELECTION FROM DOCKING SCREENS To bias the multi-billion virtual screen targeting the D2R toward
identification of novel chemotypes, we selected compounds that were dissimilar to known dopamine receptor ligands (>11,000 ChEMBL compounds). The molecular diversity was increased by
clustering the 100,000 top-scoring docked compounds (0.003% of the entire library) by topological similarity. The best-scoring molecule from each cluster was visually inspected for its
complementarity with the binding site and a set of 31 compounds were selected from the 1,000 top-ranking clusters (Supplementary Table 10 and Supplementary Data). The make-on-demand
compounds were available in in the Enamine REAL Database and were successfully synthesized in 4–5 weeks. MACHINE LEARNING-ACCELERATED VIRTUAL SCREENING PIPELINE Our workflow for combining
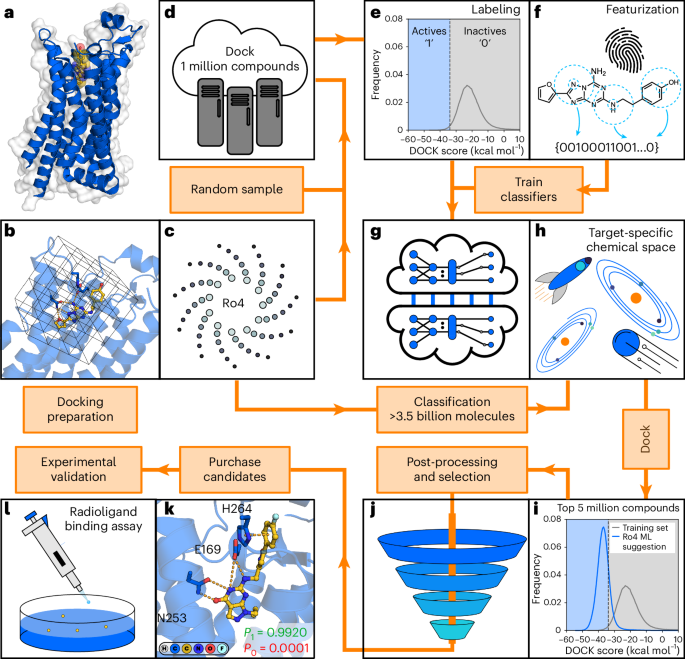
machine learning and molecular docking (Fig. 1) consists of the following consecutive steps, which are described in detail in this section and Supplementary Fig. 1. STEP 1 PREPARATION AND
DOCKING OF THE TRAINING SET A set of randomly selected molecules from an ultralarge chemical library is docked to the target protein structure (Fig. 1a–d). We recommend a training set of 1
million molecules in virtual screens of multi-billion-scale libraries. STEP 2 GENERATION AND LABELING OF THE TRAINING SET A docking score threshold (Fig. 1e) is selected to label each
compound in the training set as either virtual active (better score than the selected threshold) or inactive (equal or worse score than the selected threshold). As our CP approach is based
on aggregating predictions made by several classifiers, multiple independent training sets are generated. Our recommendation is to label the top-scoring 1% of the training set as virtual
active and generate 5 independent training sets. STEP 3 MOLECULE FEATURIZATION AND TRAINING OF THE CLASSIFIER Molecular descriptors of each molecule in the training set are generated as
input for the classifier. Each of the training sets is used to train an independent classification model to distinguish virtual actives from inactives (Fig. 1f,g). STEP 4 CONFORMAL
PREDICTION FOR THE ULTRALARGE LIBRARY The trained classification models are used to evaluate compounds from the ultralarge chemical library (Fig. 1h). Mondrian conformal predictors provide
class-specific confidence levels, which allow compounds to be categorized into one of the following four sets based on a selected significance level (_ε_): virtual active, virtual inactive,
both (virtual active or inactive) and null (no class assignment). The significance level can be tuned to control the size of the virtual active set, which is predicted to contain compounds
with a docking score better than the selected threshold. STEP 5 POST-PROCESSING AND COMPOUND SELECTION The database reduction level achieved by the workflow is target dependent, and
additional post-processing steps can be applied to identify the most promising compounds. The compounds assigned to the virtual active set are ranked by sorting them based on the quality of
information (meaning, prioritizing the predictions in which the classification model has the highest confidence) and a subset of these are docked to the target. Top-scoring molecules are
clustered by chemical similarity and representative compounds are visually inspected (Fig. 1i,j), followed by synthesis and experimental evaluation of selected compounds (Fig. 1k–l). We
recommend docking a set of 1 million to 5 million molecules selected based on the quality of information. TRAINING AND EVALUATION OF MACHINE LEARNING CLASSIFIERS CP is a QSAR method in which
an ensemble of models is used to classify molecules present in an objective set (Supplementary Fig. 1). The docking scores of the datasets were used to label molecules as virtual actives
(top 1%) and virtual inactives (bottom 99%), unless stated otherwise. The scikit-learn 0.24.2 package58 was used to perform a stratified split of the datasets into proper training sets (80%
of training set), calibration sets (20% of training set) and test sets. The ratio between virtual actives and inactives was maintained in all sets. This procedure was repeated using
different random seeds to obtain independent sets. The CatBoost 0.26 Python package was used for building and training the corresponding classifiers. The PyTorch 1.7.1 package59 combined
with the RangerLars optimizer60 was used for training the deep neural networks. The RoBERTa classifier was implemented from the simpletransformers 0.61.6 package46. The Skorch 0.10.0
package61 was used to connect the scikit-learn and PyTorch frameworks. A detailed description and analyses of the hyperparameters used in each classifier are provided in Supplementary Table
2 and Supplementary Figs. 3–5. The compounds in the test set were assigned normalized _P_ values (confidence that the sample belongs to 1 class, _P_1; and 0 class, _P_0) by each individual
classification model and its corresponding calibration set. The resulting sets of _P_1 and _P_0 values were aggregated into a single pair of _P_ values by taking the respective medians of
predictions made by individual models (Supplementary Fig. 1). On the basis of the aggregated _P_ values and the selected significance level (_ε_), the Mondrian CP framework was used to
divide the compounds into virtual active, virtual inactive, both (meaning either virtual active or inactive) and null (no class assignment) sets. Several metrics were used to assess the
performance of the conformal predictors. The sensitivity was defined as: $${{\mathrm{Sensitivity}}}=\frac{{{\mathrm{TP}}}}{{{\mathrm{AP}}}}$$ (1) where TP (true positives) were true active
molecules correctly classified by the CP framework (that is, in the predicted virtual active and both sets). AP (all positives) were all molecules with a score better than the threshold used
to define the virtual actives. The precision was defined as: $${{\mathrm{Precision}}}=\frac{{{\mathrm{TP}}}}{{{\mathrm{TP}}}+{{\mathrm{FP}}}}$$ (2) where FP (false positives) were true
inactive molecules incorrectly classified by the CP framework (that is, in the predicted virtual active and null sets). The efficiency was defined as:
$${{\mathrm{Efficiency}}}=\frac{\left\{1\right\}+\left\{0\right\}}{{{\mathrm{AP}}}+{{\mathrm{AN}}}}$$ (3) where {1} are the predicted virtual actives and {0} the predicted virtual inactives.
AN (all negatives) were all molecules with a score worse than or equal to the threshold used to define the virtual actives. The overall error rate was defined as: $${{\mathrm{Overall}}\,
{\mathrm{error}}\,{\mathrm{rate}}}=\frac{{{\mathrm{FP}}}+{{\mathrm{FN}}}}{{{\mathrm{AP}}}+{{\mathrm{AN}}}}$$ (4) where FN (false negatives) are true virtual active molecules incorrectly
classified by the CP framework (that is, in the predicted virtual inactives and null sets). The error rate for the virtual actives was defined as: $${{\mathrm{Actives}}\, {\mathrm{error}}\,
{\mathrm{rate}}}=\frac{{{\mathrm{FN}}}}{{{\mathrm{AP}}}}$$ (5) The error rate for the virtual inactives was defined as: $${{\mathrm{Inactives}}\, {\mathrm{error}}\,
{\mathrm{rate}}}=\frac{{{\mathrm{FP}}}}{{{\mathrm{AN}}}}$$ (6) COMPUTATIONAL COSTS AND HARDWARE SPECIFICATIONS To train a conformal predictor on 1 million Morgan2 fingerprints, approximately
4 GB of random access memory was required. Training and predicting were performed using 2x Intel Xeon Gold 6130 CPUs @ 2.10 GHz. The RoBERTa models were trained on 12 cores of a single
Nvidia Tesla T4 graphics processing unit with 1,844 GiB memory. The times (in seconds) required to train a conformal predictor on 1 million molecules with different architectures and
descriptors or predict 1 million molecules are reported in Supplementary Table 9. BINDING ASSAYS Screening compounds (Supplementary Tables 10 and 12) were purchased from Enamine (compound
purity >90%, which was confirmed by liquid chromatography−mass spectrometry and 1H NMR spectroscopy for identified ligands; Supplementary Figs. 19–30). Human D2R competition binding
experiments were carried out in polypropylene 96-well plates. Each well contained 20 µg of membranes from a CHO-D2R #S20 cell line (protein concentration of 4,322 µg ml−1), 1.5 nM
[3H]-spiperone (54.3 Ci mmol−1, 1 mCi ml−1, PerkinElmer NET1187250UC) and the compound studied. Non-specific binding was determined in the presence of 10 µM sulpiride (Sigma Aldrich S8010).
The reaction mixture (250 µl per well) was incubated at 25 °C for 120 min, after which 200 µl was transferred to a GF/C 96-well plate (Millipore) pretreated with 0.5% PEI and treated with
binding buffer (50 mM Tris-HCl, 1 mM EDTA, 5 mM MgCl2, 5 mM KCl, 120 mM NaCl, pH 7.4). The reaction mixture was filtered and washed 4 times with 250 µl wash buffer (50 mM Tris-HCl, 0.9%
NaCl, pH 7.4) before the addition of 30 μl Universol. The final measurement was performed in a microplate beta scintillation counter (Microbeta Trilux, PerkinElmer). Human A2AR competition
binding experiments were carried out in a multiscreen GF/C 96-well plate (Millipore) pretreated with binding buffer (Tris-HCl 50 mM, EDTA 1 mM, MgCl2 10 mM, 2 U ml−1 adenosine deaminase, pH
7.4). Each well was incubated with 5 μg of membranes from the HeLa-A2A cell line and prepared in our laboratory (lot A003/14-04-2019, protein concentration 2,058 μg ml−1), 1 nM [3H]-ZM241385
(50 Ci mmol−1, 1 mCi ml−1, ARC-ITISA 0884), and the compounds studied and standards. Non-specific binding was determined in the presence of NECA 50 μM (Sigma E2387). The reaction mixture
(total volume of 200 μl per well) was incubated at 25 °C for 30 min, then filtered and washed 4 times with 250 μl wash buffer (Tris-HCl 50 mM, EDTA 1 mM, MgCl2 10 mM, pH 7.4), and measured
in a microplate beta scintillation counter (Microbeta Trilux, PerkinElmer). Unless stated otherwise, three independent experiments were carried out to calculate _K_i values. FUNCTIONAL
ASSAYS Human D2R activity was measured by determining the amount of cAMP produced. Human D2R functional experiments were carried out in a CHO-D2 #S20 cell line. Five-thousand cells were
seeded in 30 μl of Optimem (Invitrogen 11058) + 500 μM IBMX (Sigma 17018) on a 96-well black-and-white isoplate (PerkinElmer 6005030). Test compounds and dopamine were added in their
corresponding wells and incubated for 10 min at 37 °C with gentle stirring (150 r.p.m.). Then, 10 µM forskolin (Sigma 17018) was added and incubated for 5 min at 37 °C with gentle stirring
(150 r.p.m.). Reagents from the kit (#CISBIO 62AM4PEC) were added and incubated for 1 h at room temperature with gentle stirring (90 r.p.m.) and protected from light. HTRF (excitation
wavelength: 320 nm; emission wavelengths: 620–665 nm) from each well was measured using a Tecan Infinite M1000 Pro. Two independent experiments were carried out to calculate EC50 and Emax
values. STATISTICS AND REPRODUCIBILITY Compounds for which no molecular descriptors could be generated were excluded from the analyses. No statistical method was used to predetermine the
sample size. Training and test sets were generated by taking random samples with the determined size of the virtual libraries. Proper training and calibration sets were constructed by
performing a stratified split on the parent training sets, maintaining the ratio of samples belong to the minority and majority classes. Unless stated otherwise, three independent
calculations (training and predictions) were performed to derive statistics on model performance. Unless stated otherwise, three independent experiments were performed to derive statistics
on the activities of the designed compounds. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA
AVAILABILITY The ZINC15 and Enamine REAL databases are available at https://zinc15.docking.org and https://enamine.net/compound-collections/real-compounds/real-database, respectively. The
PDB accession codes for the molecular docking calculations are 4EIY, 6DPT, 6XUE, 6CM4, 5FNU, 6W63, 6G3Y, 6X48, 8GNE and 7CMV. Associated source data are provided for Figs. 2–5. All compounds
tested are listed in the Supplementary Tables 10 and 12, and Supplementary Data. Chemical identities, purities (liquid chromatography−mass spectrometry), yields and spectroscopic analysis
(1H NMR) for active compounds are provided in the Supplementary figures. Large-scale docking datasets are deposited on Zenodo at https://doi.org/10.5281/zenodo.7953917 (ref. 62). CODE
AVAILABILITY The conformal predictor source code is freely available and can be found on the GitHub at https://github.com/carlssonlab/conformalpredictor. The original code is deposited on
Zenodo at https://doi.org/10.5281/zenodo.14709041 (ref. 63). REFERENCES * Bohacek, R. S., McMartin, C. & Guida, W. C. The art and practice of structure-based drug design: a molecular
modeling perspective. _Med. Res. Rev._ 16, 3–50 (1996). Article Google Scholar * Irwin, J. J. et al. ZINC20—a free ultralarge-scale chemical database for ligand discovery. _J. Chem. Inf.
Model._ 60, 6065–6073 (2020). Article Google Scholar * Grygorenko, O. O. et al. Generating multibillion chemical space of readily accessible screening compounds. _iScience_ 23, 101681
(2020). Article Google Scholar * Bellmann, L., Klein, R. & Rarey, M. Calculating and optimizing physicochemical property distributions of large combinatorial fragment spaces. _J. Chem.
Inf. Model._ 62, 2800–2810 (2022). Article Google Scholar * Lyu, J. et al. Ultra-large library docking for discovering new chemotypes. _Nature_ 566, 224–229 (2019). Article Google
Scholar * Luttens, A. et al. Ultralarge virtual screening identifies SARS-CoV-2 main protease inhibitors with broad-spectrum activity against coronaviruses. _J. Am. Chem. Soc._ 144,
2905–2920 (2022). Article Google Scholar * Carlsson, J. & Luttens, A. Structure-based virtual screening of vast chemical space as a starting point for drug discovery. _Curr. Opin.
Struct. Biol._ 87, 102829 (2024). Article Google Scholar * Gorgulla, C. et al. An open-source drug discovery platform enables ultra-large virtual screens. _Nature_ 580, 663–668 (2020).
Article Google Scholar * Fink, E. A. et al. Large library docking for novel SARS-CoV-2 main protease non-covalent and covalent inhibitors. _Protein Sci._ 32, e4712 (2023). Article Google
Scholar * Lyu, J., Irwin, J. J. & Shoichet, B. K. Modeling the expansion of virtual screening libraries. _Nat. Chem. Biol._ 19, 712–718 (2023). Article Google Scholar * Muratov, E. N.
et al. QSAR without borders. _Chem. Soc. Rev._ 49, 3525–3564 (2020). Article Google Scholar * Zhang, J., Norinder, U. & Svensson, F. Deep learning-based conformal prediction of
toxicity. _J. Chem. Inf. Model._ 61, 2648–2657 (2021). 2021. Article Google Scholar * Yang, Y. et al. Efficient exploration of chemical space with docking and deep learning. _J. Chem.
Theory Comput._ 17, 7106–7119 (2021). Article Google Scholar * Gentile, F. et al. Deep docking: a deep learning platform for augmentation of structure based drug discovery. _ACS Cent.
Sci._ 6, 939–949 (2020). Article Google Scholar * Graff, D. E. et al. Self-focusing virtual screening with active design space pruning. _J. Chem. Inf. Model._ 62, 3854–3962 (2022). Article
Google Scholar * Vovk, V., Gammerman, A. & Shafer, G. _Algorithmic Learning in a Random World_ (Springer Nature, 2005); https://doi.org/10.1007/b106715 * Norinder, U., Carlsson, L.,
Boyer, S. & Eklund, M. Introducing conformal prediction in predictive modeling. A transparent and flexible alternative to applicability domain determination. _J. Chem. Inf. Model._ 54,
1596–1603 (2014). Article Google Scholar * Norinder, U. & Boyer, S. Binary classification of imbalanced datasets using conformal prediction. _J. Mol. Graph. Model._ 72, 256–265 (2017).
Article Google Scholar * Norinder, U., Spjuth, O. & Svensson, F. Synergy conformal prediction applied to large-scale bioactivity datasets and in federated learning. _J. Cheminform._
13, 77 (2021). Article Google Scholar * Svensson, F., Norinder, U. & Bender, A. Improving screening efficiency through iterative screening using docking and conformal prediction. _J.
Chem. Inf. Model._ 57, 439–444 (2017). Article Google Scholar * Ahmed, L. et al. Efficient iterative virtual screening with Apache Spark and conformal prediction. _J. Cheminform._ 10, 8
(2018). Article Google Scholar * Lavecchia, A. Machine-learning approaches in drug discovery: methods and applications. _Drug Discov. Today_ 20, 318–331 (2015). Article Google Scholar *
Sadybekov, A. V. & Katritch, V. Computational approaches streamlining drug discovery. _Nature_ 616, 673–685 (2023). Article Google Scholar * Vamathevan, J. et al. Applications of
machine learning in drug discovery and development. _Nat. Rev. Drug Discov._ 18, 463–477 (2019). Article Google Scholar * Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schiöth, H. B.
& Gloriam, D. E. Trends in GPCR drug discovery: new agents, targets and indications. _Nat. Rev. Drug Discov._ 16, 829–842 (2017). Article Google Scholar * Prokhorenkova, L. et al.
CatBoost: unbiased boosting with categorical features. Preprint at https://arxiv.org/abs/1706.09516 (2013). * Chen, H. et al. The rise of deep learning in drug discovery. _Drug Discov.
Today_ 23, 1241–1250 (2018). Article Google Scholar * Liu, Y. et al. RoBERTa: a robustly optimized BERT pretraining approach. Preprint at https://arxiv.org/abs/1907.11692 (2019). * Rogers,
D. & Hahn, M. Extended-connectivity fingerprints. _J. Chem. Inf. Model._ 50, 742–754 (2010). Article Google Scholar * Riniker, S. & Landrum, G. A. Open-source platform to
benchmark fingerprints for ligand-based virtual screening. _J. Cheminform._ 5, 26 (2013). Article Google Scholar * Winter, R. et al. Learning continuous and data-driven molecular
descriptors by translating equivalent chemical representations. _Chem. Sci._ 10, 1692–1701 (2019). Article Google Scholar * Sterling, T. & Irwin, J. J. ZINC 15—ligand discovery for
everyone. _J. Chem. Inf. Model._ 55, 2324–2337 (2015). Article Google Scholar * Gaulton, A. et al. ChEMBL: a large-scale bioactivity database for drug discovery. _Nucleic Acids Res._ 40,
1100–1107 (2012). Article Google Scholar * Roth, B. L., Sheffler D. J. & Kroeze W. K. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and
schizophrenia. _Nat. Rev. Drug Discov._ 3, 353–359 (2004). Article Google Scholar * Kampen, S. et al. Structure-guided design of G-protein-coupled receptor polypharmacology. _Angew. Chem.
Int. Ed._ 60, 18022–18030 (2021). Article Google Scholar * Ballesteros, J. A. & Weinstein, H. Integrated methods for the construction of three-dimensional models and computational
probing of structure-function relations in G protein-coupled receptors. _Neurosci. Methods_ 25, 366–428 (1995). Article Google Scholar * Marin, E. et al. Regression-based active learning
for accessible acceleration of ultra-large library docking. _J. Chem. Inf. Model._ 64, 2612–2623 (2024). Article Google Scholar * Sadybekov, A. A. et al. Synthon-based ligand discovery in
virtual libraries of over 11 billion compounds. _Nature_ 601, 452–459 (2022). Article Google Scholar * Beroza, P. et al. Chemical space docking enables large-scale structure-based virtual
screening to discover ROCK1 kinase inhibitors. _Nat. Commun._ 13, 6447 (2022). Article Google Scholar * Ballante, F., Kooistra, A. J., Kampen, S., de Graaf, C. & Carlsson, J.
Structure-based virtual screening for ligands of G protein-coupled receptors: what can molecular docking do for you? _Pharmacol. Rev._ 73, 527–565 (2021). Article Google Scholar * Wu, Y.
et al. Identifying artifacts from large library docking. _J. Med. Chem._ 67, 16796–16806 (2024). Article Google Scholar * RDKit Open-Source Cheminformatics Software (2025). * Salle, A.
Terashuf: A fast external memory shuffling tool. _GitHub_ https://github.com/alexandres/terashuf (2025). * Coleman, R. G. et al. Ligand pose and orientational sampling in molecular docking.
_PLoS ONE_ 8, e7599 (2019). Google Scholar * Chithrananda, S., Grand, G. & Ramsundar, B. ChemBERTa: large-scale self-supervised pretraining for molecular property prediction. Preprint
at https://arxiv.org/abs/2010.09885 (2020). * Rajapakse, T. SimpleTransformers. _GitHub_ https://github.com/ThilinaRajapakse/simpletransformers (2025). * Schrödinger, L. & DeLano, W.
PyMOL (2020); https://www.pymol.org/pymol * Word, J. M., Lovell, S. C., Richardson, J. S. & Richardson, D. C. Asparagine and glutamine: using hydrogen atom contacts in the choice of
side-chain amide orientation. _J. Mol. Biol._ 285, 1735–1747 (1999). Article Google Scholar * Weiner, S. J. et al. A new force field for molecular mechanical simulation of nucleic acids
and proteins. _J. Am. Chem. Soc._ 106, 765–784 (1984). Article Google Scholar * Bender, B. J. et al. A practical guide to large-scale docking. _Nat. Protoc._ 16, 4799–4832 (2021). Article
Google Scholar * Gallagher, K. & Sharp, K. Electrostatic contributions to heat capacity changes of DNA–ligand binding. _Biophys. J._ 75, 769–776 (1998). Article Google Scholar *
Mysinger, M. M. & Shoichet, B. K. Rapid context-dependent ligand desolvation in molecular docking. _J. Chem. Inf. Model._ 50, 1561–1573 (2010). Article Google Scholar * Meng, E. C.,
Shoichet, B. K. & Kuntz, I. D. Automated docking with grid-based energy evaluation. _J. Comput. Chem._ 13, 505–524 (1992). Article Google Scholar * Mysinger, M. M., Carchia, M., Irwin,
J. J. & Shoichet, B. K. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. _J. Med. Chem._ 55, 6582–6594 (2012). Article Google Scholar *
Pándy-Szekeres, G. et al. GPCRdb in 2023: state-specific structure models using AlphaFold2 and new ligand resources. _Nucleic Acids Res._ 51, 395–402 (2023). Article Google Scholar *
Webb, B. & Sali, A. Comparative protein structure modeling using MODELLER. _Curr. Protoc. Protein Sci._ 86, 2.9.1–2.9.37 (2016). Article Google Scholar * Xu, P. et al. Structures of
the human dopamine D3 receptor–Gi complexes. _Mol. Cell_ 81, 1147–1159.e4 (2021). Article Google Scholar * Pedregosa, F. et al. Scikit-learn: machine learning in Python. _J. Mach. Learn.
Res._ 12, 2825–2830 (2011). MathSciNet Google Scholar * Paszke, A. et al. PyTorch: an imperative style, high-performance deep learning library. _Adv. Neural Inf. Process. Syst_. 32,
(2019). * Grankin, M. RangerLars Optimizer. _GitHub_ https://www.github.com/mgrankin/over9000 (2025). * Tietz, M., Fan, T. J., Nouri, D., & Bossan, B. skorch: A scikit-learn compatible
neural network library that wraps PyTorch. _Skorch Developers_ https://skorch.readthedocs.io (2017). * Luttens, A., Cabeza de Vaca, I., Sparring, L., Norinder, U. & Carlsson, J.
Large-scale docking datasets for machine learning. _Zenodo_ https://doi.org/10.5281/zenodo.7953917 (2023). * Luttens, A. Conformal predictor. _Zenodo_ https://doi.org/10.5281/zenodo.14709041
(2025). Download references ACKNOWLEDGEMENTS A.L. was supported by a postdoctoral scholarship from the Knut and Alice Wallenberg Foundation (KAW2022.0347). J.C. received funding from the
European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement 715052), the Swedish Cancer Society, the Swedish Research Council and
the Olle Engkvist Foundation. This research was partially supported by the project AI4Research at Uppsala University. I.C.d.V. was funded by a postdoctoral fellowship provided by the Sven
och Lilly Lawski foundation. The computations were enabled using resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) (partially funded by the
Swedish Research Council through grant agreement number 2022-06725) and the supercomputing resource Berzelius provided by the National Supercomputer Centre at Linköping University and the
Knut and Alice Wallenberg Foundation. J.B., A.L.M. and M.I.L. were funded by Agencia Estatal de Investigación (PID2020-119428RB-I00), Xunta de Galicia (ED431C 2022/20) and European Regional
Development Fund (ERDF). A.L., I.C.d.V. and J.C. thank OpenEye Scientific Software for the use of OEToolkits at no cost. We thank J. Zhang for providing the initial deep neural network code.
FUNDING Open access funding provided by Uppsala University. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Science for Life Laboratory, Department of Cell and Molecular Biology, Uppsala
University, BMC, Uppsala, Sweden Andreas Luttens, Israel Cabeza de Vaca, Leonard Sparring, Nour Aldin Kahlous & Jens Carlsson * Infectious Disease and Microbiome Program, Broad Institute
of MIT and Harvard, Cambridge, MA, USA Andreas Luttens * Institute for Medical Engineering and Science and Department of Biological Engineering, Massachusetts Institute of Technology,
Cambridge, MA, USA Andreas Luttens * Innopharma Drug Screening and Pharmacogenomics Platform, BioFarma research group, Center for Research in Molecular Medicine and Chronic Diseases (CiMUS),
Department of Pharmacology, Pharmacy and Pharmaceutical Technology, University of Santiago de Compostela, Santiago de Compostela, Spain José Brea, Antón Leandro Martínez & María Isabel
Loza * Health Research Institute of Santiago de Compostela, Santiago de Compostela, Spain José Brea, Antón Leandro Martínez & María Isabel Loza * Enamine Ltd, Kyiv, Ukraine Dmytro S.
Radchenko & Yurii S. Moroz * Taras Shevchenko National University of Kyiv, Kyiv, Ukraine Yurii S. Moroz * Chemspace LLC, Kyiv, Ukraine Yurii S. Moroz * Department of Pharmaceutical
Biosciences, Uppsala University, Uppsala, Sweden Ulf Norinder Authors * Andreas Luttens View author publications You can also search for this author inPubMed Google Scholar * Israel Cabeza
de Vaca View author publications You can also search for this author inPubMed Google Scholar * Leonard Sparring View author publications You can also search for this author inPubMed Google
Scholar * José Brea View author publications You can also search for this author inPubMed Google Scholar * Antón Leandro Martínez View author publications You can also search for this author
inPubMed Google Scholar * Nour Aldin Kahlous View author publications You can also search for this author inPubMed Google Scholar * Dmytro S. Radchenko View author publications You can also
search for this author inPubMed Google Scholar * Yurii S. Moroz View author publications You can also search for this author inPubMed Google Scholar * María Isabel Loza View author
publications You can also search for this author inPubMed Google Scholar * Ulf Norinder View author publications You can also search for this author inPubMed Google Scholar * Jens Carlsson
View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS A.L., U.N. and J.C. designed the study. A.L. performed the molecular docking, homology
modeling and machine learning calculations and selected compounds under the supervision of J.C. and U.N. A.L., I.C.d.V., L.S. and U.N. developed the protocol. A.L. and I.C.d.V. wrote the
final version of the code. N.A.K. performed sequence alignments for homology modeling, constructed the D2R homology models and provided the D2R agonist set. The binding and functional assays
were performed by the USEF screening platform under the supervision of J.B., A.L.M. and M.I.L. D.S.R. and Y.S.M. provided the Enamine REAL Database and analytical data for the synthesized
compounds. U.N. provided support with critical evaluation of the machine learning protocol. A.L., I.C.d.V. and J.C. wrote the paper with contributions from the other authors. CORRESPONDING
AUTHORS Correspondence to Andreas Luttens, María Isabel Loza, Ulf Norinder or Jens Carlsson. ETHICS DECLARATIONS COMPETING INTERESTS J.C. is a founder of DareMe Drug Discovery Consulting.
D.S.R. and Y.S.M. are employed by Enamine Ltd. Y.S.M. serves as a scientific advisor to Chemspace LLC. The other authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION
_Nature Computational Science_ thanks Mayukh Chakrabarti, Matthew O’Meara and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports
are available. Primary Handling Editor: Kaitlin McCardle, in collaboration with the _Nature Computational Science_ team. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains
neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION Supplementary Sections 1 and 2, Tables
1–14, Figs. 1–30, methods and References. REPORTING SUMMARY PEER REVIEW FILE SUPPLEMENTARY DATA Experimentally validated molecules. SMILES, identifiers and experimental activities. SOURCE
DATA SOURCE DATA FIG. 2 Source data for Fig. 2a–f. SOURCE DATA FIG. 3 Source data for Fig. 3a–c, e and f. SOURCE DATA FIG. 4 Source data for Fig. 4a. SOURCE DATA FIG. 5 Source data for Fig.
5b and c. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and
reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if
changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the
material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to
obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS
ARTICLE Luttens, A., Cabeza de Vaca, I., Sparring, L. _et al._ Rapid traversal of vast chemical space using machine learning-guided docking screens. _Nat Comput Sci_ 5, 301–312 (2025).
https://doi.org/10.1038/s43588-025-00777-x Download citation * Received: 05 June 2024 * Accepted: 04 February 2025 * Published: 13 March 2025 * Issue Date: April 2025 * DOI:
https://doi.org/10.1038/s43588-025-00777-x SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative